

The subcellular organelles within living cells are anything but static. Rather, organelles such as mitochondria undergo continual dynamic changes; they divide, they fuse and they are selected for recycling. The careful regulation of these processes is essential for cell health and survival. Malfunctions of autophagy and the systems regulating mitochondrial dynamics have been implicated in many neurodegenerative diseases. Here, we review recent progress in the understanding of autophagy, mitochondrial fusion and fission, and their roles in retinal diseases. Autophagy and other forms of mitochondrial plasticity may be important adaptive mechanisms utilized by cells to cope with the metabolic stresses that often underlie retinal disease.
1. Autophagy
Autophagy describes a system used by the cell to deliver substrates from the cytoplasm to the lysosomal compartment for degradation. This system is composed of three processes: chaperone-mediated autophagy, microautophagy and macroautophagy. This review will focus on macroautophagy, often referred to simply as autophagy. In autophagy, a double-membraned vesicle, also known as a phagophore or autophagosome, engulfs part of the cytoplasm and delivers it to the lysosome. These vesicles can transport single organelles, invading pathogens or an entire collection of proteins. The autophagosome then fuses with the lysosome and releases its contents for eventual degradation and recycling, as shown in Figure 1.
Figure 1.
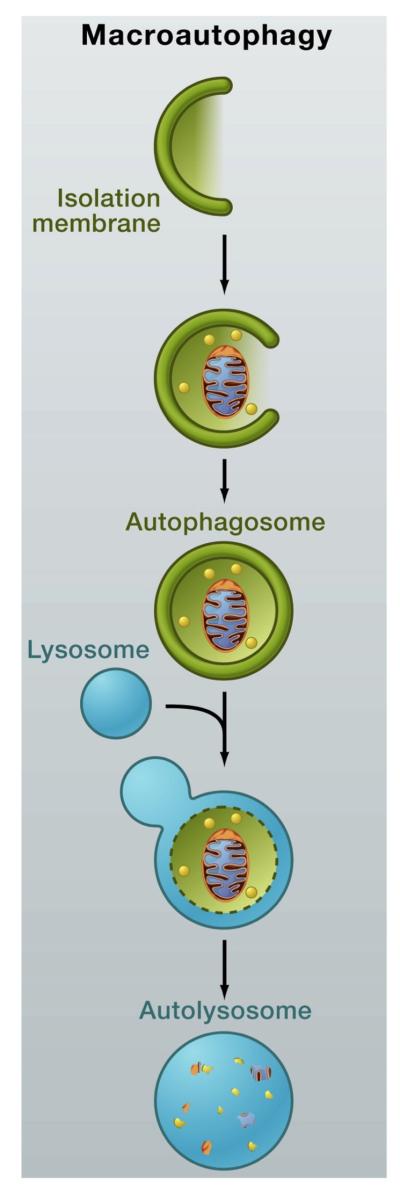
Macroautophagy: A double-membraned vesicle engulfs a part of the cytoplasm which contains single organelles, invading pathogens or an entire collection of proteins and then delivers it to the lysosome. The autophagosome fuses with the lysosome and releases its contents for degradation. Reprinted from [90] with permission from Elsevier.
Autophagy can itself be subdivided into two forms: non-selective autophagy and cargo-specific autophagy. In the former, nutrient deprivation triggers autophagy as a means of obtaining required metabolic constituents from within the cell for energy production and baseline cellular maintenance and repair, a phenomenon first discovered in the yeast Saccharomyces cerevisiae [1]. In the latter, autophagy is used by a nutrient-rich cell to remove either unnecessary or damaged organelles or protein aggregates that could prove harmful for an otherwise healthy cell (reviewed in Ref. [2]).
1.1 Non-Selective Autophagy
Non-selective autophagy allows the cell to recycle fundamental building blocks such as proteins, carbohydrates and lipids, and to re-allocate them during periods of starvation. For example, the liver relies heavily on non-selective autophagy. Lipophagy, in which lipid droplets are repurposed for the generation of free fatty acids, is used to generate additional energy via β-oxidation [3]. These fuel molecules can then be consumed by the hepatocytes themselves or exported into the circulation for utilization by other cell types, including neurons [4]. The liver also utilizes autophagy for amino acid recycling, a function that is of particular importance in times of nutritional stress [5, 6].
