

Abstract
This paper describes the activation of a biologically inert Co(III) Schiff base (SB) complex to its protein inhibitor form by photoinduced electron transfer (PET) from a colloidal PbS quantum dot (QD, radii = 1.5 – 1.7 nm) to the cobalt center, with a charge separation time constant of 125 ns. Reduction of the Co(III)-SB initiates release of the native axial ligands, promoting replacement with the histidine mimic 4-methylimidazole. The rate of ligand displacement increases by approximately a factor of eight upon exposure of the PbS QD/Co(III)-SB mixture to light of energy greater than the energy of the first excitonic state of the QDs, from which PET occurs. These results suggest an approach for the preparation of inorganic therapeutic agents that can be specifically coupled to a biologically active site by cooperative redox-binding ligation.
INTRODUCTION
Recent advances in the development of therapeutic antitumor and antiviral agents have focused on compounds that bind to the biological active site of an enzyme. Although these reversibly bound drugs are susceptible to nonspecific and potentially undesirable reactions, the success of transition metal therapeutics, such as cisplatin, has refocused efforts to investigate new complexes in this broad class.1 This research has facilitated a greater understanding of the interactions between complex biological systems and inorganic coordination complexes.2,3
To further enhance the efficacy of transition metal inhibitors in research and clinical applications, it is crucial that precise spatial and temporal control of the activity of the agent is realized. Strategies for designing prodrugs – drugs that are administered as inactive compounds but are triggered by some controllable stimulus – have exploited differences in biological environments, such as pH, redox status, and protein expression to achieve a higher level of specificity and efficacy.4 For example, the use of light to control the reactivity of a prodrug has provided an external and orthogonal route of activation that is consistent throughout a broad range of applications.5,6 Sadler et al. have shown that a non-cytotoxic platinum(IV) diazido complex undergoes photoreduction and aquation following exposure to 325 nm light to form a cytotoxic platinum(II) complex,5,7 a result that exemplifies the potential of utilizing light in prodrug strategies for transition metal-based therapeutics.
Nanomaterials have been studied extensively in prodrug applications using energy deposition strategies, including light activation.8,9 Recently, colloidal quantum dots (QDs) have been shown to initiate photoreduction of Pt(IV) complexes to form potentially cytotoxic Pt(II) complexes through photoinduced electron transfer (PET) following photoexcitation at 615 nm.10 The wavelength of PET and subsequent Pt(IV) reduction is within a favorable range (600–1300 nm) for maximum tissue depth penetration for in vivo applications.11 QDs possess highly tunable electrochemical and spectroscopic properties with excitonic transitions in the low-energy visible and near IR regions.12,13 In addition, QDs have high two-photon cross-sectional efficiencies that surpass traditional organic dyes.14,15 Properties such as water solubility, cellular uptake, and selective accumulation in malignant tumors have been tuned to achieve superior biocompatibility.16–19 These attributes make QDs favorable candidates as photosensitizers for accessing multiple redox states of metal-based therapeutics in prodrug designs.
Cobalt(III) Schiff base (Co(III)-SB) complexes of the equatorial tetradentate ligand bis(acetyleactone)ethylenediimine [acacen] are known to be potent inhibitors of a wide array of zinc-dependent proteins, including thermolysin, α-thrombin, and matrix metalloproteinase-2.20–23 Modification of the acacen backbone to incorporate biomolecular targeting moieties (such as oligonucleotides) has been shown to selectively target zinc finger transcription factors Sp1, Ci, and the Snail family.24–26 Evidence suggests the inhibition activity is due to disruption of the protein structure by coordination of Co(III) to active-site histidine residues.27–29 This coordination event occurs via a dissociative ligand exchange and is strongly dependent on the nature of the axial ligands present on the Co(III)-SB complex. Selective enzyme inhibition is observed when the axial positions are occupied by either sterically-hindered 2-methylimidazole or labile amine ligands. In contrast, complexes with substitutionally inert axial ligands, such as imidazole (Im) or 4-methylimidazole (4-MeIm), are poor protein inhibitors. In general, coordination behavior of cobalt Schiff base complexes is dependent on the oxidation state of the metal ion. Due to the redox properties of cobalt, axial ligand coordination of Co(II)-SB complexes has an increased propensity toward dissociation.27
Here we describe a substitutionally inert Co(III)-SB, Co(acacen)(Im)2 (1). Photoinduced electron transfer from a photosensitizer (a colloidal PbS QD with a radius between 1.5 and 1.7 nm, depending on the synthetic batch) reduces Co(III)-SB to Co(II)-SB, where the electron in the Co(II) occupies the anti-bonding dz2 orbital, creating a high-spin d7 electronic configuration. The high-spin Co(II) complex has higher axial ligand reactivity than the Co(III)-SB, so PET promotes axial ligand dissociation (1*).27,30 Subsequent charge recombination oxidizes the Co(II) center back to Co(III), providing an active Co(III)-SB complex with open axial coordination sites for essential His residues (Figure 1). The PbS QD-Co(III)-SB system is therefore a potential redox-activated prodrug.
