

Abstract
Hyperuricemia, excess of uric acid in the blood, is a clinical problem that causes gout and is also considered a risk factor for cardiovascular disease. The enzyme xanthine oxidase (XO) produces uric acid during the purine metabolism; therefore, discovering novel XO inhibitors is an important strategy to develop an effective therapy for hyperuricemia and gout. We found that 3,4-dihydroxy-5-nitrobenzaldehyde (DHNB), a derivative of the natural substance protocatechuic aldehyde, potently inhibited XO activity with an IC50 value of 3 μM. DHNB inhibited XO activity in a time-dependent manner, which was similar to that of allopurinol, a clinical XO inhibitory drug. DHNB displayed potent mixed-type inhibition of the activity of XO, and showed an additive effect with allopurinol at the low concentration. Structure-activity relationship studies of DHNB indicated that the aldehyde moiety, the catechol moiety, and nitration at C-5 were required for XO inhibition. DHNB interacted with the molybdenum center of XO and was slowly converted to its carboxylic acid at a rate of 10-10 mol/L/s. In addition, DHNB directly scavenged free radical DPPH and ROS, including ONOO− and HOCl. DHNB effectively reduced serum uric acid levels in allantoxanamide-induced hyperuricemic mice. Furthermore, mice given a large dose (500 mg/kg) of DHNB did not show any side effects, while 42% of allopurinol-treated mice died and their offspring lost their fur. Thus, DHNB could be an outstanding candidate for a novel XO inhibitory drug that has potent activity and low toxicity, as well as antioxidant activity and a distinct chemical structure from allopurinol.
Keywords: Hyperuricemia; Gout; Xanthine oxidase inhibitor; 3,4-Dihydroxy-5-nitrobenzaldehyde; Antioxidant
1. Introduction
Uric acid is a product of purine catabolism and is excreted in the urine. Overproduction or under excretion of uric acid in humans could lead to hyperuricemia and gout, which is caused by crystallization and deposition of uric acid in joints and surrounding tissues [1]. Unlike most other mammals, humans lack the enzyme uricase [2], which converts uric acid to the more soluble allantoin [1]. Gout affects 1-2% of adults in developed countries and represents the most common case of inflammatory arthritis in men [1,3]. Furthermore, hyperuricemia and gout are also associated with chronic diseases such as hypertension, diabetes mellitus, metabolic syndrome, and renal and cardiovascular disease [1].
Xanthine oxidase (XO) is a form of the molybdoflavin protein xanthine oxidoreductase (XOR) [4] that plays an important role in the catabolism of purines in humans. XO first catalyzes the oxidation of hypoxanthine to xanthine, and then catalyzes the oxidation of xanthine to uric acid [4, 5]. Because overproduction of uric acid is the primary cause of hyperuricemia [1], XO is considered the most promising target for therapeutic treatment of this condition. Currently, the drugs allopurinol and febuxostat are available to reduce serum uric acid levels by inhibiting XO. Allopurinol is the most commonly used therapy for chronic gout and has been used clinically for more than 40 years. However, allopurinol cannot be used when the patient is hypersensitive or intolerant to the drug, or when the treatment fails. While rare, allopurinol has life-threatening side effects such as hypersensitivity syndrome consisting of fever, skin rash, eosinophilia, hepatitis, and renal toxicity, for which the mortality rate approaches 20% [1]. More recently, febuxostat, a new non-purine XO inhibitor, has been approved for the management of gout in the European Union and USA [6]. Many side effects of febuxostat have been reported [7]. Both allopurinol and febuxostat are not recommended for the treatment of asymptomatic hyperuricemia because of concern for their potential side effects [8-10]. Beyond gout, recent studies have indicated that asymptomatic hyperuricemia is associated with or may have a causal relationship with cardiovascular disease [11]. New XO inhibitors with more specific effects and fewer side effects than allopurinol and febuxostat are needed for preventing and treating gout and cardiovascular disease associated with hyperuricemia.
Recent studies have searched for XO inhibitors in natural compounds ranging from flavonoids to a host of natural plant products [12-19]. For example, nitro-oleic acid inhibits purine oxidation more potently than allopurinol does [14]. Caffeic acid shows variable XO inhibitory effects [20-24]. In this study, we intended to develop a novel XO inhibitor that is derived from natural substances, has potent activity and low toxicity, and whose chemical structure is distinct from allopurinol and febuxostat. Our goal was to have a novel XO inhibitor that could be used to treat gout patients that failed to allopurinol and febuxostat therapies, or had hypersensitivity or intolerance to these drugs. In addition, we wanted a drug that could be used in combination with allopurinol or febuxostat in order to increase therapeutic efficacy and reduce the potential toxicity of these drugs. Furthermore, we wanted a drug that could be used to treat patients with asymptomatic hyperuricemia to prevent cardiovascular disease and other hyperuricemia-associated diseases.
Protocatechuic aldehyde can be obtained from plants such as Salvia miltiorrhiza Bunge, and leaves of Stenoloma Chusanum(L.) Ching and Ilex chinensis Sims. Protocatechuic aldehyde has an anti-inflammatory effect and increases coronary artery blood flow [5]. It is the most effective component of the leaves of Ilex chinensis Sims for treating angina, and of Herb of Common Edelweiss for treating nephritis. Protocatechuic aldehyde is an important intermediate in the synthesis of various antibiotics and anti-inflammatory drugs. In the present study, we explored the inhibitory effects of 15 catechol compounds on XO activity, and found that protocatechuic aldehyde has limited inhibitory activity. However, its 5-nitro derivative, 3,4-dihydroxy-5-nitrobenzaldehyde (DHNB), is a potent XO inhibitor in a cell-free system. In this study, we determined the potency and potential mechanism of XO inhibition by DHNB in a cell-free system and in a mouse model of hyperuricemia, as well as its toxicity in vivo. DHNB could be a potential novel therapeutic agent for the treatment of gout and hyperuricemia.
2. Materials and Methods
2.1. Chemicals and reagents
XO from bovine milk, xanthine, allopurinol, 3,4-dihydroxybenzaldehyde (DHB-CHO), gallic acid, phosphate buffered saline (PBS) solution, potassium nitrite (KNO2), polyethylene glycol 400, sodium carboxymethyl cellulose (CMC-Na), sodium phosphotungstate hydrate, dioxide manganese (MnO2), diethylene-triamine-pentaacetic acid (DTPA), EDTA, ferrous ammonium sulfate, hydrogen peroxide (H2O2), sodium hypochlorite, DPPH, 5,5’-dithio-bis(2-nitrobenzoic acid) (DTNB), sodium borohydride, potassium persulfate, ascorbic acid and (±)-α-tocopherol were obtained from Sigma Chemical Co (Saint Louis, MO). 3,4-Dihydroxy-5-nitrobenzaldehyde (DHNB), 3,4-dimethoxybenzyl alcohol (DMB-CH2OH), 3,4-dihydroxyphenyl ethanol (Hydroxytyrosol), caffeic acid, 3,4-dihydroxybenzyl alcohol (DHBA), 3,4,5-trihydroxybenzaldehyde hydrate (THB-CHO), 4-hydroxy-3-methoxybenzyl alcohol, vanillin, 3,4-dihydroxybenzoic acid (DHB-COOH) and dihydrorhodamine were obtained from VWR Inc (Radnor, PA). 3,4-Dihydroxy-6-nitrobenzaldehyde (DH6NB) was obtained from Oakwood Products Inc (West Columbia, SC). Entacapone was obtained from Toronto Research Chemicals (Ontario, Canada). 3,4,5-trihydroxybenzyl alcohol (THB-CH2OH) and 3,4-Dihydroxy-5-nitrobenzyl alcohol (DHNB-CH2OH) were prepared by reducing 3,4,5-trihydroxybenzaldehyde and 3,4-Dihydroxy-5-nitrobenzaldehyde with sodium borohydride, respectively, according to the literature [25]. Allantoxanamide (purity >98% by HPLC) was purchased from Nanjing Chemlin Chemical Industry Co.,Ltd, Nanjing, China.