1.2 Cargo-specific Autophagy: Mitophagy
Cargo-specific autophagy occurs when the autophagic process is directed specifically at a particular substrate or organelle. Included in this category are ribophagy (the elimination of ribosomes), xenophagy (elimination of intracellular pathogens), pexophagy (removal of peroxisomes) and aggrephagy (disposal of aggregate-prone proteins). Mitophagy, or the targeted removal of mitochondria, is a well-studied example of cargo-specific autophagy. Mitophagy eliminates dysfunctional mitochondria and their toxic contents, protecting the cell and thereby serving as an alternative to programmed cell death (apoptosis).
Mitochondria have 3 main interrelated functions: producing energy for the cell via adenosine triphosphate (ATP) production, controlling cellular metabolism and regulating apoptosis. Though the former is the role for which mitochondria are best known, it is the latter that requires a delicate balance and careful regulation. And all three functions are mutually dependent on one another. Mitochondria need to produce sufficient ATP in order to facilitate cellular activity. Because autophagy is so energy-demanding, it is also dependent on the availability of sufficient quantities of ATP [7]. Thus basal autophagy levels are intimately linked to the underlying functionality of the resident mitochondria.
Due to their role in both apoptosis and autophagy, mitochondria bridge a fine line. On one hand they are able to activate cell death via the release of reactive oxygen species (ROS) and proteins such as cytochrome C, which help trigger apoptosis. On the other hand, by participating in mitophagy, the cell can quarantine and recycle dysfunctional mitochondria that might otherwise result in cell death. When autophagy is inhibited, mammalian cells demonstrate an increased level of apoptosis [8]. A functional autophagic system is therefore crucial for maintaining a healthy cellular environment.
In their 2013 review, Kubli and Gustafsson clearly explain that by holding both apoptosis and mitophagy in reserve as potential responses to stress, the cell is well equipped to manage any level of injury [9]. If the injury is minor, either mitochondrial fusion or mitophagy should be sufficient, but if that method fails to manage the injury sufficiently, the cell can transition to apoptosis in order to spare the tissue or organism [9, 10]. A graphic of these outcomes is shown in Figure 2.
Figure 2.

A schematic describing the potential pathways for distressed mitochondria
Though mitophagy is essential to a cell’s ability to function normally, excessive mitophagy is not “more of a good thing.” This is exemplified in studies of proteins associated with mitophagy, including Parkin, a cytosolic E3 ubiquitin ligase known to promote the mitophagy of damaged mitochondria, and the mitochondrial kinase PTEN-induced putative kinase 1 (PINK1). Parkin is recruited to dysfunctional mitochondria which display PINK1 to indicate their decreased membrane potential [11]. Without Parkin, mitophagy cannot proceed as usual, resulting in swollen and dysfunctional mitochondria [12]. However, an excess of Parkin-mediated mitophagy can be just as toxic and if left unchecked, will facilitate the complete elimination of a cell’s mitochondria [13].
2. Mitochondrial biogenesis and dynamics
This small organelle, which comes in a variety of shapes and sizes, is thought to have originated as a free-living aerobic bacteria engulfed by an anaerobic eukaryotic cell (reviewed in [14]). In their current incarnation, the morphology of these double-membraned organelles may vary greatly, even within the same organism. For example, fibroblast mitochondria are long and cylindrical, while hepatic mitochondria are often rounded and spherical. This variation is reflected in the Greek origins of its name, as ‘mitos’ means thread and ‘chondros’ grain [15]. Mitochondria can either divide during mitosis, a natural moment for growth, or at other points in the cell cycle, if triggered by external signals. For example, mitochondrial proliferation may be prompted by myogenesis [16] or by substances such as benzodiazepines [17]. The synthesis of mitochondria requires genetic input from DNA in both the mitochondria and the nucleus (reviewed in Ref. [18]). Proteins derived from nuclear genes are imported into the matrix of the mitochondrion, where they will be used along with proteins transcribed and synthesized from mitochondrial DNA (mtDNA) (reviewed in [18]). In humans, mitochondrial genes are maternally inherited, with only 0.05-0.1% of the mitochondria in a fertilized egg originating from the father, resulting in a non-Mendelian pattern of inheritance (reviewed in Ref. [18]). Because of this, diseases caused by a mutation in mtDNA are easily identifiable in studies of affected families.
