

Abstract
Small, interfacial conformational changes occur in some antigen-antibody interactions. Using cryogenic electron microscopy (cryo-EM), we have demonstrated such changes in a major antigenic site of a poliovirus capsid protein. During cell entry, native human poliovirus (160S particle) converts to a cell-entry intermediate (135S particle) and later to an RNA-released (80S) particle. By mixing particles with Fabs of the neutralizing C3 monoclonal antibody, we labeled the external BC loop of the capsid protein VP1 (residues 95–105) in the 160S and 135S states. We then determined three-dimensional structures by cryo-EM and enhanced their interpretability by fitting high-resolution coordinates of C3 Fab and the capsid proteins into the density maps. Binding of C3 to either 160S or 135S particles caused residues of the BC loop, located on the tip of a prominent peak known as the “mesa”, to move by an estimated 5 Å. C3 antibodies are neutralizing and can bind bivalently. The orientation of the bound Fabs in our reconstructions suggests that C3 neutralizes poliovirus by binding two adjacent BC loops on the same mesa and inhibiting conformational changes in the viral capsid.
Keywords: antibodies, antibody neutralization, antigen-binding fragment (Fab), 135S cell-entry intermediate particle, 80S RNA-released particle, antibody-antigen interactions, antibody-protein interactions, C3 antibody, cryo-electron microscopy, electron cryo-microscopy, fragment antibody-binding (Fab), picornavirus, viral, virus-antibody interactions
Introduction
Antibody, antigen, or both may change conformation during antibody-antigen binding, e.g. (1). This example of an induced-fit is typically seen by comparing atomic-resolution structures. Here, by cryogenic electron microscopy (cryo-EM), we show evidence of a changed antigen conformation occurring in poliovirus.
Poliovirus capsid is composed of 60 copies each of four capsid proteins (VP1, VP2, VP3, and VP4), arranged with icosahedral symmetry (Fig. 1). The capsid surrounds an approximately 7500-nucleotide, single-stranded RNA genome. VP4 and the amino-terminal segments of VP1, VP2, and VP3 lie on the inner surface of the capsid with extended conformations. The external surface of the capsid is constituted of VP1, VP2, and VP3.
FIGURE 1. Prominent structural features of poliovirus.
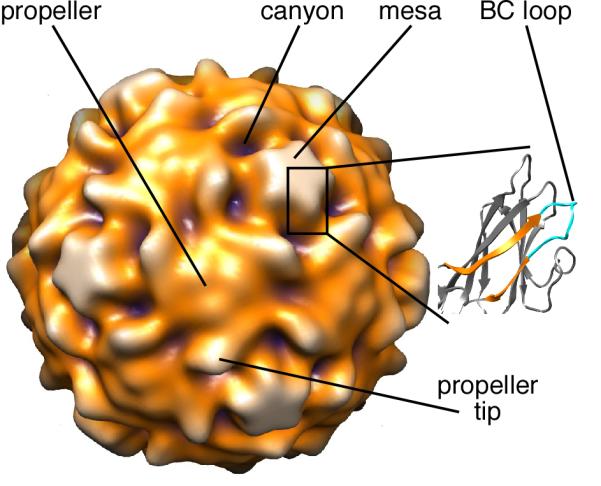
Prominent structural features on the exterior of a surface-rendered 160S poliovirus particle (17) are labeled. The expanded representation shows the core β-jellyroll subunit and BC loop of VP1 (2). The BC loop (cyan) is between β-strands B and C (orange) and is located at the tip of the star-shaped “mesa”. In the surface rendering, purple signifies features closest to the center of the particle, and white represents features farthest from the center.
A loop connecting the B and C β-strands of the VP1 β-jellyroll (the “BC loop”) is located at each tip of the star-shaped mesas that surround the particle fivefold axes (Fig. 1). As shown by several studies, this loop (residues 95–105 (2)) is a major epitope for neutralizing antibodies (3-10). One such antibody is monoclonal C3 (4). During cell-entry, poliovirus (sedimentation coefficient, 160S) binds to its receptor, CD155, which initiates conformational rearrangements and conversion to the cell-entry intermediate (135S) particle. After RNA is released, the empty capsid sediments at 80S. C3, which was raised by immunizing mice with the 80S particle (3), binds all three particles—160S, 135S, and 80S (11).
A crystal structure of the C3 Fab fragment, with a bound peptide corresponding to residues 93–103 of VP1, was determined previously (12). In this complex, the structure of the peptide differed significantly from its structure in the BC loop of the 160S particle, suggesting that the loop changes its structure upon antibody binding. This observation raised the question of whether the same phenomenon occurs when the antibody binds to an assembled particle.
To address this question, we mixed the C3 Fab with 160S and 135S particles and solved the structures of the respective complexes by cryo-EM. In both particle states, our results show that the conformation of the BC loop is affected by binding this antibody; fitting of the C3 Fab (12) and capsid protein (2, 13) coordinates into the cryo-EM density maps indicated that the Fab-bound epitopes had altered positions. The resulting structures also suggest that bivalent antibody binding occurs as the two Fab arms bind adjoining epitopes on a single pentameric mesa and that C3-Ab-induced neutralization occurs because a conformational change that is required for infection is inhibited.
Materials and Methods
Preparation of viral and Fab complex
Poliovirus virions (160S particles) and C3 Fab were prepared as described previously (12, 14, 15). 135S particles were prepared by heat treatment (50 °C for 3 min.) of 160S particles in low-salt buffer containing 2 mM CaCl2 (13, 15). Equal volumes of 160S-particle (3.9 mg/ml) and C3-Fab (1.7 mg/ml) solutions were mixed, giving a Fab-to-virus ratio of 74:1. A similar ratio was used to mix 135S particles and C3 Fabs.
Electron Microscopy
Mixtures of poliovirus (160S or 135S) and C3 Fab were suspended over holey carbon films, vitrified, and imaged as described previously (16). A CM200 electron microscope (FEI, Hillsboro, Oregon, USA) equipped with a Gatan 626 cryoholder (Pleasanton, California, USA) was used. Focal pairs of micrographs were recorded at magnifications of 38,000× at 120 kV. The electron dose was ~14 electrons per Å2.