Figure 1.

Co(acacen)(Im)2 (1) adsorbs to the PbS QD surface and PET occurs from the PbS QD to 1, increasing propensity toward axial ligand dissociation (1*). Oxidation back to Co(III) provides open coordination sites in the axial positions for the incoming ligand – here, 4MeIm – to form a mixture of three species – 1, 2, and 3 in relative abundances of 25%, 50%, and 25%, respectively.
RESULTS AND DISCUSSION
We synthesized several batches of near infrared (NIR) light-absorbing PbS QDs with a first excitonic absorption between ~900 – 1000 nm (see Supporting Information); this absorption corresponds to core radii of 1.5 – 1.7 nm.31 We selected PbS QDs for their tunable, size-dependent band-gaps in the range of 850–2100 nm, within the photo-therapeutic window of biological tissue.32,33 The Co(III)-SB complexes were synthesized and characterized according to literature procedures (see Supporting Information). To prepare the PbS QD/Co(III)-SB samples, we transferred a methanolic solution of the Co(III)-SB into a scintillation vial and dried it under nitrogen before adding 1.4×10−5 M PbS QDs in CHCl3. The vial was shaken until the Co(III)-SB complex was dissolved, and the solution was allowed to equilibrate for 24 hours before performing measurements.
Reduction of the Co(III)-SB complex requires PET from the LUMO of the QD (populated by photoexcitation) to the LUMO of the Co(III)-SB complex. Based on ultraviolet photoemission measurements of the electronic structure of the QD,13 and cyclic voltammetry of the Co(III)-SB complex,27 we estimate the driving force for the PET reaction to be ~100 meV for QDs of this range of radii. We observe a quenching of the PL of the PbS QDs (upon excitation at 850 nm) with increasing concentration of added Co(III)-SB (Figure 2A), consistent with electron transfer from the excitonic state of the QD. Figure 2B shows the PL intensity of each PbS QD/Co(III)-SB mixture relative to that of the QD sample alone (PL/PL0) as a function of the concentration of added Co(III)-SB. As expected, we observe a decrease in PL intensity with an increasing concentration of Co(III)-SB until the PL of the QDs is completely quenched.
Figure 2.
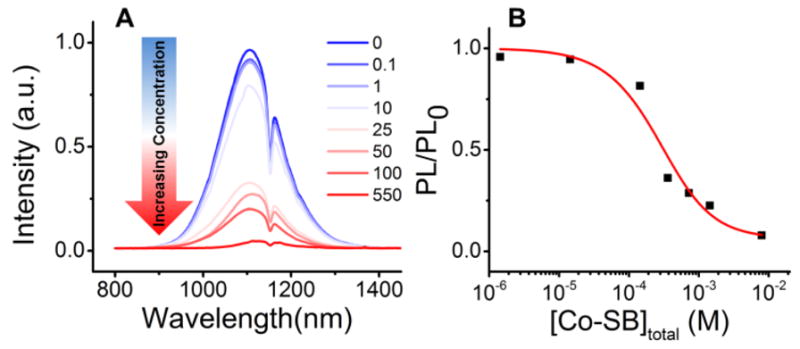
A) Photoluminescence spectra showing the decrease in emission intensity of 1.4×10−5 M PbS QDs in CHCl3 upon addition of an increasing concentration of 1. The legend indicates the number of molecules of 1 added per QD in the mixture, which we allowed to equilibrate for 24 h before acquiring the spectra. B) Fraction of PL intensity remaining in the QD sample after addition of various concentrations of 1, calculated by integrating the peaks in Figure 2A. The fit to this data (red line) yields a saturated surface coverage of 2.5 Co(III)-SB complexes per QD, as described in the text.
