

Abstract
Objective/Hypothesis
Laryngopharyngeal reflux (LPR) is thought to be a significant risk factor for laryngeal squamous cell carcinoma (SCC), but causality has never been proven. It is accepted that chronic reflux into the esophagus can induce metaplastic changes in esophageal mucosa with subsequent increased risk of esophageal adenocarcinoma, but no similar associations have been established for LPR and laryngopharyngeal SCC. The objective of this study was to test the hypothesis that reflux of pepsin into the laryngopharynx can promote carcinogenesis.
Study Design
Translational research study
Methods
Normal human laryngeal primary epithelial cell cultures and hypopharyngeal FaDu SCC cells were exposed to human pepsin and analyzed by Human Cancer PathwayFinder and miRNA Superarrays, flow cytometry and Western blot to determine the effect of pepsin on carcinogenesis. Laryngeal biopsy specimens, taken from cancer patients and normal control subjects, were analyzed for the presence of pepsin by Western blot.
Results
Microarray analysis demonstrated that pepsin significantly altered the expression of 27 genes implicated in carcinogenesis and also affected the expression of 22 microRNAs known to be altered in human head and neck cancers. Pepsin increased proliferation in both FaDu SCC cells and cultured normal laryngeal epithelial primary cells by increasing S phase distribution on flow cytometry analysis in a time and dose dependent manner. Furthermore, pepsin was detected in 60% (3/5) human laryngeal cancer biopsies, absent in all (0/5) normal control specimens.
Conclusion
These data support a role for refluxed pepsin in the promotion of epithelial proliferation and carcinogenesis of the larynx and pharynx.
Keywords: Pepsin, laryngopharynx, reflux, laryngopharyngeal reflux, LPR, proliferation, carcinogenesis, squamous cell carcinoma
INTRODUCTION
Laryngeal carcinoma accounts for about 1% of all newly diagnosed cancers in the United States. Approximately, 11,000 new cases are diagnosed every year and about 4,300 deaths per year are attributed to laryngeal carcinoma. Despite a decrease in the number of people who smoke in the United States, the incidence of laryngeal cancer actually appears to be rising. Unfortunately, the prognosis remains poor and the mortality rate high, with a 5-year survival rate of 40% 1-3. Tobacco and alcohol are well known established risk factors3. Other risk factors include human papilloma virus, radiation exposure, occupational chemical exposures and laryngopharyngeal reflux (LPR)4. The latter remains controversial and requires further investigation, especially since it has become one of the most common chronic diseases of adults in the United States. For many reasons, it is very difficult to prove that reflux is a causal agent in the development of laryngeal cancer. Many clinical studies have shown a high prevalence of LPR in patients with laryngeal cancer4,5, however, these studies are confounded by the fact that the majority of patients with laryngeal cancer have a significant smoking and alcohol history, and many lack appropriate controls. Another difficulty is the lack of uniformity in establishing the diagnosis of LPR in the literature.
It remains unclear whether reflux laryngitis is a precursor to laryngeal cancer despite years of research into this topic, as population and other clinical studies often have too many confounding variables to definitively prove an association. Gabriel and Jones were among the first to present evidence suggesting this possibility6. Many others have also suggested an association4,7-10. To further explore the association between LPR and laryngeal cancer, several investigators have examined the direct effect of the individual components of gastric refluxate – mainly acid, pepsin and bile acids – on laryngeal cell biology 4,11,12. These studies demonstrated a significant role for pepsin and bile acids in carcinogenesis, in a dose-dependent manner with greater toxicity at lower pH. Interestingly, several clinical studies evaluating patients with prior gastrectomy suggest that the components of non-acidic reflux promote the development of laryngeal cancer13-15. We have shown that pepsin, 0.1mg/ml at pH7 (and thus in non-acidic reflux) causes mitochondrial damage in vitro, induces the expression of multiple genes implicated in stress and toxicity, and induces a proinflammatory cytokine and receptor gene expression profile similar to that observed in patients with GERD16-18. Furthermore, using the hamster buccal pouch model of squamous cell carcinoma, we recently reported that exposure to a known carcinogen - 7, 12 dimethylbenzanthracene (DMBA) - plus active pepsin (pH4) results in a significant increase in tumor volume compare to DMBA control19. These data have established a role for refluxed pepsin in the promotion of laryngeal inflammation and cancer. The objective of this study was to specifically investigate the effect that non-acid pepsin has on the growth and proliferation of both normal and neoplastic laryngeal tissues.
MATERIALS AND METHODS
Human Biopsy Specimens
This study was approved by the Medical College of Wisconsin Institutional Review Board. Normal laryngeal tissue specimens (n = 5) were obtained from the postcricoid larynx in patients who had no clinical signs or symptoms of LPR. Signs of LPR were assessed by means of the reflux finding score (RFS)20, a short standardized clinical index to assess laryngeal reflux severity. Symptom severity was assessed by means of the reflux symptom index (RSI) questionnaire21. Patients with no inflammatory or neoplastic disease and a RSI score of 8 or below and RFS score 4 or below were considered normal. Tissue biopsy specimens were also obtained from the posterior cricoid epithelium and disease site of patients with cancers of the laryngopharynx (n = 5) Tissue biopsy specimens were snap frozen in liquid nitrogen until analysis.
Western Blot Analysis
Proteins were extracted from biopsy specimens and cultured cells using urea lysis buffer and protein content was measured by Bradford assay. 20-30μg total protein was loaded on 10% SDS-PAGE gels according to standard SDS-PAGE protocol. Purified human pepsin 3b (isolated from human gastric juice by ion exchange chromatography22) and human pepsinogen I (Sigma, St. Louis, MO) were run alongside clinical samples as positive and negative controls, respectively. Proteins were transferred to PVDF membrane (GE Healthcare, Piscataway, NJ) and probed with rabbit anti-pepsin antibody (1:350)23, rabbit anti-Ras antibody (1:25; USBiological, TX), or mouse anti-b-actin antibody (1:5000, EMD Chemicals, Gibbstown, NJ). Blots were then probed with appropriate peroxidase-conjugated secondary antibody diluted 1:5,000 (Dako, Copenhagen, Denmark). Blots were exposed to enhanced chemiluminescence reagents (Santa Cruz Biotechnology, Santa Cruz, CA) followed by radiographic exposure and development.