3-D reconstruction
Particle images were extracted, processed, and normalized as described (17). Focal settings ranged from 0.73 to 1.77 μm underfocus. Focal-pair images were computationally combined for orientation determination but used separately for origin determination and computation of the 3D reconstruction. A model determined via common lines (18) and a previous reconstruction (17) were used to begin iterative projection-matching to determine particle orientations and origins for the 160S-C3 and 135S-C3 reconstructions, respectively (19, 20). Bsoft routines were used to assess and correct for contrast transfer function (CTF) effects (21) via the algorithm by Conway and Steven (22), but images within a focal pair were not combined during CTF correction. However, for orientation and origin determination, only phase-flipping CTF corrections were performed. Spherically averaged density plots were used to calibrate size (21, 23) against previously solved structures (17) and a poliovirus X-ray crystal structure (2). For surface renderings of 3D maps, contour levels were designated by the number of standard deviations (σ) above the average map density.
Modeling
Atomic coordinates for the C3 Fab-peptide complex (12) (1FPT in Protein Data Bank) were first fitted “by-eye” into the 160S-C3 and 135S-C3 density maps by use of the UCSF Chimera package (24) or the program O (25). Next, a core-weighted, rigid-body fitting algorithm, implemented in CHARRM (26), was used to refine the fits. In determining placement of the coordinates in the cryo-EM map, the CHARRM routine computed a “theoretical map” from the atomic coordinates at the same resolution as the cryo-EM map and then aligned the internal (“core”) density and overall domain shape of the theoretical and cryo-EM structures. The fit was then observed graphically and was judged correct if the overall shape of the coordinates matched that of the observed map. The Fab antigen-binding or variable domain and the Fab non-antigen-binding or constant domain were fitted separately. We included peptide coordinates (corresponding to residues 97 to 103 of VP1) in our fitting of the variable domain. To fit the antigen-binding domains, five Fabs were fitted separately to the five tips of one mesa. Symmetry was applied so that a single Fab binding-site contained five overlapping sets of fitted coordinates. Then, the overlapping coordinates were averaged and used to calculate fitting errors. The root-mean-square deviation (RMSD) of the five fits was computed as the fitting error. In Chimera (24), the separately fitted constant and variable domains of the Fab were joined and elbow-angle residues were energy-minimized. The program RBOW (27) was then used to calculate elbow angles.
The C3 Fab coordinates included a peptide antigen, residues 93–103 of VP1 (12). Residues 97–103 were visible in the binding cleft of the C3 antigen-binding domain, and we compared the Cα positions of these seven residues to the positions of the corresponding residues in the 160S crystal structure (2) and the 135S pseudo-atomic model (13) placed into the 160S-C3 and 135S-C3 maps. Using model and fitted coordinates, we used the match alignment and measure rotation functions in UCSF Chimera (24) to determine the shift and twist of the BC loop between the 160S and 135S models and the shift and twist between the peptide antigen in the 160S-C3 and 135S-C3 fitted coordinates. This shift and twist was along an axis determined by the algorithm. For a pseudo-atomic model of the virus-Fab complexes, peptide residues 97–103 from the fitted Fab coordinates were used to replace corresponding residues for VP1 in the 160S crystal structure (2) and in the 135S pseudo-atomic model (13). These seven residues were joined to the unaltered B and C β-strands via energy-minimization (in UCSF Chimera (24)) of residues 95–96 and 104–105.
Results
We computed three-dimensional (3D) reconstructions of the 160S-C3 and 135S-C3 complexes by cryo-EM (Table I, Figs. 2, 3A). Their resolutions were 11 and 9 Å, respectively. As expected, C3 Fab binds to the tip of the mesa—where the BC loop resides in both the 160S (2) and 135S (13) particle states (Figs. 2,3). Five C3 Fabs surround each mesa, giving it the appearance of a five-petaled flower. C3 Fab density is as strong as the capsid protein density (Fig. 2C), indicating full or nearly full occupancy of all 60 binding sites per virus. Particularly in the 135S-C3 reconstruction, Fab antigen-binding domains show the appearance of β-sheets (for which ~10-Å resolution is needed, cf. (28)) and indicate that the bound Fabs are firmly held in place (Fig. 2C, white arrows).
Table I.
Microscopy, image reconstruction, and modeling data
| Sample | Micrograph focal pairsa |
Pixel size (Å) |
Particle imagesb |
CTF correctionc |
Resolu tiond (Å) |
EMDB IDe |
PDB IDf |
|---|---|---|---|---|---|---|---|
| 160S-C3 | 6 | 1.82 | 4184 | full | 11.1 | 5291 | 3J3O |
| 135S-C3 | 2 | 1.84 | 9810 | full | 9.1 | 5292 | 3J3P |
| 80S-C3g | 7 | 1.84 | 238 | phases only | 22 | 5293 | ---- |
Pairs of micrographs taken of the same field-of-view and used to compute 3D reconstructions, first image taken closer-to-focus, second image farther-from-focus. The 135S-C3 micrographs contained many more particles per micrograph than did micrographs of 160S-C3 complexes. Therefore, only two micrograph pairs were needed to compute the 135S-C3 structure.
Total number of particle images extracted from micrographs and used to compute the 3D reconstruction, divide by two for the number of image pairs (focal pairs).
Correction for the final, published reconstruction: full, deconvolution of CTF and correction for decay (see Materials and Methods); or, phases-only, corrected only for the phase-flipping effects of the CTF.
Data set split in two, reconstructions computed and compared via Fourier shell correlation. Resolution determined by the point at which the Fourier shell correlation value reaches 0.5, for reconstructions computed from images that were only corrected for phase-flipping of the CTF.
Identification code of density map in EM Data Bank.
Protein Data Bank identification code for coordinate modeling results.
FIGURE 2. Binding of C3 Fab to poliovirus.