The degree to which an adsorbate quenches the PL of a dispersion of QDs by PET, PL/PL0, is directly related to the average number of quenchers adsorbed in electron transfer-active geometries, if we can reasonably assume that the PET process quantitatively out-competes other decay processes for the photoexcited electron in the QD. This assumption is reasonable in the case of PbS QDs, for which the electron decays with a time constant of ~3 μs in the absence of quencher.34,35 The model relating PL/PL0 to the maximum (saturated) surface coverage of Co(III)-SB on the QD and the adsorption equilibrium constant, Kads, is detailed in the Supporting Information and elsewhere.35 The red line in Figure 2B shows the fit of this model to the PL/PL0 data. Through this analysis, we found that the maximum surface coverage of Co(III)-SB complexes per QD, θmax, is 2.5 (this number will vary with the intramolecular structure of the ligand coating on the QDs), and the equilibrium adsorption constant Kads = 1.1×103 M−1 for the QD/Co(III)-SB system in CHCl3. These results are consistent with the physisorption observed in other QD-molecule systems.36,37
Figure 3A shows the transient absorption (TA) spectrum of 1.4×10−5 M PbS QDs in CHCl3 immediately after photoexcitation at 850 nm (black) and after a 500-ns delay (red). Details of the TA experimental apparatus can be found elsewhere.37 The negative feature at 1000 nm is the bleach of the ground state absorption of the QD, which decays in time reflecting the depopulation of charge carriers (electron and/or hole) from their respective band-edges.
Figure 3.

TA spectra of PbS QDs (r = 1.7 nm) in CHCl3 immediately after excitation (black) and after a 500 ns delay (red), showing the evolution of the ground state bleach at 1000 nm for samples without added 1 (A), and for samples containing 100 molecules of 1 added per QD (C). B, D) Kinetic traces acquired at the peak of the ground state bleach for the same two samples; these traces show the dynamics of exciton decay. Dashed red lines are fits to the kinetic data, as described in the text.
Depopulation of charge carriers occurs through exciton recombination or charge transfer to available trap states or molecular redox centers, like the Co(III)-SB. Figure 3B shows that, for the PbS QD sample without added 1, a single exponential function with a time constant of 2.5 μs (convoluted with an instrument response function) is adequate to fit the kinetic trace for the ground state bleach recovery that we extracted from the TA spectrum at 1000 nm. Previous work has demonstrated that the observed rate constant for electron transfer from a QD to a molecular acceptor, keT,obs, increases linearly with the number of acceptors adsorbed per QD, n, as keT,int = n·keT,obs,37–39 where keT,int is the intrinsic electron transfer rate constant: the rate constant observed if every QD that participates in electron transfer has exactly one adsorbed quencher. We found keT,int by fitting the kinetic trace for the ground state bleach recovery in the sample of PbS QDs with added 1, Figure 3D, with eq 1, where OD is the observed optical density at the wavelength
| (1) |
of the ground state bleach (1000 nm), IRF is the instrument response function, θ is the average fractional surface coverage of Co(III)-SB on the QD (obtained from PL/PL0, Figure 2), N is the total number of potential adsorption sites (empty or filled by native oleate ligands) per QD, kCR is the concentration-independent charge recombination rate constant, and Ai is the amplitude of the relaxation pathway with rate constant ki. The bracketed term accounts for electron transfer to Co(III)-SB, which, we assert in this model, is distributed on the surface of QDs according to a binomial distribution. The exponential function outside the bracket accounts for the recovery of the bleach due to recombination of the transferred electron with a band-edge hole in the QD. The Supporting Information contains a derivation of eq 1. This process yields (keT,int)−1 = 125 ns and (kCR)−1 = 258 ns. We show that the observed rate constant reflects PET within an adsorbed QD/Co(III)-SB complex, rather than PET rate-limited by diffusion of a quencher to the QD, in the Supporting Information. The Supporting Information also contains PL and TA experiments conducted with PbS QDs bound to a Co(III)-SB in which the native Im ligands have been replaced with N-methylimidazole (NMeIm). Addition of NMeIm to the Co(III)-SB increases the potential for reduction of Co(III) to Co(II) by ~100 meV.27 Consistent with our proposed electron transfer mechanism, treatment of the QDs with the (NMeIm)2 complex quenches the PL of the QDs less efficiently, and results in a slower excitonic decay, than treatment of the QDs with the (Im)2 complex. We observe no quenching of the QDs’ PL upon addition of the acacen molecule (with no redox-active Co(III) center). While these experiments support electron transfer as the primary mechanism for accelerated excitonic decay upon addition of the Co-SB, we cannot conclusively determine whether additional charge carrier trapping pathways also become available due to reconstruction of the QD’s surface by the complex. Electron and hole trapping to surface states generally occurs on the picosecond-to-hundred-picosecond timescale, however, and we observe no exciton dynamics on that timescale.