2.2. XO inhibition assay
XO activity was determined using the method of continuous spectrophotometric rate measurements. The reaction mixture contained uric acid in 67 mM phosphate buffer (pH 7.4) and 20 nM XO with an activity of 5 mU/mL, with or without DHNB derivatives. After pre-incubating the mixture for 1 to 5 min at 25 °C, 50 μM xanthine was added to initiate the formation of uric acid, and the increase of absorption of uric acid at 295 nm was monitored. Allopurinol was used as a positive control. For the enzyme kinetic analysis, relatively low concentrations of xanthine were used. All test compounds, including allopurinol, were dissolved in H2O or in an aqueous solution, so H2O was used as the negative control.
2.3. Conversion of DHNB to products by XO
The kinetic reaction of DHNB with XO at different pH values was measured spectrophotometrically. The decay of DHNB was monitored at 327 nm in a system containing 30 nM XO with 30 μM DHNB in phosphate buffer, pH 6.5 to 8.5. The extinction coefficient of DHNB at 327 nm was measured as 15,600 M-1 cm-1. Samples for product analysis by mass spectroscopy and HPLC were prepared by mixing 0.3 U XO with 4 mg DHNB in 1 mL phosphate buffer (pH 7.4) for 3 days. The DHNB/XO samples were analyzed by HPLC (Bio-Rad BioLogic DuoFlow, Hercules, CA) equipped with a 250 × 4.6 mm, 5 micron Phenomenex C-18 (2) Luna column, with a mobile phase of 40% acetonitrile/water. DHNB and its product were monitored by the optical absorption at 327 nm and 279 nm, respectively.
2.4. Ultra Performance Liquid Chromatography / Mass Spectroscopy (UPLC/MS)
Negative electrospray ionization-mass spectrometry (ESI-MS) and tandem (MS-MS) were used to detect and confirm the reaction products of DHNB with XO. All mass spectrometric experiments were performed on an API 3200-Qtrap triple quadrupole mass spectrometer (Applied Biosystem/MDS SCIEX, Foster City, CA) equipped with a turbolonSpray™ source. The main working parameters for mass spectrometry were set as follows: ion-spray voltage, -4.5 kV; ion source temperature, 600 °C; gas 1, 40 psi; gas 2, 40 psi; curtain gas, 20 psi; and collision gas, high.
2.5. HOCl scavenging assay
HOCl was prepared immediately before use by adjusting the pH of a 1% (v/v) solution of NaOCl to pH 6.2 with 0.6 M sulfuric acid. The concentration was further determined spectrophotometrically at 235 nm using the molar extinction coefficient of 100 M-1cm-1. 5-thio-2-nitrobenzoic acid (TNB) was prepared by reducing 5,5’-dithio-bis(2-nitrobenzoic acid) (DTNB) with sodium borohydride in phosphate buffer. The HOCl scavenging assay was based on the inhibition of TNB oxidation to DTNB induced by HOCl [26].
2.6. Peroxynitrite scavenging assay
Peroxynitrite (ONOO−) was generated by mixing 5 mL acidic solution (0.6 M HCl) of H2O2 (0.7 M) and 5 mL of 0.6 M KNO2 on ice bath for 1 sec, and then quenching the reaction with 5 mL of ice-cold 1.2 M NaOH. Residual H2O2 was removed using granular MnO2 pre-washed with 1.2 M NaOH, and the reaction mixture was then left overnight at -20°C. Concentrations of ONOO− were determined before each experiment at 302 nm using a molar extinction coefficient of 1,670 M-1 cm-1. The ONOO− scavenging assay was performed by monitoring the oxidation of dihydrorhodamine (DHR 123) by ONOO− spectrophotometrically at 500 nm [26]. The abilities of the tested compounds to scavenge ONOO− were compared to that of vitamin C.
2.7. Superoxide scavenging assay
Superoxide (O2−•) scavenging activity was assayed in the xanthine-XO system by measuring the inhibition of reduction of nitro blue tetrazolium (NBT) to form blue formazan, which has absorption at 560 nm [26]. O2−• production and XO activity were measured as NBT reduction (at 560 nm) and uric acid production (at 295 nm), respectively. The abilities of the tested compounds to scavenge O2−• were compared to those of GSH and vitamin C.
2.8. DPPH scavenging assay
The abilities of the tested compounds to scavenge DPPH radicals were measured optically by monitoring the decrease of the absorption at 429 nm, according to the literature [27]. The compounds’ antioxidant activities were compared to those of vitamin C and vitamin E.
2.9. Mouse model of hyperuricemia
A hyperuricemia mouse model was adopted from the literature [28, 29]. Allantoxanamide, a potent uricase inhibitor, was used to induce hyperuricemia in mice [29]. Briefly, adult C57BL/6 mice (15-25 g, 6-8 weeks old, 6 per group, Jackson Labs) received DHNB at a concentration of 100 mg/kg in 1.0% polyethylene glycol 400 (PEG400 in a volume of 0.1 ml/10 g mouse body weight), via oral gavage. Just after the oral administration of DHNB, the mice were injected intraperitoneally (i.p.) with allantoxanamide at 200 mg/kg in 0.5% CMC-Na in a volume of 0.1 ml/10 g mouse body weight, to increase the serum uric acid level. Positive control mice received allopurinol at the same concentration as DHNB, followed by i.p. allantoxanaminde. The negative control mice received PEG400 only, followed by i.p. allantoxanaminde. Food and water were withheld overnight prior to the study. To collect whole blood samples at the end of the study, the mice were anaesthetized with diethyl ether inside a chamber and then bled from the orbit vein. The blood was allowed to clot for 1 h at room temperature and then centrifuged at 2350 × g for 4 min to obtain serum. The serum was kept on ice and assayed immediately. Serum uric acid was determined using the phosphotungstate method [30].
2.10. Acute toxicity assay in mice
C57BL/6 mice were randomized into 3 groups (12/group). Group 1, 2 and 3 received an oral vehicle solution of (PEG400), DHNB (500 mg/kg), and allopurinol (500 mg/kg), respectively. Each mouse was monitored for general health conditions on a daily basis for 28 days, recording mortality, body weight, and behavior of the mice. Mice were also bred within the treated group, and their offspring were observed for health conditions, including fur appearance.
2.11. Statistical analysis
Results are presented as mean ± standard deviation (SD). Statistical significance was determined by a Student’s t-test (two tailed). A value of P<0.05 was considered significant.
3. Results
3.1. DHNB effectively inhibits XO activity in a cell-free system
We studied 15 structurally-related compounds using the XO inhibition assay (Fig. 1). When 20 nM XO was mixed with increasing concentrations of allopurinol, DHNB, DH6NB, DHBA or THB-CHO, the initial rate of uric acid formation showed a concentration-dependent decrease compared to the control, reflecting the decrease of XO activity (Fig. 2A). DHNB significantly inhibited XO activity with an IC50 value of 3 μM, which is close to allopurinol’s IC50 value of 1.8 μM. IC50 values for DHBA and DH6NB were 76 μM and 96 μM, respectively, indicating weak inhibition of XO activity. The IC50 value for THB-CHO was too high to determine. For the enzyme kinetic analysis, relatively low concentrations of xanthine near the Km were used [31,32]. Lineweaver-Burk plot (1/v vs 1/[S], Double Reciprocal) of the steady-state kinetic study of DHNB-mediated inhibition of XO activity was performed (Fig. 2B). The initial rate of uric acid formation increased with increasing concentrations of xanthine to a maxmum (Vmax) of 0.125 μM/s. In the presence of DHNB at 1.3, 3.3, 5.0 and 6.7 μM, however, the Vmax decreased from 0.125 μM/s to 0.083, 0.52, 0.033 and 0.031 μM/s, respectively; while the Km increased from 1.8 to 2.7, 3.6, 4.9 and 6.7 μM, respectively, under the current assay condition. The inhibitory effect of DHNB on XO activity was not overcome by increasing concentrations of substrate xanthine. Clearly, DHNB displayed potent mixed-type inhibition of XO. In addition, we determined whether pH affects the inhibitory effect of DHNB on XO activity, and found that neutral or slightly acidic solutions favored the inhibition of XO by DHNB (Fig. 2C).
Fig. 1.