A) Complex of 135S particles and C3 Fab imaged via cryo-EM. Bound Fabs can be seen as fibrous extensions surrounding each round particle. One 80S-C3 complex is seen in this view (arrow). Bar, 100 nm. B,C) C3 Fab bound to 160S (left column) and 135S (right column) particles. For each image pair (same row), maps are viewed from the same vantage point—and therefore are to scale. B) In the top two rows, renderings are viewed along a fivefold symmetry axis. In the bottom two rows, views are 90° rotated from the corresponding view in the top two rows. Within each grouping, the gray-scale view shows Fabs bound at one fivefold mesa, and the colored view is a close-up view of one bound Fab (cf. Video 1). Coordinates of the Fab and bound peptide (12) were fitted (ribbon) into the cryo-EM density of the mesa-bound Fab (mesh). In the gray-scale panels, ribbon is black for both Fab and peptide. In the colorized panels, Fab (variable domain) and bound peptide (VP1 residues 97–103) ribbons are purple and green, respectively. Mesh is surface rendering of 3D reconstruction contoured at 0.5σ. The modeled 160S-C3 and 135S-C3 peptides are twisted 13° and shifted 8 Å with respect to each other, reflecting the overall twist observed in the antigen-binding domain of the Fab (cf. Videos 1,2). C) From maps viewed along a twofold symmetry axis, central density slices from cryo-EM maps of C3 complexed with 160S (left) and 135S (right). Relative density estimates: a sphere of radius = 3 pixels was centered on the designated spots (see “Fab” and “Capsid” labels in right panel) and the average density computed. The density of C3 is almost the same as the density of the poliovirus capsid in each case. β-sheet structure was observed in both the Fab (small white arrows) and capsid (small black arrows) densities. Inset (left panel), a non-central density slice of the 160S-C3 map gives a better view of β-sheets within the Fab density. The β-sheet structure is only seen if the imaged objects are well ordered. Another indication that the Fabs were rigidly ordered was an assessment of the resolution of small volumes of the maps (“local-resolution test”) (see (46)), where flexible domains have lower resolution than rigid domains. The antigen-binding domains and the adjacent rigid capsid were shown have the same high-resolution value (data not shown).
FIGURE 3. Interaction of Fabs with BC loops.

A) Elbow axes of bound Fabs. Top row, stereo view of a surface rendering of the 160S-C3 reconstruction viewed along a twofold symmetry axis. Darker regions are closer to the center of the particle. Bottom row, stereo close-up view of region near a twofold axis of 160S-C3 reconstruction (surface rendering). Lines represent elbow axes estimated for Fabs in virus-Fab complexes—showing their relative relationships: cyan (C3 Fab attached to 160S poliovirus, Fab shown), dark blue (C3 Fab attached to 135S poliovirus, Fab not shown), white (Fab1 attached to human rhinovirus 14, Fab not shown) (39), and green (Fab17-IA attached to human rhinovirus 14, Fab not shown) (36, 39). In contrast to antibodies that bind two BC loops in adjacent pentamers across each twofold axis in human rhinovirus 14 (39, 40), the binding aspect of the C3 Fab suggests that bivalent binding occurs on adjacent BC loops on the same pentamer (cf. Fig. 4). The position and orientation of the C3-Fab elbow can only allow the constant domain of the Fab to bend towards or away from an adjacent Fab on the same pentamer. In contrast, the binding aspect of Fab17-IA on human rhinovirus 14 (green line) allows bending of the elbow to bring two constant domains closer to the twofold axis, facilitating binding of an antibody across the twofold axis (40). The binding aspect of Fab1 (white line) is intermediate between Fab17-IA and C3 and, as suggested by binding experiments with antibody (39), likely means monovalent attachment of antibodies.
B) C3-Fab binding changes the conformation of poliovirus 160S (top) and 135S (bottom) BC loops. Views shown as stereo pairs. Mesh is surface rendering of cryo-EM reconstructions at relatively high contour levels (160S-C3 at 1.5σ and 135S-C3 at 2.5σ) to emphasize the core structure and the connection between Fab and virus. Fitted Fab (magenta ribbon), peptide residues 97–103 (green ball-and-stick), and VP1 (blue and cyan ribbons) coordinates are shown. Residues within the BC loop (95–105) were adjusted to the Fab-bound position and are shown in blue. The non-Fab-bound position of the BC loop (2) is shown in cyan.
C) Alignment of poliovirus (dark blue) and rhinovirus (light blue) B and C β-strands and BC loops shown as an amino-acid sequence (top) and a 3D-structure (bottom). Coordinates from the VP1 protein from crystal structures of poliovirus (dark blue) (2) and human rhinovirus 14 (light blue) (36) were aligned using the MatchMaker function in UCSF Chimera (24). (Only the BC region is shown.) The sequences were then aligned to the 3D-structure alignment (dark blue, poliovirus; light blue, HRV14). Because the poliovirus BC loop is four residues shorter than the HRV14 loop, some gaps were artificially placed in the poliovirus sequence to emphasize the alignment at the N- and C-terminal ends of the loop and align other residues within the loop (though the 3D structure shows the mid-loop sequence alignments are insignificant to the 3D structural alignment). Poliovirus secondary structure assignments are shown (top, right).
Coordinates of the C3 Fab-peptide complex (12) were fitted into the cryo-EM density maps and used to infer the position of the BC loop in Fab-bound 160S and 135S particles. For the antigen-binding (variable) domain, five regions of C3 Fab density around one fivefold vertex were used to fit coordinates and the average (after symmetry operations to bring four of the five fittings to one position) of all five fits is reported here. Only one region was used to fit the non-antigen-binding (constant) domain. The elbow angle of C3 Fab varied significantly between the 160S-C3 (168.6°) and 135S-C3 (182.7°) states and both differed from the angle in the Wien et al. (12) crystal structure (132.6°). Coordinates from a previously determined crystal structure of the 160S particle (2) and a pseudo-atomic model of the 135S particle (made by fitting 160S coordinates into a 135S cryo-EM map) (13) were placed, initially without adjustment, in the 160S-C3 and 135S-C3 maps and used for comparison.
In the 135S preparation used to make the complex, a few of the particles were 80S and were readily recognizable as such from their low-density centers (Fig. 2A, black arrow) (cf. (15)). A reconstruction of the 80S-C3 complex was also computed, although the small number of particles limited the resolution (Table I). As expected, the 80S-C3 reconstruction showed that the C3 Fab bound with high occupancy in a position and orientation similar to that seen in the higher resolution 135S-C3 complex (Supplementary Materials).