While mitophagy is one way the cell regulates mitochondria, is not the only mechanism available for the modulation of mitochondrial function. Rather, cells are constantly privy to a dance of merging and dividing mitochondria. These mitochondria use fusion and fission as a way to repair injured organelles, to separate those that are past the point of rehabilitation and to expand to keep up with the demands of the tissue and organism they help support.
In healthy tissue, mitochondria will engage in a steady state of division and fusion, with the rate varying by the requirements of their environment and by cell and tissue type [19]. The rates of fusion and fission can fluctuate in response to environmental signals, including changes in cellular energy sources [20]. In the extreme, a complete lack of nutrients has been shown to increase mitochondrial fusion [20].
The fusion process between two mitochondria is complicated, requiring a precise connection between both the outer and inner membranes of the organelle. Large guanosine triphosphatases (GTPases) are involved in the fusion of both membranes. Fusion of the outer membranes utilizes the GTPases Mitofusin 1 and 2 (MFN1, 2) while fusion of the inner membrane is regulated by a third GTPase, Optic Atrophy Protein 1 (OPA1) [21]. Fission is facilitated in part by a cytosolic dynamin protein (Drp1 in mammals) that circles the mitochondria and constricts, severing the mitochondria in a manner reminiscent of a string tightened around a bubble, as shown in Figure 3.
Figure 3.

At top, mitochondria are intermittently involved in fusion, mediated by mitofusins (MFNs) and fission, mediated by DRP1. The rates of these processes are impacted by starvation and stress, as indicated on the left and right-hand side, respectively. Both decreased fission and increased fusion will result in mitochondrial elongation, at bottom. Reprinted by permission from Macmillan Publishers Ltd: Nature Cell Biology, [91], copyright 2011.
Mitochondrial fission is critical for effective mitophagy, as the cell relies on fission proteins to divide mitochondrial networks up into sections small enough to then be engulfed by an autophagosome [22]. Twig et al. (2008) made a number of important findings in this area, including that fusion is required for fission and that fission is in turn needed for autophagy [23]. They further demonstrated the potential for selectivity in both fusion and fission. By showing that mitochondria with higher membrane potentials are more likely to fuse with one another, they defined the existence of a second, unfused and isolated class of mitochondria with lower membrane potentials [23]. Fission can also be selective, producing mitochondrial offspring that are metabolically different from one another [23]. Thus a single, large mitochondrion can divide unevenly, creating one set of daughter mitochondria which serve as a depository for any dysfunctional elements and are then targeted for mitophagy and a second, healthier set which may go on to divide and prosper [23]. A graphic of this process can be seen in Figure 4.
Figure 4.

Fission allows for the segregation of damaged elements from healthy ones, creating daughter mitochondria that are not equals. By merging with a healthier neighbor, fusion may be used to nurse a damaged mitochondria back to health. Reprinted from [92] with permission of AAAS.
Mitophagy and mitochondrial fusion and fission are essential to a cell’s ability to maintain a healthful state. Poole et al. (2008) showed that heterozygous loss-of-function mutations of drp1, a key mitochondrial fission-promoting component, is largely lethal in a PINK1 or Parkin mutant background, that is, an environment in which mitophagy is already limited [24]. This study also demonstrated that mitochondrial dynamics and mitophagy can serve as a balance for one another, as a phenotype associated with increased mitophagy is suppressed by a reduction in mitochondrial fission and enhanced by reductions in fusion [24]. In keeping with this finding, it has also been shown that the PINK1/Parkin pathway of mitophagy influences mitochondrial dynamics by either promoting fission, inhibiting fusion or some combination of the two [25].
Though it is intuitive that growing cells require sufficient mitochondrial fission in order to populate their offspring, even non-proliferative cells such as neurons require mitochondrial fusion and fission. Steketee et al. (2012) demonstrated that inhibiting mitochondrial division results in changes that extend far beyond the mitochondria, leading to modifications in the distal neurite and growth cone of neurons [26]. The role of appropriate fusion is clear in the case of autosomal dominant optic atrophy, the most common inherited optic neuropathy which is caused by a heterozygous mutation in OPA1 [27]. Without appropriate mitochondrial fusion in the optic nerve, these patients lose central vision beginning as early as childhood [28]. While the exact mechanism of disease has not been fully elucidated, reduced OPA1 levels cause structural changes in the mitochondria, as evidenced by disorganized cristae, resulting in disruption of the mitochondrial network (reviewed in Ref. [29]). Knocking down OPA1 also leads to a lowered mitochondrial membrane potential, which can then trigger mitophagy [23, 29].