RNA Processing and Polymerase Chain Reaction
Human gastric and laryngeal cDNA was reverse transcribed from 200ng-1ug DNase-treated RNA and PCR amplified using primers for human pepsinogen A (forward: ACCGTGGACAGCATCACCATG, reverse: TCTTCCTGGGAGGTGGCTG, 30 cycles, 62°C annealing). The housekeeping gene, hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1), was amplified as a positive control (forward: TGCTCGAGATGTGATGAAGG, reverse: CCTGACCAAGGAAAGCAAAG, 35 cycles, 55°C annealing). Amplicon was separated on 2% agarose.
Cell Culture
Human hypopharyngeal squamous cell carcinoma FaDu cells (ATCC, Manassas, VA) were grown in Minimum Essential Medium – Eagle with Earle’s Balanced Salt adjusted to 1.5g/L sodium bicarbonate containing 0.1mM non-essential amino acids, 1.0mM sodium pyruvate, and 10% Fetal Bovine Serum (ATCC, Manassas, VA) to a density of 70% confluence.
Tissue biopsies for epithelial cultures were harvested from the posterior cricoid of volunteer control subjects. Biopsies were immersed briefly in 70% ethanol, sliced finely with a scalpel blade and incubated in 0.25% bovine pancreas trypsin (Sigma-Aldrich, St. Louis, MO) in PBS containing 2% penicillin-streptomycin (Invitrogen, Carlsbad, CA) at room temperature with gentle agitation for 1 hour, then at 37°C for 45 minutes. Biopsy was triturated to further homogenize sample and trypsin was neutralized with an equal volume of Dulbecco’s Modified Eagle Medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 2% GlutaMax (Invitrogen, Carlsbad, CA), 2% penicillin-streptomycin, and 1%Fungizone (Invitrogen, Carlsbad, CA). Cells were centrifuged for 5 minutes at 1,000×g, resuspended in Keratinocyte Serum-Free Media (Invitrogen, Carlsbad, CA) supplemented with 25 ug/mL bovine pituitary extract, 5ng/mL epidermal growth factor, 1% Fungizone, and 1% penicillin-streptomycin, and placed in a collagen-coated T25 BioCoat flask (Benton Dickson, Bedford, MA). Cells were incubated at 37°C with 5% CO2 for three days. Media was replaced every three days thereafter. Upon confluency, cells were trypsinized using TrypleExpress (Invitrogen, Carlsbad, CA), diluted in twice the volume of media, centrifuged for 5 minutes at 1,000×g, and placed into new flasks. Primary laryngeal epithelial cells were characterized as in Rees et al. using light microscopy and immunohistochemistry24.
Immunofluorescence
Primary laryngeal epithelial cells were spun down onto cytoslides (Shandon Cytospin III) and blocked for 30 minutes with serum free protein block (DAKO, CA). Primary monoclonal antibodies to cytokeratins 5 and 8 (Sigma, MO) were incubated for 1 hour at room temperature. Cytokeratins were detected using a goat anti-mouse Alexa fluor-488 (45 minute incubation). Slides were washed and mounted with Vectashield Hard set with DAPI counterstain (Vector Labortatories, CA). Cells were viewed on a Zeiss Z1 microscope with a AxioCam (Zeiss, Germany).
Flow Cytometry Assays
Normal primary laryngeal and hypopharyngeal SCC FaDu cells were exposed to porcine pepsin (0.01, 0.1 or 1mg/ml; Sigma-Aldrich, St. Louis, MO) at pH7 for 1 hour at 37°C, washed 3 times briefly in fresh media and incubated for a further 4, 24 or 36 hours at 37°C in complete growth media. To measure the effect of irreversibly inactivated pepsin on cell proliferation, pepsin was irreversibly inactivated by prior incubation at pH8 for 15 minutes at 37°C before decreasing the pH to 7 and incubating with cells. Cells were either fixed in 70% ethanol and incubated with propidium iodide/Triton X-100 staining solution with RNase A (0.5ml PBS/BSA + 100U/ml RNase A + 0.5mg/ml propidium iodide; Sigma, MO) and analyzed by flow cytometry 25, or, assessed for cell cycle distribution using the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions.
Human Cancer PathwayFinder and miRNA SuperArrays
RNA was isolated as directed by the SuperArray RT2-qPCR-Grade RNA Isolation Kit (SuperArray, Frederick, MD) and the concentration and quality were assessed by UV spectroscopy and agarose gel electrophoresis. RNA was reverse transcribed using the SuperArray RT2 First Strand Kit. FaDu cell cDNA was diluted in RT2 SYBR Green/Fluorescein qPCR Master Mix and aliquoted into a 96-well RT2 Profiler PCR Array (Human Cancer PathwayFinder and miFinder ™ PCR Arrays, SuperArray, Frederick, MD). Real-time RT-PCR was performed in an iCycleriQ Multicolor Real Time PCR Detection System (Bio-Rad Life Science, Hercules, CA). Real-time PCR conditions were as follows: 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds, 60°C for 1 minute, immediately followed by a melt curve of 95°C for 1 minute, 65°C for 2 minutes, and sixty 10 second cycles of 0.5°C increase. Three biological replicates for each condition were performed.
Statistical Analyses
Proliferation Assays
Data from 5 biological replicates for dose response experiments were analyzed by one-way analysis of variance and Tukey’s multiple comparisons post test. Data from 5 biological replicates for time response experiments were analyzed by two-way analysis of variance and Bonferroni multiple comparisons post test. Data are expressed as mean ± standard deviation.
Human Cancer PathwayFinder and miRNA Arrays
Samples exhibiting more than one peak within the melt curve or Ct > 35 were excluded from the analysis. The means of the housekeeping genes for each replicate was subtracted from each gene value to normalize the data. Differential expression was evaluated by fitting a mixed model to the entire date set with fixed gene and gene-treatment interaction effects, and random intercept and treatment effect for each biological replicate. The p-values for the gene specific treatment effects were adjusted using the Benjamin-Hochberg procedure with a 5% cutoff.
RESULTS
Cultured hypopharyngeal FaDu cells were incubated with pepsin (0.1mg/ml) at pH7for 1 hour and analyzed by Cancer PathwayFinder SuperArray (Bioscience Corporation, MD). Of the 84 cancer-related genes analyzed, two were excluded from the analysis due to Ct > 35. The expression of three genes was increased by > 1.5 fold (p<0.05) in FaDu cells treated with pepsin at pH7 compared to control cells (Table 1A). The expression of 24 genes was reduced > 1.5 fold (p<0.05) in cells exposed to pepsin relative to control cells (Table 1B). Pepsin exposure generally induced genes consistent with an increased cellular proliferative state.