Particle Expansion During 160S-to-135S Transition
Our measurements from the 160S-C3 and 135S-C3 maps indicate that, during the 160S-to-135S transition, the BC loop moves radially to the same extent that it does in undecorated poliovirus (Table SI; Figs. 2B–C; Videos 1,2). Assuming that the peptide in the C3 Fab crystal structure (fitted into the 160S-C3 and 135S-C3 maps) represents the correct position of the BC loop, the BC loop moved radially outwards by 8 Å (5.0%) during the 160S-to-135S transition. Comparison of fitted coordinates of the five C3 densities surrounding a single fivefold vertex showed that the distance between the BC loop and the center of the mesa increased by 1 Å. These movements are consistent with previous observations of the undecorated 160S and 135S particles, where comparisons of cryo-EM reconstructions showed a 4% expansion along the fivefold symmetry axis (from top of mesa to center of particle) (17). Measurements of published models of the BC loops (2, 13) indicated that the distance from the center of the capsid increased by 7 Å (4.5%), and the distance between BC loops and the center of mesa increased by 1 Å. Therefore, positioning of the modeled BC loop in the 160S-C3 and 135S-C3 complexes is consistent with placement of the BC loop in models of 160S (2) and 135S (13) particles.
Antibody binding changes BC loop position
One of the issues encountered in fitting atomic-resolution structures into lower-resolution cryo-EM reconstructions (here, ~10 Å) is the need to minimize the number of parameters. Usually, this need is met by using rigid-body approximations for protein subunits, which limits the number to six, i.e. three translational and three rotational parameters. Here, the prominent marker provided by the Fab density and the presence of the BC-loop peptide in the C3 Fab crystal structure provided the opportunity to model in greater detail. In particular, we could account for structural changes that take place in the loop and position it more precisely in the 160S-C3 and 135S-C3 complexes. At the current resolutions, no significant changes were observed outside the region of the BC loop.
Using the Fab as a marker, our modeling places the Fab-bound BC loop in a different position with respect to the remainder of VP1 than was observed for the unbound loop in the crystal structure of the 160S particle (2) or in the model of the 135S particle (derived by rigid-body fitting of the capsid proteins to a sub-nanometer-resolution cryo-EM reconstruction) (13) (Fig. 3B). To measure the deviation, we compared the positions of seven residues in the binding cleft of the fitted C3 Fab coordinates with their corresponding position in the 160S (2) and 135S (13) models. That deviation averages to 5 (± 2) Å in both cases (Table SII). The direction of the Fab-bound deviations is the same for both 160S-C3 and 135S-C3 (Fig. 3B).
Although the C3 Fab crystal coordinates included only a peptide of residues 93–103 (12), the position of the seven BC-loop residues in the fitted antigen-binding cleft (amino acids 97– 103) should mimic the position of the BC loop when C3 Fab is bound to 160S or 135S particles. Therefore, the observed 5-Å deviations indicate that the BC loop moves upon binding of C3 Fab to either 160S or 135S particles. Our modeling is based on the fitting of the antigen-binding domain into five symmetry-related positions in the cryo-EM density. We determined the fitting error (root-mean-square deviation, RMSD, of five averaged fits) to be 0.5 and 0.4 Å for the 160S-C3 and 135S-C3 fits, respectively. We compared bound residues 97–103 in the fitted Fab structures (Cα coordinates only) to their corresponding residues in the BC loop of the 160S crystal structure (2) or 135S pseudo-atomic model (13). We found a deviation per residue between 2.5 and 9.0 Å (Table SII). Coordinates of bound peptide residues 97–103 were joined to the remainder of VP1 for coordinate models that combined capsid and Fab residues (Fig. 3B).
The Fab-bound C3 epitope—the BC loop—twists more extensively between the 160S and 135S structures than was deduced in the 135S pseudo-atomic model (13) (Fig. 2B; Videos 1,2). Comparison (via UCSF Chimera) of the 160S-to-135S BC loop change in the crystal structure of 160S particle and the 135S model showed a rotation of 4.6° and a shift of 3 Å. Comparison of the same change in the fitted coordinates of the 160S-C3 and 135S-C3 models showed a rotation of 13° and a shift of 8 Å.
Discussion
C3 Fabs bound to ~60 mesa tips per particle
In the 160S-C3, 135S-C3, and 80S-C3 reconstructions, the Fab was visualized bound to the tip of the 5-fold mesa (Figs. 2,3,S1). This was expected from previous crystal and cryo-EM studies of 160S, 135S, and 80S particles (2, 13, 17, 29, 30) and from the structure of the C3-peptide and Fab complex (12).
Because the Fab and capsid densities are similar (Figs. 2C,S1), all or almost all of the 60 epitopes per particle bound a Fab. This finding is consistent with the previous observation that approximately 60 Fabs can bind per virion (12).
BC loop shifts upon Fab binding
Residues in the fitted BC-loop peptide deviate 2.5–9.0 Å (average, 5 ±2 Å; RMSD, 5.7 Å) from their positions in the crystal structure of the 160S particle (2) and the model of the 135S particle (13) (Table SII; Fig. 3B). This shift is consistent with the previous observation that, in the Fab-peptide complex, the structure of the peptide differs significantly from its structure on the surface of the 160S particle (12). Together, these two studies indicate that the loop is sufficiently flexible to change structure upon antibody binding.
The C3 Fab coordinates fit snugly within the Fab density in the cryo-EM 160S-C3 and 135S-C3 maps (Figs. 2B,3B), giving confidence in our fitting and in the inference of Fab-induced BC loop movement. Some algorithms for fitting coordinates within cryo-EM density maps rely only on the overall shape of the macromolecule, but we used a fitting algorithm that emphasized internal density as well as the molecular envelope (26). Because Fabs have a pseudo-twofold axis along their longest dimension, fitting may appear comparably good if the domains are rotated 180 deg. about this axis. Remarkably, in our core-weighted fittings for both reconstructions (160S-C3 and 135S-C3), light chains for the separately fitted constant and variable domains were consistently placed on the same side of the Fab. (Therefore, heavy chains were also consistently placed.) This consistency suggests that our data have sufficient resolution for the core-weighting algorithm to distinguish light and heavy chains and would, therefore, place the antigen-binding cleft in the correct position.