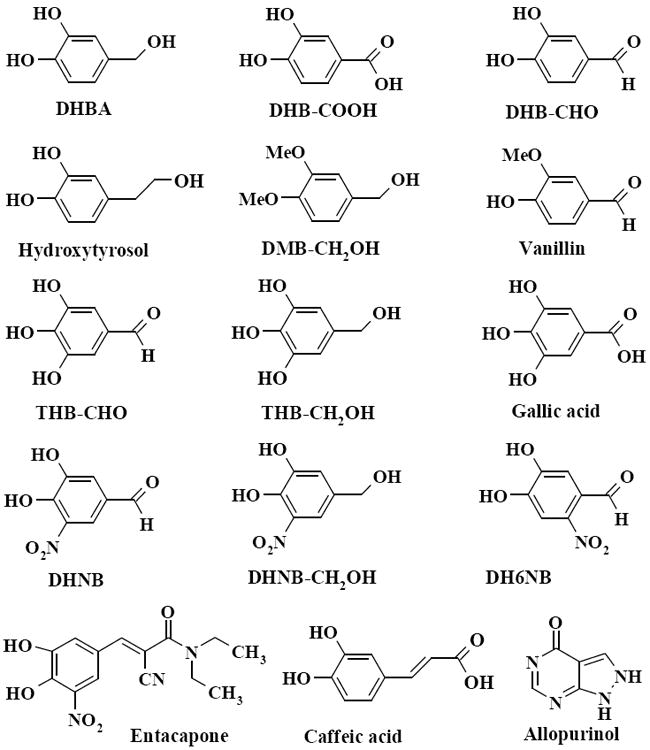
Chemical structures of catechol compounds tested in this study. Fifteen structurally-related compounds were selected to study their XO inhibitory activities. These compounds possess the same catechol skeleton in their structures, but have different functional groups.
Fig. 2.
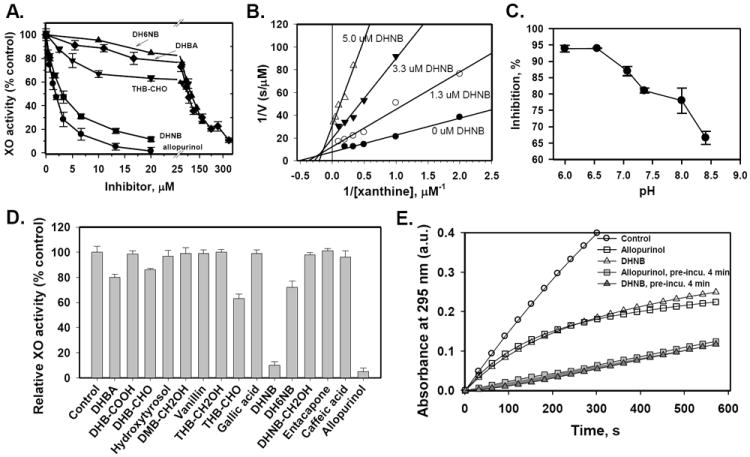
Inhibitory effects of DHNB and other compounds on XO activity in a cell free system. A. Dose dependent effects. After exposure of XO (10 milliunits/mL) to a 0-300 μM concentration of allopurinol (●), DHNB (■), DHBA (◆), DH6NB (▲) or THB-CHO (▼) in 67 mM phosphate buffer (pH 7.4, 25 °C), XO activity was determined by measuring the initial rate of formation of uric acid (λ = 295 nm). Reactions were initiated by the addition of xanthine (50 μM). IC50 values: 1.8, 3.0, 76 and 96 μM for allopurinol, DHNB, DHBA and DH6NB, respectively. IC50 is not available for THB-CHO because the inhibition did not reach 50% at concentrations up to 80 μM. B. Lineweaver-Burk plot (1/v vs 1/[xanthine], Double Reciprocal) for the inhibitory effect of DHNB on XO activity at relatively low concentrations of xanthine. C. Effect of pH on the DHNB-mediated XO inhibition. D. Comparison of the inhibitory effect of 15 catechol compounds on XO activity at the concentration of 20 μM. After pre-incubation of 20 nM XO and 20 μM inhibitor for 1 min, 50 μM xanthine was added to initiate the reaction. E. The effect of pre-incubation of XO with DHNB or allopurinol on XO activity. XO (20 nM) was pre-incubated with DHNB (6.67 μM) or allopurinal (6.67 μM) for 4 min first, and then xanthine (50 μM) was added to start the reaction. No pre-incubation of XO with DHNB or allopurinal was set for comparison. XO activity was recorded. Data represent the mean ± S.E. of at least three independent determinations.
3.2. DHNB and related compounds show a structure-activity relationship of XO inhibition
We also studied the inhibition of XO activity by several other compounds, including the drug entacapone. These compounds possess the same catechol structural skeleton; but have different functional groups. The ability of each compound to inhibit XO at a concentration of 20 μM was compared to that of allopurinol (Fig. 2D). Although these compounds have similar structures, their capacities to inhibit XO were different. Compounds containing a −CHO group such as DHNB, DH6NB, DHB-CHO and THB-CHO had an inhibitory effect on XO. However, vanillin, although it contains a −CHO group, did not inhibit XO activity. DHBA has no −CHO group, but it showed a moderate inhibition of XO. Other compounds, such as DHB-COOH, gallic acid, caffeic acid, hydroxytyrosol, DMB-CH2OH and DHNB-CH2OH, which contain −COOH or −CH2OH groups, had no inhibitory effect on XO under the current experimental condition. Entacapone, the catechol-O-methyl transferase (COMT) inhibitor, did not inhibit XO activity even though entacapone has a 3,4-dyhydroxy-5-nitrobenzyl moiety in common with DHNB, a strong XO inhibitor.
3.3. DHNB irreversibly inhibits XO in a short term time course study
Interestingly, DHNB displayed a time-dependent inhibition of XO activity, similar to that of allopurinol. When XO (20 nM) was added to the mixture of xanthine (50 μM) and DHNB or allopurinol (6.67 μM) to start the reaction up to 10 min, both DHNB and allopurinol showed a time-dependent inhibition (Fig. 2E). Under this experimental condition, XO activity was not completely inhibited by DHNB or allopurinol. Under a different experimental condition, 20 nM XO was pre-incubated with DHNB or allopurinol (6.67 μM) for 4 min first, and then xanthine was added to the reaction for 10 min; the inhibitory effect of DHNB on XO activity was substantially enhanced when comparing with that without pre-incubation of XO and DHNB or allopurinol (Fig. 2E). Both DHNB and allopurinol showed a similar inhibitory pattern. In a separate experiment, DHNB was pre-incubated with XO for 0, 0.25, 0.5, 1, 2, 3, 4 and 5 min, respectively, at 25°C, and then 50 μM xanthine was added to the reaction mixture, and XO activity was recorded for 3 min. Inhibitory effect of DHNB on XO activity was gradually enhanced with increasing pre-incubation time. For example, 6.67 μM DHNB only inhibited 20% of XO activity without pre-incubation; while a 2-min incubation of DHNB and XO, DHNB inhibited 75% of XO activity. In addition, pre-incubation also affected the inhibitory effect of other compounds including DH6NB, DHB-CHO and THB-CHO on XO activity (Fig. 3A). However, pre-incubation of XO and DHBA did not increase the inhibitory potency of DHBA (data not shown). For other compounds listed in Fig. 1, such as vanillin, DHB-COOH, hydroxytyrosol, DMB-CH2OH, THB-CH2OH, gallic acid, DHNB-CH2OH, phloretin, caffeic acid and entacapone, pre-incubation with XO for up to 5 min did not show any inhibitory effect. These results indicated that DHNB is an irreversible XO inhibitor in the tested conditions. In another experiment, XO was treated with 20 μM DHNB to induce inhibition. The reaction mixture was then treated with a high concentration of a reducing agent, including GSH (20 mM), dithiothreitol (20 mM) or 2-mercaptoethanol (20 mM). The treatment with reducing agents did not abolish the inhibitory effect of DHNB on XO activity (Fig. 3B), further indicating that this inhibition is irreversible. Furthermore, we determined whether DHNB and allopurinol have an additive effect on XO inhibition. We incubated DHNB (1 μM) or allopurinol (1 μM with 20 nM XO for 1 min, then adding 50 μM xanthine to start the reaction. DHNB (1 μM) or allopurinol (1 μM) inhibited about 21% of XO activity. When we incubated DHNB (1 μM) and allopurinol (1 μM) together with 20 nM XO for 1 min, then starting the reaction with 50 μM xanthine, DHNB (1 μM) plus allopurinol (1 μM) inhibited XO activity up to 38%, which was close to the inhibitory effect of DHNB (2 μM) or allopurinol (2 μM) alone on XO activity. These data indicate an addictive effect of DHNB and allopurinol at a relatively low concentration (1 μM) (Fig. 3C). However, at a higher concentration (2 μM), the additive effect of DHNB and allopurinol was not observed (Fig. 3C).