Previous studies have shown that 1 in the presence of 2 equivalents of 4MeIm forms a mixture of three species – 1, 2, and 3 – in relative abundances of 25%, 50%, and 25%, respectively (Figure 1). The equilibrium bond distribution is 50% Co-Im bonds and 50% Co-4MeIm bonds.28 We investigated the effect of PET on the axial ligand reactivity of 1 by monitoring the rate of the ligand displacement reaction for mixtures of 1, 4MeIm, and PbS QDs with and without photoactivation of the PET process. We added two equivalents (3.5 mM) of the competitive ligand, 4MeIm, to mixtures of PbS QDs (1.4×10−5 M) and 1 (1.7 mM) in CDCl3, and monitored the axial ligand substitution reaction – specifically the decrease in the concentration of Co-Im bonds (Figure 4) and the increase in the concentration of Co-4MeIm bonds (Figure 4, inset) – by 1H NMR spectroscopy.
Figure 4.
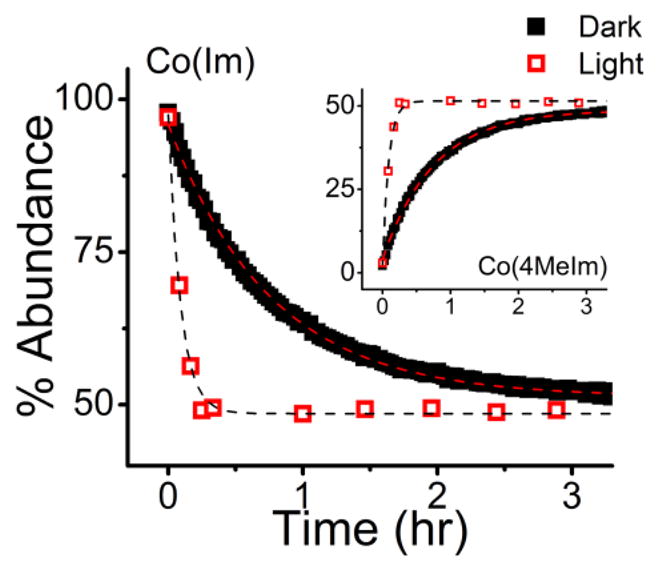
Abundance of imidazole bound to Co (“Co(Im)”) and 4MeIm bound to Co “Co(4MeIm)” (inset) within mixtures of 1.4×10−5 M PbS QDs, 1.7 mM 1, and 3.5 mM 4MeIm, where the mixtures were monitored by NMR in the dark (black solid) and under illumination (red open) with a 75 W halogen lamp passed through a 400 nm long-pass filter. PET from the photoexcited QDs decreases the time constant for axial ligand substitution from 45 minutes to 5.5 minutes.
We photoactivated the PbS QD/Co(III)-SB system by illuminating it in the NMR tube with a 75-W halogen lamp using a 400-nm long-pass filter, in order to photoexcite the PbS QDs but not the Co(III)-SB complex, with stirring. We acquired the NMR spectrum of the reaction mixture after illumination for 0, 5, 10, 15, 20, 60, 90, 120, 150, and 180 minutes. With exposure to light, the axial ligand substitution reaction achieves the expected equilibrium state with a time constant of 5.5 minutes (Figure 4, red open squares). We compared this time constant to that for a PbS QD/Co(III)-SB system with 2 equivalents of 4MeIm, prepared under identical conditions to the first sample, but monitored in the dark. In order to simulate the local heating of the first sample by the halogen lamp (which, we measured, heated the sample to 30 °C), we held this control sample at 30 °C during the NMR measurement. For the control sample, the axial ligand substitution reaction achieves its expected equilibrium state with a time constant of 45 minutes (Figure 4, black solid squares). These studies demonstrate that PET from the QD to the Co(III)-SB increases the rate of the axial ligand substitution of 1 with 4MeIm by a factor of 8.2.