The resolution of the cryo-EM maps (9–11 Å) was sufficient to allow precise fitting of the Fab coordinates within the cryo-EM density. The low error of the core-weighted fitting (26) for the antigen-binding domains implies that the 160S-C3 (RMSD uncertainty, 0.5 Å) and 135S-C3 (RMSD uncertainty, 0.4 Å) reconstructions were sufficiently detailed to allow such precise fitting and measurement of the BC-loop deviation. This may at first seem counter-intuitive, given the limited resolution of the reconstructions, but the confusion stems from a common misconception of resolution. The resolution of a structure is the minimum distance between two objects where the two objects are seen to be separate. In contrast, the fitting error is a measure of how precisely properly constrained models can be placed into a map, and is always much better than the nominal resolution of the map.
Our results are consistent with a two-step binding model proposed previously (12). Wien et al. (12) fitted their crystal structure of the C3-Fab-peptide complex onto the poliovirus crystal structure and found that their fit was incompatible with their observation that 60 C3 Fabs were capable of binding to one virus particle. Their fit showed steric overlap for adjacent Fabs in the outermost β-strands of the antigen-binding domains. They proposed a two-step binding model where initial binding of the variable region induced a conformational change in the BC loop. Our results are consistent with this model because we showed i) the loop conformation is changed (Fig. 3B) and ii) ~60 Fabs did bind per virion (Fig. 2C).
Although the BC loop is readily accessible to antibodies, its apparent flexibility may facilitate similar binding to conformationally distinct poliovirus particles. As can be inferred from antibody titer experiments, binding avidities of C3 antibodies for 160S, 135S, and 80S particles are nearly identical (11). In addition, we observed similar Fab : capsid density ratios in the 160S-C3, 135S-C3, and 80S-C3 3D maps (Figs. 2C,S1). Therefore, if the conformation of the BC loop differs among the 160S, 135S, and 80S states, the BC loop appears to have enough flexibility to change conformation to optimally bind the C3 antibody.
Paratope and Epitope Flexibility
To make the binding interaction stronger and more specific, the paratope (of the antibody) and the epitope (of the antigen) may change conformations during binding (1). For example, the binding of antibody to lysozyme induced conformational changes in the antigen at a demonstrably flexible region (1). For the interaction of HIV-1 capsid protein p24 with Fab13B5, conformational changes were observed in both antibody and antigen (31). In the Fab, shifts were observed in complementarity-determining regions (CDRs) and in an 8° relative rotation between the heavy and light chain variable domains. In the epitope, Pro207 and Ala208, located in a turn between two helices, moved after Fab binding. In the current study, we showed epitope flexibility by cryo-EM.
The deviation of the fitted, bound epitope is identical (within experimental error) for the 160S-C3 and 135S-C3 structures (Table SII), further strengthening the conclusion that binding of C3 Fab causes the BC loop to change conformation. Although the Fab-induced movement of the BC loop is not to the same relative position for the two particle states (data not shown), the movement is in the same general direction (Fig. 3B). The consistent deviation and direction for the 160S-C3 and 135S-C3 structures suggests that the Fab interacts in a similar manner with the two particle states and that the neighboring VP1 structure is also similar for the 160S and 135S particles.
The C3 paratope may also shift, but we cannot determine such a shift from our experiment. The previously published crystal structure of C3 Fab with a bound antigenic peptide also did not explore the question of paratope shifts because the only structure determined was that of Fab with antigen bound (12). Of course, differences may exist between the way the Fab binds free peptide and the constrained BC loop (on a virus particle). Such differences, if they occur, are probably found in minor stabilizing interactions, and the binding cleft in the Fab is unlikely to be in a different position. Because the major Fab-virus interaction is between the binding cleft and the epitope, any major Fab changes, either in the CDRs or between heavy and light chain variable domains (e.g. (31)), should be represented in the model of C3 Fab bound to the BC-loop peptide (12).
However, our fitting results suggest that the BC loop has a slightly different environment in the 160S and 135S particles. Deviations of the fitted BC loop from the previously modeled loops (2, 13) are slightly different in the 160S-C3 and 135S-C3 structures (Table SII, Fig. 3B). (For the 135S-C3 experiment, C3 Fab was not added until 135S particles had been made.) For example, during the 160S-to-135S transition, the BC loop in 160S-C3 and 135S-C3 complexes rotates 13° and shifts 8 Å, but in the unbound models (2, 13), the BC loop only rotates 4.6° and shifts 3 Å between the 160S and 135S structures.
The observed shift of the BC loop suggests that the loop is not fixed in a certain conformation, but, given the appropriate conditions, can easily adjust. The B-factors (temperature factors) for the BC loop are relatively high, suggesting that the loop is flexible (Table SII). However, this flexibility is probably not random, but is dictated by interactions with other proteins. Binding of Fab to the BC loops may simply trap the BC loop at one extreme of its normal deviation. In our structures, the conformational change appears to be induced by the antibody-antigen binding interaction, cf. (1). This inducible flexibility may be important in other viral processes, including cell entry. For example, poliovirus receptor (CD155) is known to also interact with the mesa tip (32-35) and a flexible BC loop may contribute to that interaction.
BC loop appears inflexible in related rhinovirus
Curiously, as shown in a crystal structure (36), the BC loop of the closely related human rhinovirus 14 (Fig. 3C) did not change conformation when bound to a Fab: only the paratope conformation changed significantly. This functional difference may result from differing binding aspects and interactions (data not shown). However, for most of the residues observed in contact with the Fabs, the B-factors are significantly (>20 Å2) higher for poliovirus VP1 (residues 97–102) (2) than for human rhinovirus 14 VP1 (residues 91–95) (37). Because increased flexibility correlates with higher B-factors, the poliovirus BC loop appears to be significantly more flexible and, therefore, more inclined to change conformation.
Mechanism of Neutralization
C3 antibody binds bivalently to poliovirus, and that binding is neutralizing, even at low antibody-to-virus ratios (12, 38). The binding of only four antibodies per virion caused a 99% loss in infectivity, but did not affect cell attachment. Therefore, the mechanism for neutralization does not involve blocking of receptor-binding sites. In contrast, C3 Fab only partially neutralizes the virus at very high Fab-to-virus ratios (≥30 copies per virion) and the extent of neutralization correlates well with the ability of the Fab to block cell attachment (38). Given that i) C3 Fab and antibody bind with nearly equal affinity to 160S and 135S particles; ii) at low copy number, antibodies are strongly neutralizing and do not block attachment; and iii) at high copy number, Fabs neutralize weakly and block attachment; neutralization by the C3 antibody most likely stems from the ability of bivalently attached antibodies to inhibit a post-attachment, receptor-mediated conformational change—perhaps by inhibiting expansion of the capsid shell (Fig. 4). This inference was proposed by Wien et al. (12), but in the absence of structural constraints, they were not able to deduce a detailed mechanism for how bivalent attachment could block structural transitions. We have used the observed structures of the Fab-labeled complexes to model how bivalent attachment occurs (Fig. 4A) and its role in inhibiting structural changes.