Fig. 3.
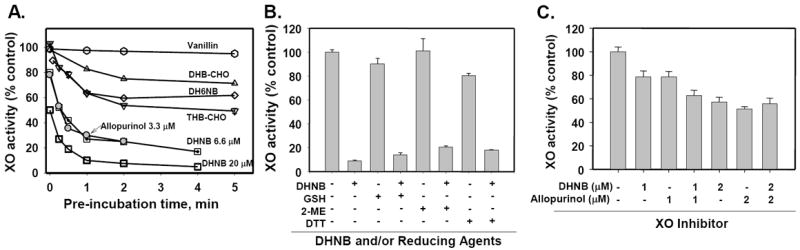
Influence of pre-incubation time, reducing agents and combination of XO inhibitors on the XO inhibitory activity of DHNB and other compounds. A. Pre-incubation time. XO activity was determined by the steady-state rate of formation of uric acid (λ = 295 nm) by pre-incubation of 20 nM XO and 20 μM testing compound (unless specified) for 0-5 min followed by the addition of 50 μM xanthine to start the reaction. DHNB (20 μM and 6.6 μM), allopurinol (3.3 μM), THB-CHO, DH6NB, DHB-CHO and vanillin were tested. B. Reducing agents. XO (10 mU/mL) was pre-incubated with 20 μM DHNB for 10 min at 25°C in phosphate buffer (100 mM pH 7.4); and then, GSH (20 mM), 2-mercaptoethanol (2-ME, 20 mM) or dithiothreitol (DTT, 20 mM) was added for 15 min. XO activity was analyzed by measuring the production of uric acid. C. Combination of DHNB and allopurinol. XO (20 nM) was pre-incubated with DHNB, allopurinol or combination of DHNB and allopurinol for 1 min in phosphate buffer (100 mM pH 7.4) followed by the addition of 50 μM xanthine to start the reaction. Two concentrations (1 and 2 μM) of DHNB and allopurinol were used. XO activity was analyzed by measuring the production of uric acid. Data represent the mean ± S.E. of at least three independent determinations.
3.4. XO slowly oxidizes DHNB to its acid form
In order to understand the mechanism of DHNB-mediated inhibition of XO activity, we incubated 30 μM DHNB with 15 mU/mL (or 30 nM) XO in phosphate buffer (pH 7.4), and then added xanthine to initiate the reaction. We found that DHNB inhibited XO activity for up to 20 h. After that, the enzymatic activity of XO was recovered, indicating that DHNB did not permanently destroy XO enzyme. To study the mechanism, we measured the optical spectral change of DHNB in a system without xanthine, containing 30 μM DHNB with 30 nM XO in phosphate buffer for 12 h. The reaction sample was taken every hour for spectophomotric analysis. We observed that the absorption of DHNB at 327 nm decreased with time and a new peak appeared at 279 nm (Fig. 4A). The decay rate was in the range of 10-10 mol/L/s and it was pH dependent, i.e., the higher the pH value was, the faster DHNB decayed (Fig. 4B). Without XO, however, DHNB itself was very stable. This suggests that, at room temperature, DHNB is converted by XO to a product which has no inhibitory effect on XO, and, consequently, XO recovers its activity as the concentration of DHNB decreases. The UV-vis spectrum of the product was different from that of DHNB. HPLC analysis of DHNB/XO showed a new peak, which was more polar than DHNB (Fig. 4C). Mass spectrometric analysis revealed that the product had a molecular ion ([M-H]-) peak at m/z 198 in the EI mass spectrum, while DHNB showed a [M-H]- peak at m/z 182 (Fig. 4D, E). The MS/MS of m/z 182 of DHNB produced several typical fragments, such as m/z 165 ([M-H-OH]-), 152 ([M-H-CHO-H]-) and 135 (m/z 152-OH). However, the MS/MS of molecular ion at m/z 198 of the product produced its first main fragment at m/z 154, a mass difference of 44 that indicates a loss of CO2, which further loses a −OH to give a fragment at m/z 137. Based on the mass spectrum, we concluded that the product is 3,4-dihydroxy-5-nitrobenzoic acid, implying that XO oxidizes DHNB to an acid. It has been reported that unhydrated acetaldehyde inhibited XO where the enzyme catalyzed the oxidation of aldehyde to its carboxylic acid [33].
Fig. 4.
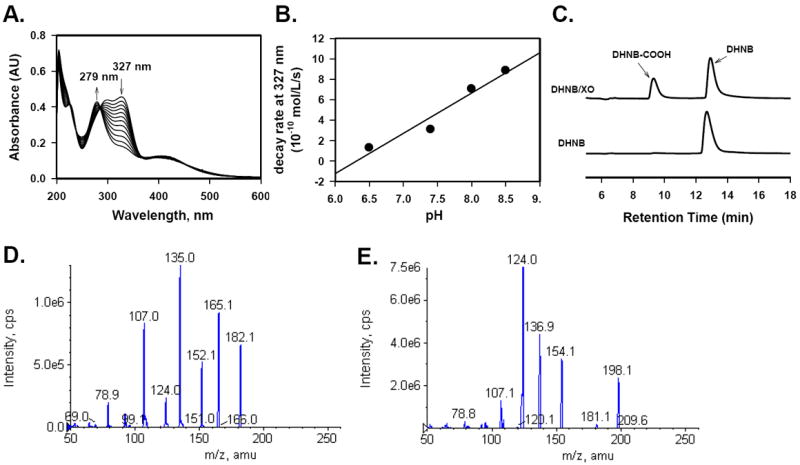
Product analysis of the reaction of DHNB with XO enzyme. A. Time course of absorption change of DHNB (327 nm, arrow indicates decrease) and the formation of the product (279 nm, arrow indicates increase). XO (30 nM) was mixed with DHNB (30 μM) in 0.1 M phosphate buffer (pH 7.4) and the reaction sample was taken for spectrophotometric analysis every hour for 12 h. B. Effect of pH on the conversion of DHNB by XO enzyme. XO (30 nM) was mixed with DHNB (30 μM) in the phosphate buffer with different pH (6.5, 7.4, 8.0 and 8.5), respectively. The reaction sample was taken for spectrophotometric analysis every hour for 12 h and the decay rate of DHNB was calculated. C. HPLC profile of DHNB (control) and DHNB/XO mixture after incubation for 3 days. D. MS/MS spectrum of DHNB (control) and DHNB/XO mixture after incubation for 3 days. E. MS/MS spectrum of reaction product (DHNB-COOH) from DHNB/XO mixture after incubation for 3 days.
3.5. DHNB has direct antioxidant activities in a cell free system
We determined whether DHNB has direct ROS scavenging properties. DHNB strongly scavenged DPPH, ONOO− and HOCl with low IC50 values, although it did not scavenge superoxide ion under the current assay condition (Fig. 5). However, allopurinol did not show any direct scavenging activities for these ROS. Thus, the antioxidant property of DHNB is one of its advantages as an XO inhibitor over allopurinol. DH6NB, DHB-CHO, THB-CHO, DHBA also showed strong direct scavenging activities for these ROS.
Fig. 5.