Table 1A.
Genes increased by > 1.5 fold (p<0.05) in FaDu cells treated with 0.1mg/ml pepsin at pH7 for 1 hour at 37∙C relative to control cells
| Gene | Symbol | Fold Change |
p-Value | Function |
|---|---|---|---|---|
| CASP8 and FADD-like apoptosis regulator |
CFLAR | 2.89 | <0.0001 | Apoptosis regulator protein; inhibitor of TNFRSF6 mediated apoptosis |
| V-Ets erythroblastosis virus E26 oncogene homolog 2 (avian) |
ETS2 | 1.87 | 0.0137 | Regulates genes involved in cell death and tunorigenesis. May be an important transcription factor for driving inflammation in acute as well as chronic inflammatory disease |
| Interleukin 8 | IL8 | 3.82 | <0.0001 | Chemokine; key parameter in localized inflammation |
Table 1B.
Genes reduced by > 1.5 fold (p<0.05) in FaDu cells treated with 0.1mg/ml pepsin at pH7 for 1 hour at 37∙C relative to control cells.
| Gene | Symbol | Fold Change |
Value | Function |
|---|---|---|---|---|
| V-akt murine thymoma viral oncogene homolog 1 |
AKT1 | 0.48 | 0.0034 | Regulates cell growth and survival |
| Ataxia telangiectasia mutated | ATM | 0.46 | 0.0017 | Activation of DNA damage checkpoint |
| BCL2-associated agonist of cell death | BAD | 0.35 | <0.0001 | Positively regulates cell apoptosis* |
| BCL2-associated X protein | BAX | 0.52 | 0.01 | Regulates apoptosis; can promote and inhibit apoptosis* |
| B-cell CLL/lymphoma 2 | BCL2 | 0.27 | 0.0038 | Regulates apoptosis; can promote and inhibit apoptosis* |
| BCL2-like 1 | BCL2L1 | 0.47 | 0.0023 | Regulates apoptosis; can promote and inhibit apoptosis* |
| Breast cancer 1, early onset | BRCA1 | 0.29 | <0.0001 | Tumor suppressor; DNA damage repair |
| Cyclin E1 | CCNE1 | 0.33 | <0.0001 | Cell cycle control; DNA damage checkpoint |
| Cell division cycle 25 homolog A (S. pombe) |
CDC25A | 0.46 | 0.0018 | Cell cycle control; DNA damage checkpoint |
| Cyclin-dependent kinase 4 | CDK4 | 0.32 | <0.0001 | Cell cycle control; tumorigenesis |
| Collagen, type XVIII, alpha 1 | COL18A1 | 0.38 | 0.0001 | Inhibits angiogenesis |
| E2F transcription factor | E2F1 | 0.45 | 0.0012 | Cell cycle control; tumor suppressor |
| V-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian) |
ERBB2 | 0.42 | 0.0005 | Involved in signal transduction leading to cell growth |
| Fibroblast growth factor receptor 2 | FGFR2 | 0.29 | <0.0001 | Mitogenesis and differentiation |
| Integrin alpha 1 | ITGA1 | 0.55 | 0.0159 | Cell-cell adhesion; may play a role in inflammation |
| Matrix metallopeptidase 1 | MMP1 | 0.50 | 0.0058 | Breakdown of extracellular matrix; metastasis |
| Metastasis associated 1 | MTA1 | 0.41 | <0.0001 | Regulates transcription; invasion/metastasis |
| Metastasis associated 1 family, member 2 |
MTA2 | 0.47 | 0.0023 | Regulates transcription; inhibits p53 |
| Non-metastatic cells 1, protein (NM23A) expressed in |
NME1 | 0.50 | 0.0055 | Metastasis suppressor |
| Phosphoinositide-3-kinase, regulatory subunit 1 (alpha) |
PIK3R1 | 0.42 | 0.0005 | Regulates cell proliferation, survival and apoptosis |
| Retinoblastoma 1 | RB1 | 0.43 | 0.0007 | Tumor suppressor |
| Spleen tyrosine kinase | SYK | 0.32 | <0.0001 | Tumor suppressor |
| Transforming growth factor, beta receptor 1 |
TGFBR1 | 0.51 | 0.0077 | Decreased expression predisposes to tumors |
| Tumor necrosis factor receptor superfamily 1A |
TVFRSF1A | 0.42 | 0.0005 | Mediates apoptosis; regulator of inflammation |
The network of signal transduction pathways that regulate cell growth and apoptosis is very complex involving many different genes and proteins. A fine balance in the expression of these genes/proteins controls the switch between cell growth, transformation and apoptosis.
The ability of non-acidic pepsin to alter cellular proliferation was assessed via growth curve analysis. FaDu cells exposed to pepsin at pH7 for 1 hour demonstrated a significant time and dose dependent increase in cell number (Figure 1A and B). Cell cycle analysis of propidium iodide stained FaDu cells treated with pepsin revealed a statistically significant increase in the percentage of cells in S phase following pepsin exposure with a clear dose response (Figure 1C and D). An increase in the percentage of cells in S phase was not observed in cells exposed to pepsin which had been irreversibly inactivated (Figure 1C and D).
Figure 1. Pepsin increases growth and proliferation of FaDu SCC cells.