As is true of so many cellular processes, a healthy cell requires not only fusion and fission but rather a balance between the two. For example, the knockdown of OPA1 in Drosophila cardiomyocytes inhibits normal mitochondrial fusion and leads to the development of cardiomyopathy and a 25% reduction in lifespan [30]. Unchecked fusion or fission is therefore damaging to the cells, disrupting the dynamic state that allows mitochondria to interact with their neighbors and thus impairing the function of both the individual mitochondrion and the entire cell [31].
3. Autophagy, mitochondrial dynamics and neurodegenerative diseases
Neurons are metabolically active cells with a high requirement for energy. The human brain, which only comprises about 2% of body mass, uses 20% of the oxygen consumption of a resting body [32]. Thus it is no surprise that oxidative stress and mitochondrial dysfunction (chronic or acute) are associated with a number of neurodegenerative diseases such as Alzheimer’s disease, Huntington’s disease and Parkinson’s disease, and have been examined as potential therapeutic targets in limiting disease progression (reviewed in Refs. [33-35]). As a result, antioxidants intended to reduce the damage caused by ROS have been trialed in animal models of all three diseases. Studies in an Alzheimer’s disease animal model have demonstrated the ability of mitochondrially-targeted antioxidants (including MitoQ - mitoquinone mesylate: [10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cycloheexadienl-yl) decyl triphenylphosphonium methanesulfonate]) to prevent cognitive decline [33]. In a mouse model of Huntington’s disease, the use of a synthetic antioxidant, XJB-5-131, was associated with preserving mtDNA integrity and copy number, improving mitochondrial function, and decreasing weight loss and motor dysfunction in the animals [34]. In a Parkinson’s disease animal model, mice that overexpress peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α), a regulatory protein for oxidative stress, are more resistant to neurodegeneration when challenged with MPTP, a neurotoxin [35].
A subset of Parkinson’s disease can serve as a prime example of the connection between defective mitophagy and neurodegenerative disease. Mutations in PINK1 and Parkin were found in autosomal recessive forms of the disease [36]. Studies in Drosophila demonstrated that loss-of-function mutations in these genes led to a loss of both dopaminergic neurons and flight muscle (reviewed in Ref. [2, 37]). Because the inheritance of these genes in Parkinson’s disease does is not typically Mendelian, their involvement highlights the likely contribution of mitochondria and mitochondrial dysfunction in the general pathophysiology of the disease.
Autophagic dysfunction has been associated with the same set of neurodegenerative diseases that are implicated in mitochondrial dysfunction. Not coincidentally, these diseases are all characterized by protein inclusions within the cytosol, inclusions which are known substrates of autophagy [38]. Without autophagy, not only will cells have an increased amount of aggregate formation within their cells but they will also have an increased number of dysfunctional mitochondria, mitochondria which would ordinarily have been delivered to a lysosome for recycling [2, 39].
4. Autophagy, mitochondrial dynamics and retinal diseases
4.1 Autophagy and Mitochondria in the Eye
Autophagy in the visual cells of a vertebrate was first described in 1977 by Remé and Young while looking at the retinas of hibernating 13-lined ground squirrels [40]. Subsequently, Remé went on to describe the presence of autophagic vacuoles in the retinas of five other species. At that time, Remé noted that the vacuoles contained “all sorts of cytoplasmic constituents, including…mitochondria” and suggested that autophagy was a mechanism used by cells to maintain the balance between “formation and degradation in the over-all process of renewal” [41].
Over 30 years later, this hypothesis has largely been borne out as we have come to understand that autophagy is necessary for proper cell functioning. This is best illustrated by mouse models of autophagy-related gene (Atg) knock-outs in which the loss of genes required for autophagy leads to a variety of cellular abnormalities, including deformed mitochondria and the accumulation of ubiquitin aggregates [42].