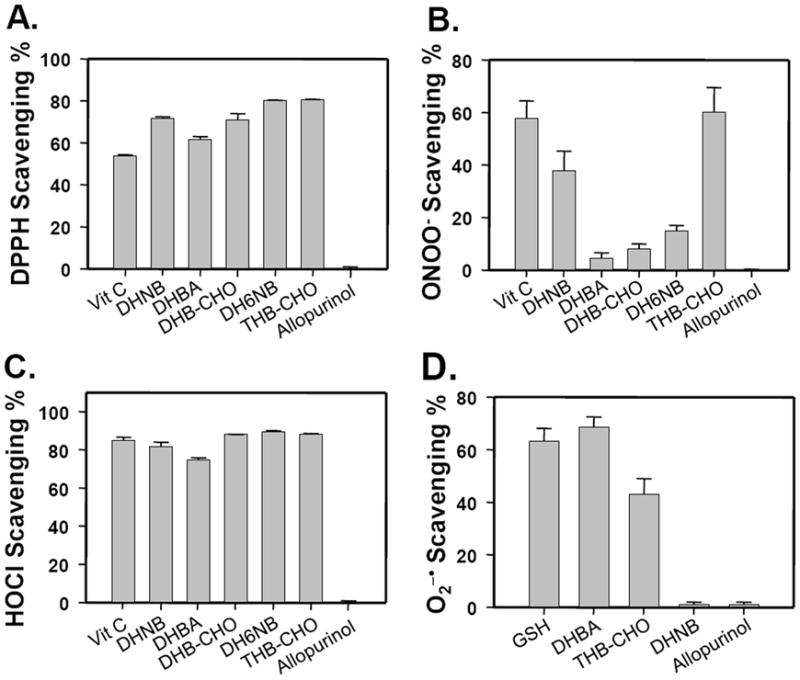
Antioxidant activities of DHNB, DH6NB, DHBA, DHB-CHO and THB-CHO on the scavenging of free radicals and reactive oxygen species. A. Free radical DPPH; B. Hypochlorous acid (HOCl); C. peroxynitrite (ONOO−); and D. superoxide ion (O2−•). A concentration of 10 μM of each compound was used. Vitamin C was used as a control, and the antioxidant activity of DHNB was compared to that of allopurinol. Data represent the mean ± S.E. of at least three independent determinations.
3.6. DHNB effectively reduces serum uric acid levels in allantoxanamide-induced hyperuricemic mice
Both allantoxanamide and potassium oxonate have been used as uricase inhibitors in mouse models, however, the hyperuricemic effect of allantoxanamide is stronger and lasts longer than that of oxonate in rats [28,29]. In this study, a single i.p. injection of 100 or 200 mg/kg of allantoxanamide in mice progressively increased their serum uric acid levels during a 4-hour experiment. Serum uric acid levels were elevated from 2 mg/dL (normal mice) to 5.4, 9.5, 13.3 and 16.4 mg/dL, 1, 2, 3 and 4 hours after the allantoxanamide i.p. injection, respectively (Fig. 6A, n = 4; Fig. 6B, n=9). By contrast, if the mice received 100 mg/kg DHNB orally before the allantoxanamide injection, the serum uric acid levels significantly decreased in 2 hours and remained at a relatively low level up to 4 hours. For a positive control, allopurinol (100 mg/kg) was used under the same condition as that of DHNB. As expected, allopurinol significantly lowered serum uric acid levels in allantoxanamide-treated mice.
Fig. 6.

Hypouricemic effect of DNHB on the allantoxanamide-induced hyperuricemic mice. A. The mouse model of allantoxanamide-induced hyperuricemia. Allantoxanamide (100 or 200 mg/kg) was given to mice (n=4 for each group) via intraperitoneal (I.P.) injection, and the serum uric acid levels were determined at 2 hours. B. Effects of DNHB on serum uric acid levels. DHNB or allopurinol (100 mg/kg) was given to mice via oral administration and then allantoxanamide (200 mg/kg) was given to the mice via I.P. injection (n=9 for each group). Serum uric acid levels were determined at 1, 2, 3 and 4 hours after allantoxanamide administration.
3.7. DHNB has no or less toxicity than allopurinol in mice
There are no reports about the general toxicity of DHNB except that the lowest lethal dose in the mouse is 312 mg/kg (oral administration, once) [34, 35]. In this study, we determined the potential toxicity of DHNB in mice, and compared it to allopurinol. Twelve mice were assigned to each group. Mice received DHNB (100, 200 or 500 mg/kg) or allopurinol (500 mg/kg), via oral gavage. Control mice received the vehicle solution. The animals were observed daily for up to 28 days. None of the DHNB-treated mice showed any symptoms of general toxicity. There were no differences in body weight and behavior between DHNB-treated mice and control mice. After 28 days, a few mice were sacrificed to perform a histological analysis of major organs including liver, kidney, heart, and lung. We found no abnormalities in the organs of DHNB-treated mice compared to those of control mice. The remaining mice were kept alive for breeding. Their offspring were all normal, including their fur condition. However, 5 out of 12 mice treated with allopurinol (500 mg/kg) died within 3 days of treatment (mortality 42%). Although the surviving 7 mice (mixed male and female) treated with allopurinol gave birth to a total 19 offspring pups, 8 died in 2 days. More interestingly, the surviving offspring pups started to lose their fur after two weeks and lost most of the back fur at 3 to 4 weeks (Fig. 7). After these pups were separated from their parents, they started to grow fur again and looked normal at 6 to 7 weeks. The same surviving parent mice that received allopurinol gave birth two times and the fur loss rate of these offspring pups was 100% and 50% in 1st and 2nd births, respectively (Table 1). We continued monitoring these parents for the birth of new pups up to 6 times and observed that pups lost fur at the average rate of 60%. Fur loss in pups was confirmed three times in separate parent mouse groups treated with allopurinol (oral 500 mg/kg once). Thus, DHNB is much safer than allopurinol tested in the mouse model.
Fig. 7.
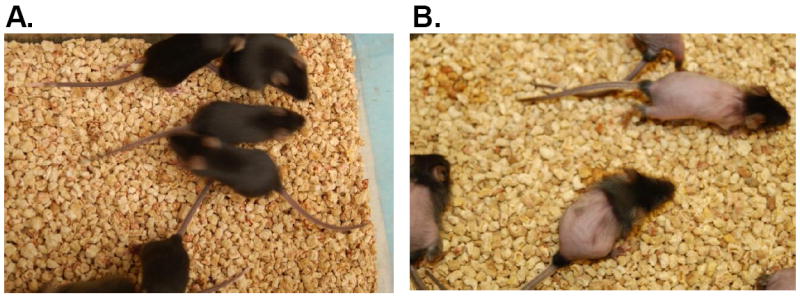
Toxicity of DHNB and allopurinol in mice. 500 mg/kg of DHNB or allopurinol was orally administrated to mice. A. DHNB treatment (n=12). All DHNB-treated mice did not show any symptom of general toxicity for more than 28 days and these mice produced healthy offspring (3 wks age). B. Allopurinol treatment (n=12). Five out of 12 mice treated with allopurinol died within 3 days (mortality 42%). The surviving mice produced offspring with hair loss (3 wks age).
Table 1.
Summary of in vivo toxicity of DHNB in mice
| 12 Mice / group | Behavior | Organs | Mortality | Offspring Mice
|
|
|---|---|---|---|---|---|
| Mortality | Fur Loss | ||||
| Vehicle solution | Normal | Normal | None | None | None |
| DHNB 100 mg/kg | Normal | Normal | None | None | None |
| DHNB 200 mg/kg | Normal | Normal | None | None | None |
| DHNB 500 mg/kg | Normal | Normal | None | None | None |
| Allopurinol 500 mg/kg | Normal for survivors | N/A | Average 42% | Average 42% | 1st birth: 100% |
| 2nd birth: 50% | |||||
4. Discussion
DHNB is derived from the natural substance protocatechuic aldehyde (DHB-CHO, 3,4-dihydroxybenzaldehyde). In this study, we demonstrated that DHNB significantly inhibits XO activity with potency close to that of allopurinol. Kinetic analysis revealed that the inhibitory effect of DHNB on XO activity is mixed-type inhibition. Meanwhile, DHBA, THB-CHO, and DH6NB were relatively weak inhibitors of XO. All these compounds have potent antioxidant activities. Most importantly, DHNB showed no notable toxicity in mice under the experimental conditions used, and effectively reduced serum uric acid levels in allantoxanamide-induced hyperuricemic mice.