FIGURE 4.

Model for inhibition of 160S-to-135S transition by C3 antibody. (A) Models of C3 Ab (blue ribbon) bound to 160S (left) and 135S (right) particles. Fitted coordinates for Fab (from (12)) were placed in adjacent epitopes, and their non-antigen-binding (constant) domains were rotated towards each other about their respective elbow angles to allow the two Fabs to be joined to coordinates from the Fc portion of an IgG crystal structure (47). The antigen-binding (variable) domains were not moved. The IgG model was made with the use of UCSF Chimera (24). The 160S (17) and 135S (13) structures are from previously published studies and are shown as surface renderings in multiple colors. Darker hues show portions of poliovirus capsid that are closer to the particle center. According to our modeling, an IgG must bind to two adjacent BC loops. A single IgG molecule cannot bind two loops that are farther apart. Pictures are from the same view direction. (See also Video 4.) (B) Expansion model of poliovirus. Pentagon represents the mesa on the poliovirus capsid with blue loops corresponding to the BC loop. Top row, the 160S-to-135S transition has a hypothesized transient intermediate state in which the BC loops are separated by a relatively larger distance compared to the final 4% expansion observed in the 135S particle. Bottom row, according to the model presented on the top row, binding of the C3 antibody prevents the transition from 160S to 135S particle because the antibody prevents expansion beyond 4%. Note that C3 antibody can bind to preformed 135S particles (bottom row, right) (11). 160S particles bound to C3 Fabs would still be able to make the 160S-to-135S conversion because the Fab regions are not linked.
Antibodies to picornavirus BC loops could bind bivalently through inter- or intra-pentamer interactions; our modeling suggests the latter for C3 antibody and poliovirus. The binding aspect of the Fabs, and therefore the position of the elbow axis, is the key factor in distinguishing between the two ways (Fig. 3A): 1) C3 could bind twofold-related copies of the BC loop on adjacent pentamers. This mode of binding was proposed for the 12-IA and 17-1A monoclonal antibodies to human rhinovirus 14 (39, 40) (Fig. 3A). However, attempts to model interpentamer C3 binding, based on the poliovirus C3-Fab complexes reported here, were unsuccessful. Despite the flexibility of the hinge and elbow regions, the C3 antibody simply cannot bind to one epitope and then reach across the twofold axis and contact a second binding site (cf. Fig. 3A). 2) C3 antibody could bind two fivefold-related BC loops within a single pentamer. This could involve binding to two adjacent loops or two loops separated by one neighboring loop. In our modeling of the antibody-virus complex, an antibody easily bound two adjacent fivefold related BC loops in both the 160S-Fab complex and the 135S-Fab complex (Fig. 4A), but no permissible bending of the elbow or hinge regions was consistent with bivalent attachment to nonadjacent loops (data not shown).
If a C3 antibody binds bivalently to a given fivefold mesa, then the binding may prevent expansion of that mesa (to the 135S state) because the two Fab arms are constrained by linkages in the intact antibody. Adjacent BC loops (on the same mesa) separate by an additional 1 Å in the 160S-to-135S transition (Table SI). Although the antibody can clearly bind to either state (11) (Fig. 4A), the energy required to change the hinge linking the Fabs to the Fc domain may be sufficient to inhibit that slight expansion (4%) and twist (see Results section above). On the other hand, flexible portions of the antibody may be able to accommodate these slight conformational changes (Fig. 4A, Video 4), and the bivalently bound antibody may neutralize by inhibiting the formation of a transient state (between 160S and 135S) in which the particle expands beyond the modest 160S-to-135S expansion (Fig. 4B). Indeed, because the expansion observed in the 135S particle does not indicate a clear path for the N-terminus of VP1, initially inside, to reach its location on the outside surface of the 135S particle (41), a transition state that is larger than the 135S state is plausible. In any case, our data and modeling suggest that the 160S-to-135S expansion is not simply a radial shift of a static pentameric mesa (Video 4).
Exit of N-terminus of VP1
During the cell-entry process, receptor binding induces native poliovirus (160S) to convert to the 135S intermediate particle (11, 42). In this transition, the virion changes conformation, externalizing the membrane-binding entities VP4 and the N-terminus of VP1. Competing models have suggested that the N-terminus of VP1 exits through a channel along the fivefold axis (43-45) or at the base of the canyon (13, 17). Our results support the latter model.
Although the BC loop moves as 160S particle transitions to the 135S particle form, the extent of this movement is inconsistent with the “fivefold exit” model for VP1 and VP4 (43-45). The BC loop of VP1 moves 1 Å from the center of the mesa (Table SI). If the N-terminus of VP1 exits the capsid through the fivefold axis, the BC loop of VP1, and hence the position of C3 Fab, must change significantly. The lack of significant movement supports a model in which the N-terminus of VP1 does not exit through the fivefold axis.
Supplementary Material
Acknowledgments
We thank Eduardo Sanz-Garcia and Bernard Heymann for computer help; Peter Shen, Giovanni Cardone, and Gregory F. Burton for helpful discussions; Thomas J. Smith for providing data from references (36, 39) and for helpful suggestions; David J. Filman for help with initial pseudo-atomic modeling of the C3 Fab bound to poliovirus; and Emily, Rebekah, and Holly Belnap for help with selection of particle images. We also thank Radu Crainic, Francis Delpeyroux, and Bruno Blondel (Pasteur Institute, Paris) for providing the C3 antibody. The UCSF Chimera (24) and Bsoft (21) packages were used to prepare images of modeled coordinates and cryo-EM reconstructions.