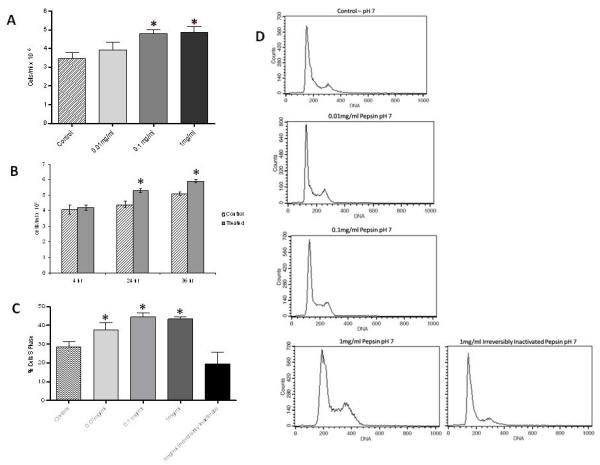
(A) FaDu cells exposed to pepsin at pH7 for 1 hour at 37∙C, washed 3 × briefly and incubated in fresh media for 24 hours at 37∙C. Cells stained with trypan blue and counted using a hemocytometer. Pepsin exposure (0.1mg/ml and 1mg/ml) results in a significant dose response increase in cell number (p<0.01). (B) Cells exposed to 0.1mg/ml pepsin at pH7 for 1 hour at 37∙C, washed 3 × briefly and incubated in fresh media for 4, 24, or 36 hours as indicated. A significant time response increase (24hr and 36hr) in cell number is observed in cells exposed to pepsin (p<0.001). Data from 5 biological replicates for dose response experiments were analyzed by one-way analysis of variance and Tukey’s multiple comparisons post test. Data from 5 biological replicates for time response experiments were analyzed by two-way analysis of variance and the Bonferroni multiple comparisons post test. Bar graphs show mean and standard deviation. (C) FaDu cells exposed to pepsin at pH7 for 1 hour at 37∙C, washed 3 × briefly, and incubated in fresh media for 24 hours at 37∙C. Cells were fixed, stained with propidium iodide and analyzed by flow cytometry. Data from 5 biological replicates were analyzed by one-way analysis of variance and Tukey’s multiple comparisons post test. Bar chart shows mean and standard deviation. There is a dose response increase in % cells in S phase when treated with 0.01mg/ml pepsin relative to control (p<0.05), with a further increase in % cells in S phase when exposed to pepsin at 0.1 and 1mg/ml (p<0.001). An increase in % cells in S phase is not seen when cells are treated with irreversibly inactivated pepsin. (D) Representative histograms from cell cycle analysis.
Since the hypopharyngeal FaDu cells are transformed, primary normal human laryngeal epithelial cell cultures were established from normal human laryngeal biopsy specimens taken from the posterior cricoid of reflux-free and neoplasia-free control subjects who have no inflammatory disease but were undergoing surgery for structural/functional procedures as described in the Methods section. These cell cultures were established and characterized following the protocol reported by Rees et al. (Figure 2A)24. Primary laryngeal epithelial cells formed a characteristic epitheloid shape (Figure 2Ai) showing a pavement-like appearance (Figure 2Aii) by phase contrast microscopy. The epithelial phoenotype was also confirmed by expression of cytokeratins 5 and 8 on cell cytospins by immunofluorescence (Figure 2Aiii). Primary cells were incubated with complete growth media ± pepsin (0.1mg/ml, pH7, 37°C) for 1 hour, washed and processed for analysis by the Human Cancer PathwayFinder PCR array. Of the 84 cancer-related genes analyzed, two were excluded from the analysis due to Ct > 35. The expression of one gene was increased by > 1.5 fold (p<0.05) in primary laryngeal cells treated with pepsin at pH7 compared to control cells and the expression of 30 genes was reduced > 1.5 fold (p<0.05) in cells exposed to pepsin relative to control cells (Table 2). Many of the genes affected by pepsin in FaDu cells were also changed in primary cell cultures - their function suggesting increased cell proliferation. As done using the FaDu cells, normal human primary laryngeal epithelial cells were exposed to pepsin at pH7 and assessed for overall cell growth. A significant time and dose dependent increase in cell number was seen following exposure to pepsin (Figure 2B and 2C). An increase in proliferation of these primary epithelial cells was also confirmed using the rigorous Click-iT EdU proliferation assay (Figure 2D).
Figure 2. Pepsin increases growth and proliferation of normal laryngeal primary epithelial cells.
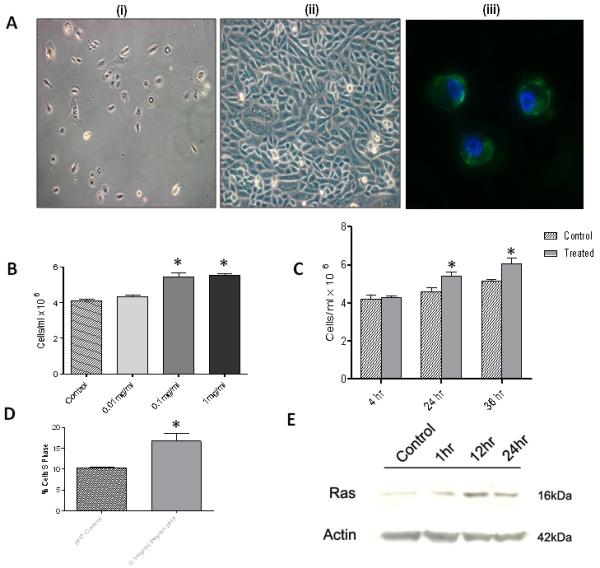
(A) Isolation and culture of primary human laryngeal epithelial cells from normal biopsy specimens. Characterization as in Rees et al. using light microscopy and immunohistochemistry24. i) Phase contrast microscopy of primary laryngeal epithelial cells at 5 days, 10× magnification. ii) 15 days, 20× magnification. Characteristic epithelioid shape showing a pavement-like arrangement. iii) The epithelial phenotype of cells was also confirmed by expression of cytokeratins 5 and 8 (green fluorescence) on cell cytospins by immunofluorescence. Cell nuclei were stained with DAPI (blue fluorescence), 40× magnification (45). (B) Primary epithelial laryngeal cells exposed to pepsin at pH7 for 1 hour at 37∙C, washed 3 × briefly and incubated in fresh media for (i) 24 hours at 37∙C. Cells stained with trypan blue and counted using a hemocytometer. Pepsin exposure (0.1mg/ml and 1mg/ml) results in a significant dose response increase in cell number (p<0.01). (C) Cells exposed to 0.1mg/ml pepsin at pH7 for 1 hour at 37∙C, washed 3 × briefly and incubated in fresh media for 4, 24, or 36 hours as indicated. A significant time response increase (24hr and 36hr) in cell number is observed in cells exposed to pepsin (p<0.001). (D) Click-iT EdU proliferation assay. Mean ± SEM % cells in S phase. Pepsin (0.1mg/ml at pH7 for 1 hour, washed 3 times and incubated in growth media for 24 hours at 37∙C) significantly increases proliferation of primary epithelial cells (p<0.001). (E) Western blot analysis for Ras protein levels in normal human laryngeal epithelial primary cells exposed to pepsin (0.1mg/ml, pH7) for 1, 12 and 24 hours at 37∙C and pH7 control. Ras protein levels increased following exposure to pepsin.
Table 2.