Since XO plays a critical role in hyperuricemia and gout as well as in cardiovascular disease, XO inhibitors are potentially effective drugs to control these uric acid-related health problems. Due to the limitations of currently available XO inhibitory drugs, the development of new ones with more potency, different pharmacological mechanisms, and less toxicity is an active field of research. We have compared 15 compounds with a catechol moiety on their ability to inhibit XO activity, and found that DHNB is the most effective. DHNB inhibited XO activity with an IC50 value of 3 μM, which is comparable to that of allopurinol. Several other compounds, including DHBA, DH6NB, THB-CHO and DHB-CHO, inhibited XO activity weakly in a cell-free system.
In general, the potency of an enzyme inhibitor could be affected by its reactive functional groups, such as aldehydes, nitrogen mustards, haloalkanes, and/or alkenes. These groups may covalently modify the enzyme by reacting with amino acid side chains containing nucleophiles, such as amino, hydroxyl or sulfhydryl groups, to form covalent adducts and causing irreversible inhibition [36]. In this study, we tested compounds containing the electrophilic group −CHO, such as DHNB, DH6NB, DHB-CHO, and THB-CHO, and they showed a time dependent-inhibition. We observed that reduction of the −CHO group of DHNB to −CH2OH abolished DHNB’s ability to inhibit XO activity. This observation indicates that the −CHO group binds covalently to XO. On the other hand, DHBA, which has no −CHO group, showed a moderate inhibitory effect on XO. Thus, other functional groups of DHNB may also play a role in XO inhibition.
It has been reported that several other XO inhibitors contain a nitro group, suggesting that this group is important for the inhibitory activity [14, 17-19]. In our study, four compounds, DHNB, DH6NB, DHB-CHO and THB-CHO, have the same base structure, 3,4-dihydroxybenzaldehyde. DHNB and DH6NB have an additional −NO2 group, which enhances their inhibitory effects on XO. The inhibitory effect of DHNB was much stronger than that of THB-CHO, since DHNB has a −NO2 at C-5, while THB-CHO has a −OH at the same position. Moving −NO2 away from C-5, replacing it with a −OH or a −H, or reducing the aldehyde to an alcohol, may strongly alter DHNB-mediated inhibition of XO activity. Our results indicate that DHNB’s ability to inhibit XO is highly associated with its specific structure, implying possible electrostatic interactions between DHNB and particular domains on XO.
The position of nitro group (−NO2) in DHNB is important for its XO inhibitory activity. Moving −NO2 from C-5 to C-6 significantly decreased DHNB’s inhibitory effect. DHNB has a −NO2 group at the meta position of −CHO, which may make this −CHO more electrophilic than the ortho −NO2 group of DH6NB does. Therefore, the reaction of DHNB with the molybdenum (Mo) center of XO may be easier, and DHNB’s inhibitory effect on XO is stronger than that of DH6NB. On the other hand, vanillin has −CHO, but no inhibitory effect on XO, possibly because one of its −OH groups is methylated, and the electron-donating ability of methoxyl group may make −CHO less electrophilic, thus abolishing the binding to XO. DHNB-CH2OH does not inhibit XO at all, although it has −OH at C-3 and C-4, and −NO2 at C-5.
DHNB is one of the metabolites of entacapone, a drug that functions as a catechol-O-methyl transferase (COMT) inhibitor, and is used in the treatment of Parkinson’s disease [37]. DHNB is also a potent COMT inhibitor [38]. In their study, kinetic data showed that all dihydroxynitrobenzaldehydes tested were potent inhibitors of COMT, and their activities were dependent on the position of the nitro group. Having the nitro group in a position ortho relative to one hydroxyl group, increased DHNB’s inhibitory effect on COMT [38]. Thus, DHNB may have a clinical application for the treatment of Parkinson’s disease. On the other hand, we have shown that entacapone does not inhibit XO directly. However, if entacapone is used in humans or in animal models, it may inhibit XO through its metabolite DHNB. Thus, entacapone could be considered a pre-drug of the XO inhibitor, which could be used for the treatment of hyperuricemia and gout.
XO contains a FAD domain, two iron sulfur centers, and a molybdopterin-binding domain with the four redox centers aligned in an almost linear fashion per enzymatic unit. The Mo atoms are contained as molybdopterin cofactors and are the active sites of the enzyme [35]. The active site is also coordinated by terminal oxygen, sulfur atoms, and a terminal hydroxide. In the reaction with xanthine to form uric acid, an oxygen atom is transferred from Mo to xanthine, and several intermediates are assumed to be involved [39]. Allopurinol is a clinical drug that inhibits XO. It is hydroxylated by XO to oxypurinol at the Mo cofactor, where oxypurinol then blocks the catalytic activity by coordinating tightly to the reduced form of the Mo center and replacing specifically the Mo-OH group of the native enzyme [13]. Thus, allopurinol gradually inhibits the activity of XO, and finally reaches a steady state due to the irreversibility of the inhibition, as shown in the time course in this study and several other studies [13,40]. In this study, the time course of DHNB-mediated inhibition of XO activity had a pattern similar to that of allopurinol-mediated inhibition, with or without pre-incubation of XO and the inhibitor. Other aldehydes, DH6NB and THB-CHO, also showed a time-dependent XO inhibition, although their effects were weaker than that of DHNB. Furthermore, the inhibitory effect of DHNB was not reversed by exposing it to high-doses of reducing agents, including GSH, MTT, and 2-ME. This indicates that DHNB irreversibly inhibits XO under the experimental conditions. In other cases, thiol-containing reducing agents, such as GSH or DTT, can restore the enzymatic activity of nitroalkene-inhibited enzyme, indicating reversibility [38].
Whether DHNB interacts with the Mo cofactor of XO or with any amino acids is not clear, and its specific function remains unknown. Structurally, it might be that the electron-withdrawing ability of the nitro group increases the electrophilicity of benzaldehyde, thus increasing its reactivity toward the nucleophilic oxygen of Mo-O, or nitrogen of an amino group of XO. It is possible that a functional group at 5-position interacts electrostatically with a group near the Mo cofactor of XO and enhances the binding of DHNB to XO. For example, in a separate study, a nitro group was very crucial in the inhibition of XO activity by nitro-oleic acid [14]. Our results reveal that DHNB’s ability to inhibit XO activity is highly structure specific, indicating that electrostatic interactions between DHNB and particular domains on the XO protein may be crucial.
The kinetic analysis indicates that the inhibitory effect of DHNB on XO is mixed-type inhibition, which is similar to febuxostat [42]; but may be different with allopurinal. Previous study indicates that allopurinal shows competitive-type inhibition of XO at relatively low concentrations [43]. For the mixed-type inhibition, the inhibitor can bind to the active site as the enzyme’s substrate or to an allosteric site of the enzyme, which may affect the binding of the substrate. In this study, DHNB decreased the Vmax and increased the Km of XO; the inhibitory effect of DHNB was not reversed by reducing agents or overcome by increasing concentrations of substrate xanthine. Most importantly, based on our product analysis of the reaction of DHNB and XO, DHNB is slowly oxidized by XO to carboxylic acid; and XO activity can be recovered once DHNB is oxidized. Thus, DHNB does not permanently damage XO. DHNB binds to most possibly the Mo center, the active site of XO enzyme. The oxidation of DHNB to its carboxylic acid might be similar to that of xanthine to uric acid. We propose a mechanism for the inhibitory effect of DHNB on XO activity based on the strong inhibition, product analysis, and pH effect on the inhibition (Fig. 8). Aldehyde DHNB could be converted to carboxylic acid, and this process is relatively slow and pH dependent. An active base glutamate may abstract the proton from the Mo-OH group, which could initiate a nucleophilic attack on the carbonyl group of DHNB and a concomitant hydride transfer to Mo=S group, yielding an intermediate product coordinated to the reduced Mo center via the Mo-O group. This intermediate may break down by deprotanation and electron transfer to other redox-active centers, such as [2Fe-2S] cluster and FAD site in the enzyme, forming a new intermediate. Displacement of the product by hydroxide from solvent follows, to return to the starting state. In our study, we found that a light acidic solution greatly decreased XO activity, but enhanced the inhibitory effect of DHNB. An acidic solution may favor the protonation of −CHO group leading to an increase of its electrophilicity. The subsequent replacement by the hydroxide may be not favored, thus DHNB may be bound tightly with Mo center. On the other hand, a basic solution may favor the hydrogen abstraction by glutamate and the replacement of product, thus reducing DHNB-mediated inhibition of XO activity. Therefore, the structure of DHNB as an XO inhibitor is unique, because changing any of the functional groups on DHNB will change its ability to inhibit XO activity.