Abbreviations
- 3D
three-dimensional
- 80S
RNA-released poliovirus particle
- 135S
cell-entry intermediate poliovirus particle
- 160S
native poliovirus virion
- BC loop
a loop connecting the B and C β-strands of the poliovirus protein VP1
- C3
a monoclonal antibody to poliovirus VP1 protein
- cryo-EM
cryogenic electron microscopy
- CTF
contrast transfer function
- RMSD
root-mean-square deviation
- VP1–4
virus proteins 1–4, four poliovirus capsid proteins
- σ
standard deviations above the average
Footnotes
This work was partially supported by grants R15AI084085 (to DMB) and R01AI020566 (to JMH) from the National Institute of Allergy and Infectious Diseases. This work was also supported by BYU institutional funds (to DMB), by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (DMB, NC, ACS), and by the Health Sciences Center and the Department of Biology at the University of Utah (DMB). Jun Lin was partially supported by a fellowship from the BYU Department of Chemistry and Biochemistry.
References
- 1.Davies DR, Cohen GH. Interactions of protein antigens with antibodies. Proc Natl Acad Sci U S A. 1996;93:7–12. doi: 10.1073/pnas.93.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wien MW, Curry S, Filman DJ, Hogle JM. Structural studies of poliovirus mutants that overcome receptor defects. Nature Struct. Biol. 1997;4:666–674. doi: 10.1038/nsb0897-666. [DOI] [PubMed] [Google Scholar]
- 3.Blondel B, Akacem O, Crainic R, Couillin P, Horodniceanu F. Detection by monoclonal antibodies of an antigenic determinant critical for poliovirus neutralization present on VP1 and on heat-inactivated virions. Virology. 1983;126:707–710. doi: 10.1016/s0042-6822(83)80027-0. [DOI] [PubMed] [Google Scholar]
- 4.Wychowski C, van der Werf S, Siffert O, Crainic R, Bruneau P, Girard M. A poliovirus type 1 neutralization epitope is located within amino acid residues 93 to 104 of viral capsid polypeptide VP1. EMBO J. 1983;2:2019–2024. doi: 10.1002/j.1460-2075.1983.tb01694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Werf S, Wychowski C, Bruneau P, Blondel B, Crainic R, Horodniceanu F, Girard M. Localization of a poliovirus type 1 neutralization epitope in viral capsid polypeptide VP1. Proc. Natl. Acad. Sci. USA. 1983;80:5080–5084. doi: 10.1073/pnas.80.16.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minor PD, Schild GC, Bootman J, Evans DMA, Ferguson M, Reeve P, Spitz M, Stanway G, Cann AJ, Hauptmann R, Clarke LD, Mountford RC, Almond JW. Location and primary structure of a major antigenic site for poliovirus neutralization. Nature. 1983;301:674–679. doi: 10.1038/301674a0. [DOI] [PubMed] [Google Scholar]
- 7.Emini EA, Jameson BA, Wimmer E. Priming for and induction of anti-poliovirus neutralizing antibodies by synthetic peptides. Nature. 1983;304:699–703. doi: 10.1038/304699a0. [DOI] [PubMed] [Google Scholar]
- 8.Evans DMA, Minor PD, Schild GS, Almond JW. Critical role of an eight-amino acid sequence of VP1 in neutralization of poliovirus type 3. Nature. 1983;304:459–462. doi: 10.1038/304459a0. [DOI] [PubMed] [Google Scholar]
- 9.Chow M, Yabrov R, Bittle J, Hogle J, Baltimore D. Synthetic peptides from four separate regions of the poliovirus type 1 capsid protein VP1 induce neutralizing antibodies. Proc Natl Acad Sci U S A. 1985;82:910–914. doi: 10.1073/pnas.82.3.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horaud F, Crainic R, Vanderwerf S, Blondel B, Wichowski C, Akacem O, Bruneau P, Couillin P, Siffert O, Girard M. Identification and characterization of a continuous neutralization epitope (C3) present on type 1 poliovirus. Prog Med Virol. 1987;34:129–155. [PubMed] [Google Scholar]
- 11.Fricks CE, Hogle JM. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 1990;64:1934–1945. doi: 10.1128/jvi.64.5.1934-1945.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wien MW, Filman DJ, Stura EA, Guillot S, Delpeyroux F, Crainic R, Hogle JM. Structure of the complex between the Fab fragment of a neutralizing antibody for type 1 poliovirus and its viral epitope. Nature Struct. Biol. 1995;2:232–243. doi: 10.1038/nsb0395-232. [DOI] [PubMed] [Google Scholar]
- 13.Bubeck D, Filman DJ, Cheng N, Steven AC, Hogle JM, Belnap DM. The structure of the poliovirus 135S cell entry intermediate at 10-Angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J. Virol. 2005;79:7745–7755. doi: 10.1128/JVI.79.12.7745-7755.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rueckert RR, Pallansch MA. Preparation and characterization of encephalomyocarditis (EMC) virus. In: Pestka S, editor. Interferons. Academic Press; New York: 1981. pp. 315–325. [PubMed] [Google Scholar]
- 15.Curry S, Chow M, Hogle JM. The poliovirus 135S particle is infectious. J. Virol. 1996;70:7125–7131. doi: 10.1128/jvi.70.10.7125-7131.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zlotnick A, Cheng N, Conway JF, Booy FP, Steven AC, Stahl SJ, Wingfield PT. Dimorphism of Hepatitis B Virus Capsids Is Strongly Influenced by the C-Terminus of the Capsid Protein. Biochemistry. 1996;35:7412–7421. doi: 10.1021/bi9604800. [DOI] [PubMed] [Google Scholar]
- 17.Belnap DM, Filman DJ, Trus BL, Cheng N, Booy FP, Conway JF, Curry S, Hiremath CN, Tsang SK, Steven AC, Hogle JM. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J. Virol. 2000;74:1342–1354. doi: 10.1128/jvi.74.3.1342-1354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuller SD, Butcher SJ, Cheng RH, Baker TS. Three-dimensional reconstruction of icosahedral particles--the uncommon line. J. Struct. Biol. 1996;116:48–55. doi: 10.1006/jsbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 19.Baker TS, Cheng RH. A model-based approach for determining orientations of biological macromolecules imaged by cryo-electron microscopy. J. Struct. Biol. 1996;116:120–130. doi: 10.1006/jsbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 20.Sanz-García E, Stewart AB, Belnap DM. The random-model method enables ab initio three-dimensional reconstruction of asymmetric particles and determination of particle symmetry. J. Struct. Biol. 2010;171:216–222. doi: 10.1016/j.jsb.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heymann JB, Belnap DM. Bsoft: image processing and molecular modeling for electron microscopy. J. Struct. Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Conway JF, Steven AC. Methods for reconstructing density maps of ‘single’ particles from cryoelectron micrographs to subnanometer resolution. J. Struct. Biol. 1999;128:106–118. doi: 10.1006/jsbi.1999.4168. [DOI] [PubMed] [Google Scholar]