Genes reduced/increased by > 1.5 fold (p<0.05) in normal human primary laryngeal epithelial cells treated with 0.1mg/ml pepsin at pH7 for 1 hour at 37∙C relative to control cells. Genes also significantly reduced in FaDu cells are highlighted.
| Gene | Fold Change | p Value |
|---|---|---|
| FOS | 1.7 | 0.0101 |
| IFNB1 | 0.4 | <.0001 |
| E2F1 | 0.5 | 0.0003 |
| PDGFB | 0.5 | 0.0047 |
| BRCA1 | 0.5 | 0.0047 |
| NFKB1 | 0.5 | 0.0047 |
| RAF1 | 0.5 | 0.0047 |
| PDGFA | 0.6 | 0.0052 |
| TWIST1 | 0.6 | 0.0052 |
| CDKN1A | 0.6 | 0.0052 |
| SYK | 0.6 | 0.0076 |
| NME4 | 0.6 | 0.0102 |
| BCL2L1 | 0.6 | 0.0106 |
| BCL2 | 0.6 | 0.0106 |
| BAD | 0.6 | 0.0106 |
| AKT1 | 0.6 | 0.0106 |
| FGFR2 | 0.6 | 0.0106 |
| MAP2K1 | 0.6 | 0.0106 |
| MTSS1 | 0.6 | 0.0148 |
| ETS2 | 0.6 | 0.0148 |
| MTA2 | 0.6 | 0.0197 |
| NFKBIA | 0.6 | 0.0197 |
| TGFBR1 | 0.6 | 0.0197 |
| ITGB3 | 0.6 | 0.0283 |
| PNN | 0.7 | 0.0371 |
| ANGPT1 | 0.7 | 0.0371 |
| TNFRSF1A | 0.7 | 0.0371 |
| CDKN2A | 0.7 | 0.0469 |
| CDK2 | 0.7 | 0.0469 |
| CCNE1 | 0.7 | 0.0469 |
| ITGAV | 0.7 | 0.0469 |
A human miFinder miRNA PCR Array (Bioscience Corporation, MD) was used to examine the effect of pepsin, at neutral pH, on the expression of the 88 most abundantly expressed and best characterized miRNA sequences. Normal human primary laryngeal epithelial cultured cells were incubated with complete growth media ± pepsin (0.1mg/ml) at pH7, 37°C, for 1 hour, washed and processed for PCR. Data from three biological replicates for each condition were analyzed. Of the 88 genes analyzed, three were excluded from the analysis due to Ct > 35. The expression of 9 genes was increased by > 1.5 fold (p<0.05) in primary cells treated with pepsin at pH7 for 1 hour compared to control cells (Table 3A). The expression of 13 genes was reduced > 1.5 fold (p<0.05) in cells exposed to pepsin relative to control cells (Table 3B). Western blot analysis was performed to confirm the down-stream targets of these miRNAs. For example, let miRNAs negatively regulates Ras protein. Pepsin was found to decrease let miRNA expression levels in normal human laryngeal epithelial cells on gene array analysis. In support of this, pepsin exposure increased Ras protein levels in normal human laryngeal epithelial cells (Figure 2E).
Table 3A.
miRNA’s increased by > 1.5 fold (p<0.05) in human normal laryngeal epithelial primary cells treated with 0.1mg/ml pepsin at pH7 for 1 hour at 37∙C relative to control cells. Highlighted miRNA’s: expression change consistent with ratio in tumor:normal analysis of HNSCC by Hui et al. and Min Liu et al.38,39
| Gene | Fold Change | p-Value |
|---|---|---|
| miR-130a | 3.4 | 0.0047 |
| miR-141 | 5.7 | <0.0001 |
| miR-15a | 2.4 | 0.0479 |
| miR-185 | 2.7 | 0.0216 |
| miR-222 | 2.6 | 0.0285 |
| miR-29b | 6.5 | <0.0001 |
| miR-32 | 8.6 | <0.0001 |
| miR-423-3p | 2.4 | 0.0422 |
| miR-423-5p | 3.7 | 0.0029 |
Table 3B.
miRNA’s reduced by > 1.5 fold (p<0.05) in human normal laryngeal epithelial primary cells treated with 0.1mg/ml pepsin at pH7 for 1 hour at 37∙C relative to control cells. Highlighted miRNA’s: expression change consistent with ratio in tumor:normal analysis of HNSCC by Hui et al. and Min Liu et al.38,39
| Gene | Fold Change | p-Value |
|---|---|---|
| Let-7a | 0.3 | 0.0047 |
| Let-7b | 0.3 | 0.004 |
| Let-7c | 0.2 | 0.0002 |
| Let-7e | 0.3 | 0.0076 |
| miR-128 | 0.4 | 0.0325 |
| miR-151-5p | 0.4 | 0.0163 |
| miR-155 | 0.4 | 0.0188 |
| miR-223 | 0.2 | 0.0012 |
| miR-23a | 0.3 | 0.0104 |
| miR-23b | 0.3 | 0.0121 |
| miR-25 | 0.4 | 0.0216 |
| miR-430a | 0.4 | 0.0479 |
| miR-7 | 0.2 | 0.0007 |
Pepsin was detected in posterior cricoid tissue biopsy specimens from 3/5 cancer patients, but not in any (0/5) of the specimens from normal control subjects (Figure 3). Pepsin was also found in all 3 corresponding laryngeal cancer biopsies of those patients with pepsin-positive post-cricoid tissue.
Figure 3. Pepsin is detected in human laryngeal cancer biopsies, absent in normal laryngeal control specimens.
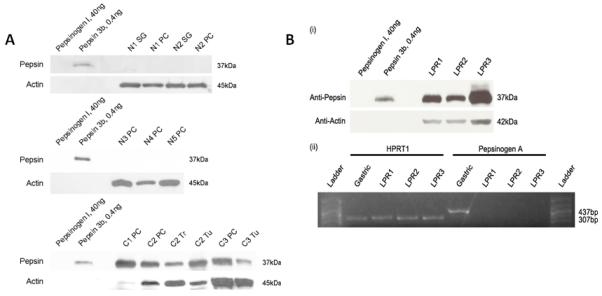
(A) Western blot analysis for pepsin in human tissue biopsy specimens. Pepsin was detected in 3/5 cancer patients (C1-C3), but not in any of the normal control subjects (0/5; N1-N5). SG = subglottic, PC = posterior cricoids, Tr – trachea, Tu = tumor. 30ug total protein was loaded for all except C1PC (0.6ug), C2PC (1.5ug) and C2Tu (10ug). (B) Presence of pepsin in LPR patient laryngeal biopsies and corresponding absence of pepsin precursor mRNA. (i) Pepsin was detected in all samples. To verify that pepsin observed by western blot was not synthesized locally, a section of the biopsy was analyzed for presence of pepsinogen mRNA via reverse transcriptase polymerase chain reaction (RT-PCR) (ii). Pepsinogen mRNA control testing was negative. Amplicon corresponding to pepsinogen A (437bp, expected size) was detected in human gastric tissue, but not in laryngeal specimens. HPRT1 was detected in all samples (307bp, expected size).