The high energy photons with which we photoexcited the samples for the NMR experiment create electronically “hot” (above-band-edge) carriers that relax to the band-edge states through several mechanisms including phonon emission, which may cause local heating in the sample. The ligand substitution rate for Co-SB complexes does increase with temperature;28 however, given the known temperature dependence of the Im-4MeIm equilibration rate in water,28 the difference in energy between the average excitation photon used in the NMR experiment (2.07 eV) and the band-edge of the QDs (1.24 eV), and the heat capacity of lead sulfide, we estimate that perfect heat transfer from the QD to the Co(III)-SB would only increase the temperature of the PbS lattice by ~3 °C, and increase the ligand substitution rate by a factor of 2 – 3. This value represents an upper bound, assuming that all excess energy is converted to heat, that the heat is transferred to the Co(III)-SB without loss, and that the Co(III)-SB does not dissipate heat itself. We are therefore confident that electron transfer to Co(III) is the primary mechanism for acceleration of the axial ligand ejection.
CONCLUSION
In summary, we have shown that selective photoexcitation of PbS QDs within mixtures of the QDs and Co(acacen)(Im)2 (Co(III)-SB) increases the axial ligand reactivity of Co(III)-SB, such that substitution of Im ligands with the His mimic 4MeIm occurs with a rate that is more than a factor of eight faster than in reaction mixtures that have not been photoactivated. We propose that the mechanism for this observation is electron transfer from the PbS QDs to Co(III), given that (i) the ejection of axial ligands is a documented and well-understood consequence of reduction of Co(III) to Co(II) in this Co(III)-SB complex,27 (ii) electron transfer from the bottom of the conduction band of PbS QDs of this size to Co(III) is energetically favorable by ~100 meV, while energy transfer is not thermodynamically possible, (iii) addition of Co(III)-SB to the QDs quenches their PL, while exposure of the QDs to the acacen molecule without the redox-active Co(III) center does not quench their PL, and (iv) increasing the reduction potential of the Co(III) center within the Co(III)-SB by changing the axial ligand makes the Co(III)-SB a less efficient quencher of the PL of the QDs. Although, for convenience, we used a broad-spectrum light source in this proof-of-principle experiment, the creation of excitons in PbS QDs requires only near-infrared light.
Our measured time constant for axial ligand exchange of 5.5 minutes for the illuminated sample does not represent an intrinsic limit to the efficiency of ligand exchange for two main reasons. (i) In our study, the sample geometry was optimized ad hoc for the NMR experiment, but we can increase the axial ligand reactivity by optimizing illumination conditions such that all regions of the sample receive the maximum photon flux without photo-degradation of the material. (ii) The PbS QDs used in this study are coated with a layer of oleate ligands that maximize the dispersability of the QDs in organic solvents, in order to probe the physical parameters between the QD and the Co(III)-SB complex. We can improve the electronic coupling between the QD and the Co(III)-SB by using a QD coating that minimizes steric repulsion at the surface of the QD or by functionalizing the Co(III)-SB such that it can approach the QD at a shorter distance. For example, the Supporting Information describes a QD/Co(III)-SB system in which the Co(III)-SB can chemisorb to the surface of the QDs with an carboxyl group; for this system, we measured an intrinsic PET time of 2.2 ns – a factor of 50 faster than the QD/Co(acacen)(Im)2 system described above. The labile axial ligands of this complex make it unsuitable for prodrug applications; however, the system represents a strategy for increasing PET yield in these systems, if necessary. We have not yet determined the effect of the QD or its coating on the ability of a protein to bind to the axial position of the Co(III)-SB, but previous studies have shown that CdSe/ZnS QDs functionalized with peptides bind efficiently to streptavidin and other proteins.40,41
Our results offer a unique route for light activation of a Co(III)-SB protein inhibitor via near-infrared excitation, and suggest that the development of inorganic therapeutic agents may be specifically coupled to a biologically active site by cooperative redox-binding ligation. We expect that that mechanisms of the phenomena described in this text are also applicable in biologically relevant conditions. Translation of this system to aqueous conditions involves exchanging the ligands on the particles for water-solubilizing ligands, which can be designed to maintain the absorption cross-section of the QDs16 and the lifetime of the QD excited state;42 thus, we expect electron transfer to outcompete alternative relaxation mechanisms in aqueous conditions, as it does in organic solvent. Additionally, exchange of electrons between the QD and the complex apparently only requires physisorption of the donor and acceptor, which is achievable in aqueous conditions by adjusting the surface chemistry of the particles. Future studies will focus on fine-tuning the system to achieve biocompatibility through improving water solubility, cellular uptake, and selective accumulation in malignant tumors.