Fig. 8.
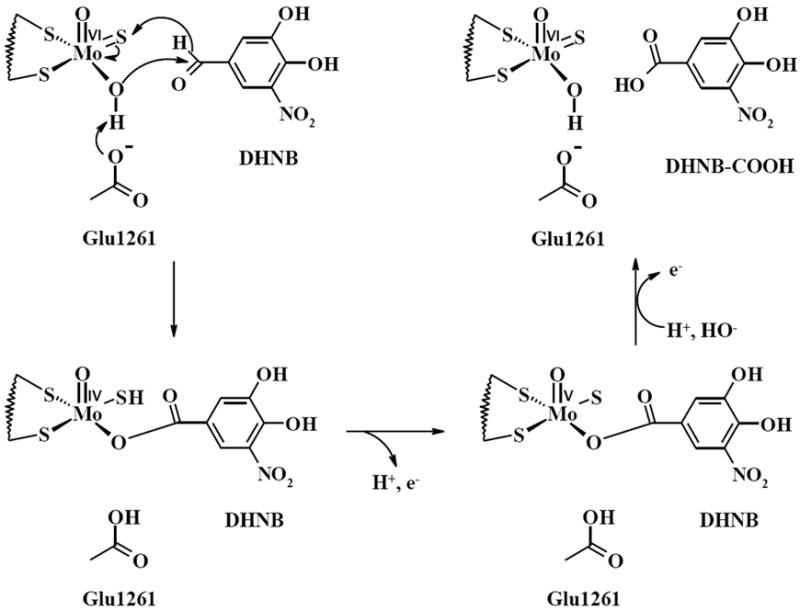
Proposed mechanism of the inhibitory effect of DHNB on XO enzyme. Aldehyde DHNB could be converted to carboxylic acid at the molybdenum (Mo) active center of XO. The reaction between DHNB and XO is relatively slow and pH dependent.
To study the hypouricemic effect of DHNB in vivo, we used a hyperuricemic mouse model using allantoxanamide, an inhibitor of uricase. Allantoxanamide elevated serum uric acid progressively to a much higher level than that maintained by repeatedly dosing with potassium oxonate [28,35,44]. Indeed, we have shown that a single i.p. injection of 100 or 200 mg/kg of allantoxanamide substantially increased serum uric acid levels in mice for 4 hours. Intragastric administration of DHNB dose-dependently reduced serum uric acid levels in the hyperuricemic mice, although the hypouricemic action of DHNB was slightly less than that of allopurinol. The hypouricemic action of DHNB began less than 1 h after the administration; this was as fast as that of allopurinol.
More importantly, DHNB, as a derivative of a natural product, might be much safer than allopurinol. Indeed, we showed that 12 mice that received each 500 mg/kg DHNB orally, did not present any toxicity and neither did their offspring. In contrast, 42% of the mice treated with oral administration of 500 mg/kg allopurinol died after 3 days, and the offspring of the surviving mice showed fur loss. Therefore, DHNB is worthy of further development as a potential XO inhibitor.
In addition to being an XO inhibitor, DHNB is a potent antioxidant. Our current data showed that DHNB directly scavenged several high reactive free radicals, including ONOO−, HOCl, and DPPH in a cell-free system; while allopurinol had no direct free radical scavenging activity. There are no previous reports describing this antioxidant activity of DHNB. Our previous study demonstrated that DHBA significantly reduced oxidative stress and endothelial dysfunction in human endothelial cells and porcine coronary arteries treated with the antiretroviral drug ritonavir [45]. It is well known that oxidative stress plays a critical role in the development of cardiovascular disease, and XO is one important source of the superoxide anion, which can be converted to ONOO− and HOCl [46,47]. Hyperuricemia is also considered a risk factor for cardiovascular disease [8,11,47]. Therefore, in addition to the treatment of hyperuricemia and gout, DHNB may have a potential application in the prevention and treatment of cardiovascular disease by its XO inhibition and antioxidant activities.
Acknowledgments
This work is supported by a research grant from the National Institutes of Health (R21 AR-063017 to C. Chen) and a Pilot Project in Experimental Therapeutics from the Baylor College of Medicine (to JM Lu). The authors would like to thank Dr. Ming Hu and Dr. Zheng Yang from the University of Houston, Houston, Texas, for their technique assistance in the Mass Spectroscopy analysis. The authors would also like to thank Dr. Ana María Rodríguez for her critical reading and editing of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Richette P, Bardin T. Gout. Lancet. 2010;375:318–28. doi: 10.1016/S0140-6736(09)60883-7. [DOI] [PubMed] [Google Scholar]
- 2.Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40:437–42. doi: 10.1038/ng.106. [DOI] [PubMed] [Google Scholar]
- 3.Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801–8. [PubMed] [Google Scholar]
- 4.Hille R. Molybdenum-containing hydroxylases. Arch Biochem Biophys. 2005;433:107–16. doi: 10.1016/j.abb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med. 2002;33:774–97. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- 6.Gray CL, Walters-Smith NE. Febuxostat for treatment of chronic gout. Am J Health Syst Pharm. 2011;68:389–98. doi: 10.2146/ajhp100394. [DOI] [PubMed] [Google Scholar]
- 7.Love BL, Barrons R, Veverka A, Snider KM. Urate-lowering therapy for gout: focus on febuxostat. Pharmacotherapy. 2010;30:594–608. doi: 10.1592/phco.30.6.594. [DOI] [PubMed] [Google Scholar]
- 8.Chao J, Terkeltaub R. A critical reappraisal of allopurinol dosing, safety, and efficacy for hyperuricemia in gout. Curr Rheumatol Rep. 2009;11(2):135–40. doi: 10.1007/s11926-009-0019-z. [DOI] [PubMed] [Google Scholar]
- 9.Dubchak N, Falasca GF. New and improved strategies for the treatment of gout. Int J Nephrol Renovasc Dis. 2010;3:145–66. doi: 10.2147/IJNRD.S6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khanna D, Khanna PP, Fitzgerald JD, Singh MK, Bae S, Neogi T, Pillinger MH, et al. American College of Rheumatology guidelines for management of gout. part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res. 2012;64(10):1447–61. doi: 10.1002/acr.21773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agabiti-Rosei E, Grassi G. Beyond gout: uric acid and cardiovascular diseases. Curr Med Res Opin. 2013;(Suppl 3):33–9. doi: 10.1185/03007995.2013.790804. [DOI] [PubMed] [Google Scholar]
- 12.Niu Y, Zhu H, Liu J, Fan H, Sun L, Lu W, Liu X, Li L. 3,5,2’,4’-Tetrahydroxychalcone, a new non-purine xanthine oxidase inhibitor. Chem Biol Interact. 2011;189:161–6. doi: 10.1016/j.cbi.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Pauff JM, Hille R. Inhibition studies of bovine xanthine oxidase by luteolin, silibinin, quercetin, and curcumin. J Nat Prod. 2009;72:725–31. doi: 10.1021/np8007123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelley EE, Batthyany CI, Hundley NJ, Woodcock SR, Bonacci G, Del Rio JM, Schopfer FJ, Lancaster JR, Jr, Freeman BA, Tarpey MM. Nitro-oleic acid, a novel and irreversible inhibitor of xanthine oxidoreductase. J Biol Chem. 2008;283:36176–84. doi: 10.1074/jbc.M802402200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Chen R, Shang Y, Jiao B, Huang C. Lithospermic acid as a novel xanthine oxidase inhibitor has anti-inflammatory and hypouricemic effects in rats. Chem Biol Interact. 2008;176:137–42. doi: 10.1016/j.cbi.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Murata K, Nakao K, Hirata N, Namba K, Nomi T, Kitamura Y, Moriyama K, Shintani T, Iinuma M, Matsuda H. Hydroxychavicol: a potent xanthine oxidase inhibitor obtained from the leaves of betel, Piper betle. J Nat Med. 2009;63:355–9. doi: 10.1007/s11418-009-0331-y. [DOI] [PubMed] [Google Scholar]
- 17.Sato T, Ashizawa N, Iwanaga T, Nakamura H, Matsumoto K, Inoue T, Nagata O. Design, synthesis, and pharmacological and pharmacokinetic evaluation of 3-phenyl-5-pyridyl-1,2,4-triazole derivatives as xanthine oxidoreductase inhibitors. Bioorg Med Chem Lett. 2009;19:184–7. doi: 10.1016/j.bmcl.2008.10.122. [DOI] [PubMed] [Google Scholar]