DISCUSSION
Common clinical manifestations of LPR are primarily attributed to mucosal inflammation. Inflammation and other immune responses initiated within airway mucosa are a fundamental determinant of tissue damage during reflux and are predicted to give rise to the diverse phenotypes characteristic of reflux-attributed disease26. In fact, while obvious factors such as frequency, duration, or volume of refluxate vary widely relative to the severity of reflux disease27, the expression profile of proinflammatory cytokines within esophageal mucosa is highly correlative with the grading of endoscopic findings and risk of relapse in patients diagnosed with the related GERD28,29. We have recently reported that exposure of hypopharyngeal cells to pepsin in a nonacidic environment induces the expression of several pro-inflammatory cytokines and receptors, including those known to be involved in inflammation of esophageal epithelium in response to reflux and which contribute to the pathophysiology of reflux esophagitis18. These data indicate that refluxed pepsin may contribute to laryngeal inflammation, even in nonacidic refluxate.
Nonacidic gastric reflux is a recently discovered phenomenon that occurs frequently in healthy persons as well as patients with GERD and LPR30, but is also suspected to contribute to inflammatory signs and symptoms of LPR in some individuals who are sensitized to this inflammation. Research using combined multi-channel intraluminal impedence (MII) and pH probe technology has demonstrated that the majority of episodes of gastric reflux into the laryngopharynx are nonacidic and that weakly and nonacidic gastric reflux events are associated with persistent laryngeal symptoms observed in acid-suppressed patients31-35. This evidence has prompted investigation into mucosal injury by components of refluxate beyond acid.
Although it has been well established that the proteolytic gastric enzyme pepsin contributes significantly to mucosal damage caused by acid reflux4, our recent work indicates that pepsin can also induce laryngeal mucosal damage during nonacidic reflux. While pepsin is enzymatically inactive at neutral pH, we have shown that pepsin is actively taken up by laryngeal and hypopharyngeal epithelial cells in a nonacidic environment by endocytosis, in a receptor mediated manner, and is retained in intracellular vesicles of low pH in which the enzyme’s proteolytic activity could be restored22. By analysis of cellular morphology, mitochondrial function, and the expression of stress response genes in pepsin-treated laryngeal biopsies or cultured hypopharyngeal cells, we confirmed that endocytosed inactive but stable pepsin at pH7 causes cell damage which may contribute to laryngoscopic findings and symptoms associated with weakly and nonacidic LPR16-18. We propose that inactive but stable pepsin taken up by the cell may cause damage by becoming reactivated inside the cell. We have documented the presence of pepsin in late endosomes and in the trans-reticular Golgi using markers for these intracellular compartments. The pH of these compartments is well documented in the literature – pH536. Pepsin is known to have approximately 40% of its maximal activity at this pH22,37. Thus, inactive but stable pepsin at pH7 taken up by the cell could become reactivated inside these intracelular compartments of lower pH and cause cell/tissue damage and disease. Alternatively, it may be that binding of pepsin to a receptor or the activation of a receptor on the surface of laryngeal epithelial cells initiates a cell signaling event ultimately having a negative effect on normal cell function16. Signal transduction, whereby binding of a ligand to its receptor initiates a cell signaling cascade, is often dysregulated in disease. It seems unlikely that there is a specific biologic receptor for pepsin, but perhaps more plausible that pepsin exploits/’piggy-backs’ another receptor-ligand complex, dysregulating its normal function. Interestingly, we have recently shown that irreversible, but not reversible, inhibition of peptic activity prevents cell damage by pepsin protein at pH7 supporting the former hypothesis that pepsin at pH7 causes damage by becoming reactivated inside the cell16. We report the same effect in this study when FaDu cell cultures were incubated with pepsin which had been irreversibly inactivated. Pepsin was incubated at pH8.5 for 15 minutes at 37°C to irreversibly inactivate enzymatic activity prior to incubation with cultured cells at pH7. Irreversible inhibition of pepsin prevented pepsin-mediated increased cell proliferation.
Gastro-esophageal reflux is known to cause a metaplastic change in the esophagus (Barrett’s esophagus) which confers an increased risk of developing esophageal adenocarcinoma. Laryngeal mucosa is thought to be more sensitive to the effects of gastric refluxate compared to the esophagus. Thus, it is plausible that chronic LPR could cause neoplastic changes and contribute to oncogenic transformation. The results of this study demonstrate that reflux of pepsin (at pH7 – in nonacidic reflux), present in posterior cricoid biopsies from cancer patients but not in normal control subjects, increases cell proliferation and alters the expression of multiple genes implicated in carcinogenesis, including multiple miRNA’s specifically known to play a role in HNSCC38,39. The genes and miRNAs were significantly up/down-regulated in a manner consistent with the ratio reported in the analysis of HNSCC by Hui et al and Min Liu et al. again supporting a potential role for refluxed pepsin in carcinogenesis of the laryngopharynx38,39. Furthermore, we have identified four of the let miRNA family members to be significantly dysregulated in laryngeal epithelial cells exposed to nonacid pepsin and demonstrated a corresponding increase in the expression of the let target oncoprotein, Ras. Increased Ras is known to lead to an increase in cell proliferation and its dysregulation in laryngeal cancer is prevalent40.
One of the limitations of this study is the small number of normal and cancer biopsy samples analyzed for the presence of pepsin. Access to such clinical material is limited. To better compare the prevalence of pepsin in these two populations, we intend to use laryngeal disease spectrum tissue microarrays containing squamous cell carcinoma and normal tissues, in addition to recruiting more patients in our clinic. Analysis of pepsin in laryngeal inflammatory diseases and head and neck cancer patients is becoming more commonplace at our institution. We recently reported that reflux of pepsin may also induce post-surgical complications in laryngectomy patients41.