Supplementary Material
Acknowledgments
Research supported as part of the Argonne-Northwestern Solar Energy Research Center (ANSER), an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under Award #DE-SC0001059 (electron transfer and NMR studies), and by the National Institutes of Health’s Centers of Cancer Nanotechnology Excellence initiative of the National Cancer Institute under award U54CA151880, and the Rosenberg Cancer Foundation (synthesis and NMR studies). The authors thank Ms. Marie C. Heffern and Dr. Natsuho Yamamoto for helpful discussions.
Footnotes
The authors declare no competing financial interests.
Supporting Information Available. Experimental details, formulation and justification for eq 1, additional electron transfer-related calculations, data from additional control experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Zhang CX, Lippard SJ. Curr Opin Chem Biol. 2003;7:481. doi: 10.1016/s1367-5931(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 2.Hambley TW. Science. 2007;318:1392. doi: 10.1126/science.1150504. [DOI] [PubMed] [Google Scholar]
- 3.Wang D, Lippard SJ. Nat Rev Drug Discovery. 2005;4:307. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 4.van Rijt SH, Sadler PJ. Drug Disc Today. 2009;14:1089. doi: 10.1016/j.drudis.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farrer NJ, Woods JA, Munk VP, Mackay FS, Sadler PJ. Chem Res Toxicol. 2009;23:413. doi: 10.1021/tx900372p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graf N, Lippard SJ. Adv Drug Delivery Rev. 2012;64:993. doi: 10.1016/j.addr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farrer NJ, Woods JA, Salassa L, Zhao Y, Robinson KS, Clarkson G, Mackay FS, Sadler PJ. Angew Chem, Int Ed. 2010;49:8905. doi: 10.1002/anie.201003399. [DOI] [PubMed] [Google Scholar]
- 8.Caruthers SD, Wickline SA, Lanza GM. Curr Opin Biotechnol. 2007;18:26. doi: 10.1016/j.copbio.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Nie S, Xing Y, Kim GJ, Simons JW. Annu Rev Biomed Eng. 2007;9:257. doi: 10.1146/annurev.bioeng.9.060906.152025. [DOI] [PubMed] [Google Scholar]
- 10.Blanco NG, Maldonado CR, Mareque-Rivas JC. Chem Commun. 2009;0:5257. doi: 10.1039/b910000h. [DOI] [PubMed] [Google Scholar]
- 11.Szaciłowski K, Macyk W, Drzewiecka-Matuszek A, Brindell M, Stochel G. Chem Rev. 2005;105:2647. doi: 10.1021/cr030707e. [DOI] [PubMed] [Google Scholar]
- 12.Hines MA, Scholes GD. Adv Mater. 2003;15:1844. [Google Scholar]
- 13.Jasieniak J, Califano M, Watkins SE. ACS Nano. 2011;5:5888. doi: 10.1021/nn201681s. [DOI] [PubMed] [Google Scholar]
- 14.Pu SC, Yang MJ, Hsu CC, Lai CW, Hsieh CC, Lin SH, Cheng YM, Chou PT. Small. 2006;2:1308. doi: 10.1002/smll.200600157. [DOI] [PubMed] [Google Scholar]
- 15.Makarov NS, Drobizhev M, Rebane A. Optics Express. 2008;16:4029. doi: 10.1364/oe.16.004029. [DOI] [PubMed] [Google Scholar]
- 16.Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Science. 2003;300:1434. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 17.Gao X, Cui Y, Levenson RM, Chung LWK, Nie S. Nat Biotech. 2004;22:969. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 18.Duan H, Nie S. J Am Chem Soc. 2007;129:3333. doi: 10.1021/ja068158s. [DOI] [PubMed] [Google Scholar]