- 18.Sato T, Ashizawa N, Matsumoto K, Iwanaga T, Nakamura H, Inoue T, Nagata O. Discovery of 3-(2-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole, FYX-051 - a xanthine oxidoreductase inhibitor for the treatment of hyperuricemia [corrected] Bioorg Med Chem Lett. 2009;19:6225–9. doi: 10.1016/j.bmcl.2009.08.091. [DOI] [PubMed] [Google Scholar]
- 19.Yu Z, Fong WP, Cheng CH. The dual actions of morin (3,5,7,2’,4’-pentahydroxyflavone) as a hypouricemic agent: uricosuric effect and xanthine oxidase inhibitory activity. J Pharmacol Exp Ther. 2006;316:169–75. doi: 10.1124/jpet.105.092684. [DOI] [PubMed] [Google Scholar]
- 20.Chiang HC, Lo YJ, Lu FJ. Xanthine oxidase inhibitors from the leaves of Alsophila spinulosa (Hook) Tryon. J Ezny Inhib. 1994;8:61–71. doi: 10.3109/14756369409040777. [DOI] [PubMed] [Google Scholar]
- 21.Chan WS, Wen PC, Chiang HC. Structure-activity relationship of caffeic acid analogues on xanthine oxidase inhibition. Anticancer Res. 1995;15:703–7. [PubMed] [Google Scholar]
- 22.Beyer G, Melzig MF. Effects of selected flavonoids and caffeic acid derivatives on hypoxanthine-xanthine oxidase-induced toxicity in cultivated human cells. Planta Med. 2003;69:1125–9. doi: 10.1055/s-2003-45194. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen MT, Awale S, Tezuka Y, Tran QL, Watanabe H, Kadota S. Xanthine oxidase inhibitory activity of Vietnamese medicinal plants. Biol Pharm Bull. 2004;27:1414–21. doi: 10.1248/bpb.27.1414. [DOI] [PubMed] [Google Scholar]
- 24.Flemmig J, Kuchta K, Arnhold J, Rauwald HW. Olea europaea leaf (Ph.Eur.) extract as well as several of its isolated phenolics inhibit the gout-related enzyme xanthine oxidase. Phytomedicine. 2011;18:561–6. doi: 10.1016/j.phymed.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 25.Adams C, Gold V, Reuben DME. Reduction of carbonyl compounds by sodium borohydride (tetrahydridoborate) in water, dimethyl sulphoxide, and their mixtures as solvents: products and kinetics. J Chem Soc Perkin Trans. 1977;2:1466–72. [Google Scholar]
- 26.Medina-Campos ON, Barrera D, Segoviano-Murillo S, Rocha D, Maldonado PD, Mendoza-Patino N, Pedraza-Chaverri J. S-allylcysteine scavenges singlet oxygen and hypochlorous acid and protects LLC-PK(1) cells of potassium dichromate-induced toxicity. Food Chem Toxicol. 2007;45:2030–9. doi: 10.1016/j.fct.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Zhao F, Liu ZQ, Wu D. Antioxidative effect of melatonin on DNA and erythrocytes against free-radical-induced oxidation. Chem Phys Lipids. 2008;151:77–84. doi: 10.1016/j.chemphyslip.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 28.Yonetani Y, Iwaki K, Ogawa Y. Decreasing effect of allantoxanamide, a hyperuricemic agent on renal functions in rats. Jpn J Pharmacol. 1987;45:37–43. doi: 10.1254/jjp.45.37. [DOI] [PubMed] [Google Scholar]
- 29.Johnson WJ, Chartrand A. Allantoxanamide: a potent new uricase inhibitor in vivo. Life Sci. 1978;23:2239–44. doi: 10.1016/0024-3205(78)90210-2. [DOI] [PubMed] [Google Scholar]
- 30.Carroll JJ, Coburn H, Douglass R, Babson AL. A simplified alkaline phosphotungstate assay for uric acid in serum. Clin Chem. 1971;17:158–60. [PubMed] [Google Scholar]
- 31.Spector T, Hall WW, Krenitsky TA. Human and bovine xanthine oxidases inhibition studies with oxipurinol. Biochem Pharmacol. 1986;35:3109–14. doi: 10.1016/0006-2952(86)90394-1. [DOI] [PubMed] [Google Scholar]
- 32.Krenitsky TA, Spector T, Hall WW. Xanthine oxidase from human liver: Purification and characterization. Arch Biochem Biophys. 1986;247:108–19. doi: 10.1016/0003-9861(86)90539-4. [DOI] [PubMed] [Google Scholar]
- 33.Aversa ML, Meany JE. The inhibition of xanthine oxidase by acetaldehyde in aqueous solution. Physiol Chem Phys Med NMR. 1996;28:153–62. [PubMed] [Google Scholar]
- 34.Borgulya J, Bruderer H, Bernauer K, Zürcher G, Da Prada M. Catechol-O-methyltransferase-Inhibiting Pyrocatechol Derivatives: Synthesis and Structure-Activity Studies. Helvetica Chimica Acta. 1989;72:952–68. [Google Scholar]
- 35.Enroth C, Eger BT, Okamoto K, Nishino T, Pai EF. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: structure-based mechanism of conversion. Proc Natl Acad Sci USA. 2000;97:10723–8. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lundblad RL. Chemical Reagents for Protein Modification. CRC Press Inc; 2004. [Google Scholar]
- 37.Wikberg T, Vuorela A, Ottoila P, Taskinen J. Identification of major metabolites of the catechol-O-methyltransferase inhibitor entacapone in rats and humans. Drug Metab Dispos. 1993;21:81–92. [PubMed] [Google Scholar]
- 38.Perez RA, Fernandez-Alvarez E, Nieto O, Piedrafita FJ. Dihydroxynitrobenzaldehydes and hydroxymethoxynitrobenzaldehydes: synthesis and biological activity as catechol-O-methyltransferase inhibitors. J Med Chem. 1992;35:4584–8. doi: 10.1021/jm00102a011. [DOI] [PubMed] [Google Scholar]
- 39.Metz S, Thiel W. A combined QM/MM study on the reductive half-reaction of xanthine oxidase: substrate orientation and mechanism. J Am Chem Soc. 2009;131:14885–902. doi: 10.1021/ja9045394. [DOI] [PubMed] [Google Scholar]
- 40.Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem. 2003;278:1848–55. doi: 10.1074/jbc.M208307200. [DOI] [PubMed] [Google Scholar]
- 41.Batthyany C, Schopfer FJ, Baker PR, Duran R, Baker LM, Huang Y, Cervenansky C, Branchaud BP, Freeman BA. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281:20450–63. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takano Y, Hase-Aoki K, Horiuchi H, Zhao L, Kasahara Y, Kondo S, Becker MA. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 2005;76:1835–47. doi: 10.1016/j.lfs.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 43.Pacher P, Nivorozhkin A, Szabó C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114. doi: 10.1124/pr.58.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schroder K, Vecchione C, Jung O, Schreiber JG, Shiri-Sverdlov R, van Gorp PJ, Busse R, Brandes RP. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in ApoE knockout mice fed a Western-type diet. Free Radic Biol Med. 2006;41:1353–60. doi: 10.1016/j.freeradbiomed.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 45.Weakley SM, Jiang J, Lü JM, Wang X, Lin PH, Yao Q, Chen C. Natural antioxidant dihydroxybenzyl alcohol blocks ritonavir-induced endothelial dysfunction in porcine pulmonary arteries and human endothelial cells. Med Sci Monit. 2011;17:BR235–41. doi: 10.12659/MSM.881926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–73. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 47.Higgins P, Dawson J, Walters M. The potential for xanthine oxidase inhibition in the prevention and treatment of cardiovascular and cerebrovascular disease. Cardiovasc Psychiatry Neurol. 2009;2009:282059. doi: 10.1155/2009/282059. [DOI] [PMC free article] [PubMed] [Google Scholar]