We previously used the hamster buccal pouch in vivo model of mucosal carcinogenesis to determine the effect of active pepsin (pH4) on tumor growth19. Acidic pepsin significantly increased tumor growth (p<0.0001). Our findings using this in vivo model implicate pepsin in acid reflux as a cofactor in tumorigenesis, but do not prove that pepsin is a causal factor because the promoter, DMBA, was used. Given the data from this in vitro study, we now intend to use the hamster buccal pouch model to determine whether pepsin, in non-acid reflux (pH7), is a promoter and initiator (± DMBA respectively) of cancer in vivo.
CONCLUSION
In conclusion, our data demonstrate that pepsin induces a dose and time-dependent promotion of proliferation in both normal and transformed epithelial cultures. This induction of proliferation is associated with gene and microRNA expression changes that are consistent with promotion of neoplasia. Pepsin is also often detectable in the larynges of cancer patients, but absent in patients without clinical signs of reflux. Further work is needed to elucidate the exact role that pepsin plays in the promotion of laryngopharyngeal neoplasia, and to develop targeted pharmacologic inhibitors of pepsin activity and/or receptor antagonists in the upper airway.
Acknowledgments
This research study was sponsored by the Department of Otolaryngology and Communication Sciences, Medical College of Wisconsin, Milwaukee, Wisconsin, USA
Footnotes
Conflict of Interest: None
REFERENCES
- 1.Berrino F. Survival of cancer patients in Finland, 1955-1994. Acta oncologica (Stockholm, Sweden) 1999;38:275–277. doi: 10.1080/028418699431320. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA: a cancer journal for clinicians. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 3.Rothman KJ, Cann CI, Flanders D, Fried MP. Epidemiology of laryngeal cancer. Epidemiologic reviews. 1980;2:195–209. doi: 10.1093/oxfordjournals.epirev.a036223. [DOI] [PubMed] [Google Scholar]
- 4.Koufman JA. The otolaryngologic manifestations of gastroesophageal reflux disease (GERD): a clinical investigation of 225 patients using ambulatory 24-hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. The Laryngoscope. 1991;101:1–78. doi: 10.1002/lary.1991.101.s53.1. [DOI] [PubMed] [Google Scholar]
- 5.Copper MP, Smit CF, Stanojcic LD, Devriese PP, Schouwenburg PF, Mathus-Vliegen LM. High incidence of laryngopharyngeal reflux in patients with head and neck cancer. The Laryngoscope. 2000;110:1007–1011. doi: 10.1097/00005537-200006000-00023. [DOI] [PubMed] [Google Scholar]
- 6.Gabriel CE, Jones DG. The importance of chronic laryngitis. The Journal of laryngology and otology. 1960;74:349–357. doi: 10.1017/s0022215100056693. [DOI] [PubMed] [Google Scholar]
- 7.El-Serag HB, Hepworth EJ, Lee P, Sonnenberg A. Gastroesophageal reflux disease is a risk factor for laryngeal and pharyngeal cancer. The American journal of gastroenterology. 2001;96:2013–2018. doi: 10.1111/j.1572-0241.2001.03934.x. [DOI] [PubMed] [Google Scholar]
- 8.Glanz H, Kleinsasser O. Chronic laryngitis and carcinoma (author’s transl) Archives of oto-rhino-laryngology. 1976;212:57–75. doi: 10.1007/BF00456363. [DOI] [PubMed] [Google Scholar]
- 9.Morrison MD. Is chronic gastroesophageal reflux a causative factor in glottic carcinoma? Otolaryngol Head Neck Surg. 1988;99:370–373. doi: 10.1177/019459988809900403. [DOI] [PubMed] [Google Scholar]
- 10.Ward PH, Hanson DG. Reflux as an etiological factor of carcinoma of the laryngopharynx. The Laryngoscope. 1988;98:1195–1199. doi: 10.1288/00005537-198811000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Ling ZQ, Mukaisho K, Hidaka M, Chen KH, Yamamoto G, Hattori T. Duodenal contents reflux-induced laryngitis in rats: possible mechanism of enhancement of the causative factors in laryngeal carcinogenesis. The Annals of otology, rhinology, and laryngology. 2007;116:471–478. doi: 10.1177/000348940711600613. [DOI] [PubMed] [Google Scholar]
- 12.Sung MW, Roh JL, Park BJ, et al. Bile acid induces cyclo-oxygenase-2 expression in cultured human pharyngeal cells: a possible mechanism of carcinogenesis in the upper aerodigestive tract by laryngopharyngeal reflux. The Laryngoscope. 2003;113:1059–1063. doi: 10.1097/00005537-200306000-00027. [DOI] [PubMed] [Google Scholar]
- 13.Cammarota G, Galli J, Cianci R, et al. Association of laryngeal cancer with previous gastric resection. Annals of surgery. 2004;240:817–824. doi: 10.1097/01.sla.0000143244.76135.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cianci R, Galli J, Agostino S, et al. Gastric surgery as a long-term risk factor for malignant lesions of the larynx. Arch Surg. 2003;138:751–754. doi: 10.1001/archsurg.138.7.751. discussion 755. [DOI] [PubMed] [Google Scholar]
- 15.Galli J, Cammarota G, Calo L, et al. The role of acid and alkaline reflux in laryngeal squamous cell carcinoma. The Laryngoscope. 2002;112:1861–1865. doi: 10.1097/00005537-200210000-00030. [DOI] [PubMed] [Google Scholar]
- 16.Johnston N, Wells CW, Samuels TL, Blumin JH. Rationale for targeting pepsin in the treatment of reflux disease. The Annals of otology, rhinology, and laryngology. 119:547–558. doi: 10.1177/000348941011900808. [DOI] [PubMed] [Google Scholar]
- 17.Johnston N, Wells CW, Samuels TL, Blumin JH. Pepsin in nonacidic refluxate can damage hypopharyngeal epithelial cells. The Annals of otology, rhinology, and laryngology. 2009;118:677–685. doi: 10.1177/000348940911800913. [DOI] [PubMed] [Google Scholar]
- 18.Samuels TL, Johnston N. Pepsin as a causal agent of inflammation during nonacidic reflux. Otolaryngol Head Neck Surg. 2009;141:559–563. doi: 10.1016/j.otohns.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 19.Pearson JP, Parikh S, Orlando RC, et al. Review article: reflux and its consequences--the laryngeal, pulmonary and oesophageal manifestations. Conference held in conjunction with the 9th International Symposium on Human Pepsin (ISHP) Kingston-upon-Hull, UK, 21-23 April 2010. Alimentary pharmacology & therapeutics. 33(Suppl 1):1–71. doi: 10.1111/j.1365-2036.2011.04581.x. [DOI] [PubMed] [Google Scholar]