As an extended part of the brain, the retina has similar, if not even greater, energy demands to neurons, secondary to the remarkable energy demands of this tissue. For example, in the fly, the retina alone comprises 10% of the resting ATP consumption of the entire organism [43]. The reason for such a high metabolic rate is mostly attributed to cellular components such as the second messenger system in the visual cycle, synapses and ion pumps, which are critical for the function of the retina [44]. Consequently, many retinal diseases, such as diabetic retinopathy [45], age-related macular degeneration (AMD) [46], and secondary cone photoreceptor death in retinitis pigmentosa [47] have been associated with metabolic stress and/or malfunctions of mitochondria [48]. In immunohistochemical studies of normal mouse retina, it has been demonstrated that autophagy-related proteins, including microtubule-associated protein 1 light-chain 3 (LC3), are concentrated in areas of the retina with a high metabolic demand and an increased likelihood of mitochondrial damage, including the inner and outer nuclear layer, the retinal pigment epithelium (RPE) and the ganglion cell layer [49, 50].
Autophagy within the RPE is particularly important because of the need to regulate the amount of all-trans-retinal (atRAL), a chromophore created when 11-cis-retinal is photo-isomerized, that is toxic to the retina in excessive amounts (reviewed in Ref. [51]). In mouse studies, delayed clearance of atRAL from photoreceptors caused by a knock-out of both ATP-binding cassette transporter 4 (Abca4) and enzyme retinol dehydrogenase 8 (Rdh8) will cause severe RPE and photoreceptor dystrophy [51]. This system can therefore function as a model of light-dependent retinal degeneration [51]. In a recent extension of this model, Chen et al. demonstrated that Abca4−/−Rdh8−/− mice have increased levels of LC3B-II, used as a proxy for autophagy, and Park2, a marker of mitophagy [52]. In addition, they showed that in animals with insufficient autophagy, there was an increased amount of photoreceptor cell death and an increased susceptibility to retinal degeneration [52]. By circling back on this issue, they have strengthened the argument that deficient autophagy and mitophagy may play a large role in the development of light-induced retinal degeneration. A defect in mitophagy and autophagy is only one of the ways in which the RPE may be impacted by light-dependent toxicity, but it is emerging as a growing area of interest within the field.
In diabetic retinopathy, mitochondrial dysfunction is thought to result from oxidative damage within the cell [53]. The oxidative damage in diabetic tissue may be secondary to either increased ROS production or a decreased ability of the retina to combat oxidative stress. This second hypothesis is supported by a study of diabetic rats in which lower retinal levels of free radical scavengers, including glutathione peroxidase and ascorbic acid, was associated with increased oxidative damage relative to control animals [54]. Interestingly, in early stages of diabetic retinopathy mitochondria appear to be resistant to superoxide damage, only becoming damaged after a prolonged period of disease. This has been shown in rodent models of diabetes in which mitochondria in the retina are intact and functional two-months post-diabetes induction, but demonstrate clear dysfunction by six months post-induction [55]. One theory explaining this observation proposes that mitochondrial DNA is initially protected by short-lived compensatory mechanisms which eventually become overwhelmed by the consequences of persistent hyperglycemia [56].
Mitochondrial dynamics are also impacted by other types of stress. In an animal model of glaucoma, increased intraocular pressure (IOP) induced mitochondrial fission and resulted in a reduction of OPA1 mRNA in the optic nerve head. The altered mitochondrial morphology (decreased lengths, increased cristae numbers) and decreased ATP production suggest that glaucomatous stress can negatively affect mitochondrial function [57]. This group subsequently demonstrated that an increase in IOP induces mitochondrial fission and results in a reduction of OPA1 mRNA in the optic nerve head in this glaucoma mouse model [58]. They later expanded on the influence of OPA1 by showing that ganglion cells may increase OPA1 expression as a means of warding off the ravages of increased IOP, thus improving the likelihood of their own survival [59]. Munemasa et al. have examined the impact of IOP on mitochondria and concluded that increased IOP damages otherwise normally functioning mitochondria and results in an absolute reduction of mitochondria within the cell [60].