- 23.Belnap DM, Grochulski WD, Olson NH, Baker TS. Use of radial density plots to calibrate image magnification for frozen-hydrated specimens. Ultramicroscopy. 1993;48:347–358. doi: 10.1016/0304-3991(93)90110-j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 25.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 26.Wu X, Milne JL, Borgnia MJ, Rostapshov AV, Subramaniam S, Brooks BR. A core-weighted fitting method for docking atomic structures into low-resolution maps: application to cryo-electron microscopy. J Struct Biol. 2003;141:63–76. doi: 10.1016/s1047-8477(02)00570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J. Mol. Biol. 2006;357:1566–1574. doi: 10.1016/j.jmb.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 28.Conway JF, Watts NR, Belnap DM, Cheng N, Stahl SJ, Wingfield PT, Steven AC. Characterization of a conformational epitope on hepatitis B virus core antigen and quasiequivalent variations in antibody binding. J. Virol. 2003;77:6466–6473. doi: 10.1128/JVI.77.11.6466-6473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9 Å resolution. Science. 1985;229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- 30.Levy HC, Bostina M, Filman DJ, Hogle JM. Catching a virus in the act of RNA release: a novel poliovirus uncoating intermediate characterized by cryo-electron microscopy. J Virol. 2010;84:4426–4441. doi: 10.1128/JVI.02393-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monaco-Malbet S, Berthet-Colominas C, Novelli A, Battaï N, Piga N, Cheynet V, Mallet F, Cusack S. Mutual conformational adaptations in antigen and antibody upon complex formation between an Fab and HIV-1 capsid protein p24. Structure. 2000;8:1069–1077. doi: 10.1016/s0969-2126(00)00507-4. [DOI] [PubMed] [Google Scholar]
- 32.Belnap DM, McDermott BM, Jr., Filman DJ, Cheng N, Trus BL, Zuccola HJ, Racaniello VR, Hogle JM, Steven AC. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. USA. 2000;97:73–78. doi: 10.1073/pnas.97.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He YN, Bowman VD, Mueller S, Bator CM, Bella J, Peng XH, Baker TS, Wimmer E, Kuhn RJ, Rossmann MG. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. USA. 2000;97:79–84. doi: 10.1073/pnas.97.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing L, Tjarnlund K, Lindqvist B, Kaplan GG, Feigelstock D, Cheng RH, Casasnovas JM. Distinct cellular receptor interactions in poliovirus and rhinoviruses. EMBO Journal. 2000;19:1207–1216. doi: 10.1093/emboj/19.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang P, Mueller S, Morais MC, Bator CM, Bowman VD, Hafenstein S, Wimmer E, Rossmann MG. Crystal structure of CD155 and electron microscopic studies of its complexes with polioviruses. Proc. Natl. Acad. Sci. USA. 2008;105:18284–18289. doi: 10.1073/pnas.0807848105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith TJ, Chase ES, Schmidt TJ, Olson NH, Baker TS. Neutralizing antibody to human rhinovirus 14 penetrates the receptor-binding canyon. Nature. 1996;383:350–354. doi: 10.1038/383350a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arnold E, Rossmann MG. Analysis of the structure of a common cold virus, human rhinovirus 14, refined at a resolution of 3. J. Mol. Biol. 1990;211:763–801. doi: 10.1016/0022-2836(90)90076-x. [DOI] [PubMed] [Google Scholar]
- 38.Wien MW. Recognition of Poliovirus: Structural Studies of Neutralization and Receptor Specificity. Harvard University; 1996. [Google Scholar]
- 39.Che Z, Olson NH, Leippe D, Lee W-M, Mosser AG, Rueckert RR, Baker TS, Smith TJ. Antibody-mediated neutralization of human rhinovirus 14 explored by means of cryoelectron microscopy and X-ray crystallography of virus-Fab complexes. J. Virol. 1998;72:4610–4622. doi: 10.1128/jvi.72.6.4610-4622.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith TJ, Olson NH, Cheng RH, Chase ES, Baker TS. Structure of a human rhinovirus-bivalently bound antibody complex: implications for viral neutralization and antibody flexibility. Proc. Natl. Acad. Sci. USA. 1993;90:7015–7018. doi: 10.1073/pnas.90.15.7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin J, Cheng N, Chow M, Filman DJ, Steven AC, Hogle JM, Belnap DM. An externalized polypeptide partitions between two distinct sites on genome-released poliovirus particles. J. Virol. 2011;85:9974–9983. doi: 10.1128/JVI.05013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hogle JM. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annual Reviews of Microbiology. 2002;56:677–702. doi: 10.1146/annurev.micro.56.012302.160757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giranda VL, Heinz BA, Oliveira MA, Minor I, Kim KH, Kolatkar PR, Rossmann MG, Rueckert RR. Acid-induced structural changes in human rhinovirus 14: possible role in uncoating. Proc Natl Acad Sci U S A. 1992;89:10213–10217. doi: 10.1073/pnas.89.21.10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hewat EA, Blaas D. Cryoelectron microscopy analysis of the structural changes associated with human rhinovirus type 14 uncoating. J Virol. 2004;78:2935–2942. doi: 10.1128/JVI.78.6.2935-2942.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolatkar PR, Bella J, Olson NH, Bator CM, Baker TS, Rossmann MG. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 1999;18:6249–6259. doi: 10.1093/emboj/18.22.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen PS, Enderlein D, Nelson CDS, Carter WS, Kawano M, Xing L, Swenson RD, Olson NH, Baker TS, Cheng RH, Atwood WJ, Johne R, Belnap DM. The structure of avian polyomavirus reveals variably sized capsids, non-conserved inter-capsomere interactions, and a possible location of the minor capsid protein VP4. Virology. 2011;411:142–152. doi: 10.1016/j.virol.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris LJ, Larson SB, Hasel KW, McPherson A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry. 1997;36:1581–1597. doi: 10.1021/bi962514+. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.