- 20.Belafsky PC, Postma GN, Koufman JA. The validity and reliability of the reflux finding score (RFS) The Laryngoscope. 2001;111:1313–1317. doi: 10.1097/00005537-200108000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Belafsky PC, Postma GN, Koufman JA. Validity and reliability of the reflux symptom index (RSI) J Voice. 2002;16:274–277. doi: 10.1016/s0892-1997(02)00097-8. [DOI] [PubMed] [Google Scholar]
- 22.Johnston N, Dettmar PW, Bishwokarma B, Lively MO, Koufman JA. Activity/stability of human pepsin: implications for reflux attributed laryngeal disease. The Laryngoscope. 2007;117:1036–1039. doi: 10.1097/MLG.0b013e31804154c3. [DOI] [PubMed] [Google Scholar]
- 23.Johnston N, Knight J, Dettmar PW, Lively MO, Koufman J. Pepsin and carbonic anhydrase isoenzyme III as diagnostic markers for laryngopharyngeal reflux disease. The Laryngoscope. 2004;114:2129–2134. doi: 10.1097/01.mlg.0000149445.07146.03. [DOI] [PubMed] [Google Scholar]
- 24.Rees LE, Gunasekaran S, Sipaul F, Birchall MA, Bailey M. The isolation and characterisation of primary human laryngeal epithelial cells. Molecular immunology. 2006;43:725–730. doi: 10.1016/j.molimm.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 25.Darzynkiewicz Z, Gong J, Traganos F. Analysis of DNA content and cyclin protein expression in studies of DNA ploidy, growth fraction, lymphocyte stimulation, and the cell cycle. Methods in cell biology. 1994;41:421–435. doi: 10.1016/s0091-679x(08)61732-x. [DOI] [PubMed] [Google Scholar]
- 26.Fitzgerald RC, Onwuegbusi BA, Bajaj-Elliott M, Saeed IT, Burnham WR, Farthing MJ. Diversity in the oesophageal phenotypic response to gastro-oesophageal reflux: immunological determinants. Gut. 2002;50:451–459. doi: 10.1136/gut.50.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Avidan B, Sonnenberg A, Schnell TG, Sontag SJ. Acid reflux is a poor predictor for severity of erosive reflux esophagitis. Digestive diseases and sciences. 2002;47:2565–2573. doi: 10.1023/a:1020580630594. [DOI] [PubMed] [Google Scholar]
- 28.Isomoto H, Inoue K, Kohno S. Interleukin-8 levels in esophageal mucosa and long-term clinical outcome of patients with reflux esophagitis. Scandinavian journal of gastroenterology. 2007;42:410–411. doi: 10.1080/00365520600931469. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida N, Uchiyama K, Kuroda M, et al. Interleukin-8 expression in the esophageal mucosa of patients with gastroesophageal reflux disease. Scandinavian journal of gastroenterology. 2004;39:816–822. doi: 10.1080/00365520410006729. [DOI] [PubMed] [Google Scholar]
- 30.Oelschlager BK, Quiroga E, Isch JA, Cuenca-Abente F. Gastroesophageal and pharyngeal reflux detection using impedance and 24-hour pH monitoring in asymptomatic subjects: defining the normal environment. J Gastrointest Surg. 2006;10:54–62. doi: 10.1016/j.gassur.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Gatta L, Vaira D, Sorrenti G, Zucchini S, Sama C, Vakil N. Meta-analysis: the efficacy of proton pump inhibitors for laryngeal symptoms attributed to gastro-oesophageal reflux disease. Alimentary pharmacology & therapeutics. 2007;25:385–392. doi: 10.1111/j.1365-2036.2006.03213.x. [DOI] [PubMed] [Google Scholar]
- 32.Mahieu HF. Review article: The laryngological manifestations of reflux disease; why the scepticism? Alimentary pharmacology & therapeutics. 2007;26(Suppl 2):17–24. doi: 10.1111/j.1365-2036.2007.03474.x. [DOI] [PubMed] [Google Scholar]
- 33.Sharma N, Agrawal A, Freeman J, Vela MF, Castell D. An analysis of persistent symptoms in acid-suppressed patients undergoing impedance-pH monitoring. Clin Gastroenterol Hepatol. 2008;6:521–524. doi: 10.1016/j.cgh.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 34.Tamhankar AP, Peters JH, Portale G, et al. Omeprazole does not reduce gastroesophageal reflux: new insights using multichannel intraluminal impedance technology. J Gastrointest Surg. 2004;8:890–897. doi: 10.1016/j.gassur.2004.08.001. discussion 897-898. [DOI] [PubMed] [Google Scholar]
- 35.Tutuian R, Vela MF, Hill EG, Mainie I, Agrawal A, Castell DO. Characteristics of symptomatic reflux episodes on Acid suppressive therapy. The American journal of gastroenterology. 2008;103:1090–1096. doi: 10.1111/j.1572-0241.2008.01791.x. [DOI] [PubMed] [Google Scholar]
- 36.Pastan I, Willingham MC. In: The pathway of endocytosis. Pastan I, Willingham MC, editors. Plenum Publishing Corp.; New York: 1985. pp. 1–44. [Google Scholar]
- 37.Piper DW, Fenton BH. pH stability and activity curves of pepsin with special reference to their clinical importance. Gut. 1965;6:506–508. doi: 10.1136/gut.6.5.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hui AB, Lenarduzzi M, Krushel T, et al. Comprehensive MicroRNA profiling for head and neck squamous cell carcinomas. Clin Cancer Res. 16:1129–1139. doi: 10.1158/1078-0432.CCR-09-2166. [DOI] [PubMed] [Google Scholar]
- 39.Liu M, Wu H, Liu T, et al. Regulation of the cell cycle gene, BTG2, by miR-21 in human laryngeal carcinoma. Cell research. 2009;19:828–837. doi: 10.1038/cr.2009.72. [DOI] [PubMed] [Google Scholar]
- 40.Paluszczak J, Krajka-Kuzniak V, Malecka Z, et al. Frequent gene hypermethylation in laryngeal cancer cell lines and the resistance to demethylation induction by plant polyphenols. Toxicol In Vitro. 25:213–221. doi: 10.1016/j.tiv.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Bock JM, Brawley MK, Johnston N, et al. Analysis of pepsin in tracheoesophageal puncture sites. The Annals of otology, rhinology, and laryngology. 119:799–805. doi: 10.1177/000348941011901203. [DOI] [PubMed] [Google Scholar]