- 19.Cai W, Shin DW, Chen K, Gheysens O, Cao Q, Wang SX, Gambhir SS, Chen X. Nano Lett. 2006;6:669. doi: 10.1021/nl052405t. [DOI] [PubMed] [Google Scholar]
- 20.Harney AS, Sole LB, Meade T. J Biol Inorg Chem. 2012;17:853. doi: 10.1007/s00775-012-0902-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeuchi T, Bottcher A, Quezada CM, Meade TJ, Gray HB. Bioorg Med Chem. 1999;7:815. doi: 10.1016/s0968-0896(98)00272-7. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi T, Böttcher A, Quezada CM, Simon MI, Meade TJ, Gray HB. J Am Chem Soc. 1998;120:8555. [Google Scholar]
- 23.Heffern MC, Yamamoto N, Holbrook RJ, Eckermann AL, Meade TJ. Curr Opin Chem Biol. doi: 10.1016/j.cbpa.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harney AS, Lee J, Manus LM, Wang P, Ballweg DM, LaBonne C, Meade TJ. Proc Natl Acad Sci U S A. 2009;106:13667. doi: 10.1073/pnas.0906423106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harney AS, Meade TJ, LaBonne C. PLoS One. 2012;7:e32318. doi: 10.1371/journal.pone.0032318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurtado RR, Harney AS, Heffern MC, Holbrook RJ, Holmgren RA, Meade TJ. Mol Pharm. 2012;9:325. doi: 10.1021/mp2005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Böttcher A, Takeuchi T, Hardcastle KI, Meade TJ, Gray HB, Cwikel D, Kapon M, Dori Z. Inorg Chem. 1997;36:2498. [Google Scholar]
- 28.Manus LM, Holbrook RJ, Atesin TA, Heffern MC, Harney AS, Eckermann AL, Meade TJ. Inorganic Chemistry. 2013;52:1069. doi: 10.1021/ic302379j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matosziuk LM, Holbrook RJ, Manus LM, Heffern MC, Ratner MA, Meade TJ. Dalton Trans. 2013 doi: 10.1039/c2dt32565a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ware DC, Wilson WR, Denny WA, Rickard CEF. Chem Commun. 1991:1171. [Google Scholar]
- 31.Cademartiri L, Montanari E, Calestani G, Migliori A, Guagliardi A, Ozin GA. J Am Chem Soc. 2006;128:10337. doi: 10.1021/ja063166u. [DOI] [PubMed] [Google Scholar]
- 32.Tromberg BJ, Shah N, Lanning R, Cerussi A, Espinoza J, Pham T, Svaasand L, Butler J. Neoplasia. 2000;2:26. doi: 10.1038/sj.neo.7900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boulnois JL. Lasers in Medical Science. 1986;1:47. [Google Scholar]
- 34.Tachiya M. Chem Phys Lett. 1975;33:289. [Google Scholar]
- 35.Morris-Cohen AJ, Vasilenko V, Amin VA, Reuter MG, Weiss EA. ACS Nano. 2011;6:557. doi: 10.1021/nn203950s. [DOI] [PubMed] [Google Scholar]
- 36.Donakowski MD, Godbe JM, Sknepnek R, Knowles KE, Olvera de la Cruz M, Weiss EA. J Phys Chem C. 2010;114:22526. [Google Scholar]
- 37.Morris-Cohen AJ, Frederick MT, Cass LC, Weiss EA. J Am Chem Soc. 2011;133:10146. doi: 10.1021/ja2010237. [DOI] [PubMed] [Google Scholar]
- 38.Song N, Zhu H, Jin S, Zhan W, Lian T. ACS Nano. 2010;5:613. doi: 10.1021/nn1028828. [DOI] [PubMed] [Google Scholar]
- 39.Rodgers MAJ, Da Silva E, Wheeler MF. Chem Phys Lett. 1978;53:165. [Google Scholar]
- 40.Pinaud F, King D, Moore HP, Weiss S. J Am Chem Soc. 2004;126:6115. doi: 10.1021/ja031691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldman ER, Anderson GP, Tran PT, Mattoussi H, Charles PT, Mauro JM. Anal Chem. 2002;74:841. doi: 10.1021/ac010662m. [DOI] [PubMed] [Google Scholar]
- 42.Wuister SF, de Mello Donegá C, Meijerink A. J Phys Chem B. 2004;108:17393. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.