While there is not yet a single definitive gene known to cause glaucoma, there have been a number of studies looking at candidate genes known to associate with the disease, including latent transforming growth factor-beta binding protein 2 (LTBP2) [61] and a gene within the family of cytochrome P450, P4501B1 (CYP1B1) [62]. Many continue to use genome-wide association studies (GWAS) to elucidate this issue [63]. Genetic evidence available today does however suggest a link between glaucoma and mitochondrial dysfunction. One study established a 6-fold to 8-fold increase in the likelihood that primary open angle glaucoma is maternally inherited [64], a finding in keeping with a pattern of mitochondrial inheritance. This was further supported by a 2006 study of 27 patients with primary open angle glaucoma (POAG) that demonstrated 22 concomitant, potentially pathogenic, nonsynonymous mtDNA changes in this cohort [65]. More recently, Kong et al. reported increased mtDNA mutations and decreased mitochondrial oxidative phosphorylation enzyme expression in an aging mouse model with impaired mitochondrial polymerase γ (PolG) [66]. They went on to report that these mice were even more susceptible to retinal dysfunction when exposed to increased intraocular pressure [66].
As researchers consider ways to circumvent the mitochondrial damage found in disease states, they have aimed to either restore normal mitochondrial function in the face of increasing oxidative stress or to reduce the amount of oxidative stress incurred. The latter has been considered by those who suggest transporting antioxidants into mitochondria in order to assist in the fight against ROS-mediated damage. A recent review looked at, amongst others, SkQ1 (10-(6′-plastoquinonyl) decyltriphenylphosphonium), ergothioneine and MitoQ as contenders for this role [67]. There have also been attempts to link lipophilic cations with antioxidants in order to facilitate the mitochondrial uptake of the latter [68].
4.2 Autophagy, aging and age-related macular degeneration (AMD)
Recently, it has been suggested that increased autophagy may be a mechanism used by tissues or organisms to extend their life span [69]. This is intuitively pleasing, as we know that autophagy prevents the accumulation of damaged molecules within the cell, including molecules such as lipofuscin, which is known to accelerate the aging process [70]. In addition, the inhibition of macroautophagy in mammalian tissues has been shown to induce changes that resemble those associated with aging, supporting the theory that autophagy is aging-protective [71].
As cells age, they seem to produce less ATP. This in turn results in a decrease in the ratio of ATP to adenosine diphosphate (ADP), which subsequently triggers mitochondrial production of ROS. In cells such as the RPE and other long-lived, post-mitotic cells such as neurons, the lowered ATP production and elevated ROS generation associated with aging can increase the probability of pathogenic changes [72]. In susceptible tissues such as the retina, these changes may contribute to aging-related diseases such as AMD, glaucoma, and diabetic retinopathy [72]. He et al. described this aging-associated decline in ATP levels when they reported a 30% reduction in the intracellular levels of ATP in aging RPE cells [19]. They examined the mitochondria within RPE cells cultured from donors and found that there were many fewer mitochondria in the older patients, with a greater than 2-fold difference between the youngest patient (9 years old) and the eldest (76 years old) [19]. When the baseline insults due to aging are combined with any additional mitochondrial dysfunction, whether due to lowered enzymatic activity, reduced mitochondria numbers or increased mitochondrial DNA mutations, the result is the augmentation and acceleration of disease pathogenesis.
This theory of aging accelerated by mitochondrial dysfunction may apply particularly well to AMD. AMD is a progressive disease that impacts the central, cone-dominant macula and is the leading cause of legal blindness in white individuals in the United States [73]. As the global population ages, AMD is poised to be increasingly important throughout the world. Genome-wide association studies have implicated the possible involvement of specific polymorphisms in the likelihood of developing the disease [74-76]. A mutation at chromosome 10q26 has the highest significance ratio, though mutations in 1q32, 2p, 3p and 16 have also been shown to be significant [74]. Another mutation, located in the mitochondrial protein age-related maculopathy susceptibility 2 (ARMS2), or LOC387715, is known to confer risk for progression to advanced forms of AMD [77]. The function of the ARMS2 protein is as yet unknown, but it has been localized to mitochondria, suggesting that mitochondrial dysfunction may play a role in the development of AMD [78]. The relationship between mitochondria and AMD pathogenesis is supported by other findings as well. Proteomic analyses of human RPE revealed that the expression of proteins associated with mitochondrial trafficking and refolding were altered in tissue from AMD eyes when compared with age-matched controls [79]. Age-related decreases in cristae and matrix density in human RPE mitochondria, as observed by electron microscopy, were also more pronounced in AMD eyes relative to age-matched controls [80].
5. Potential Animal Model
There are currently a number of model systems used to study mitochondria and their role in neurologic and ophthalmic diseases. These include in vitro cell-based systems [26, 72, 81], Caenorhabditis elegans models [82, 83], and Drosophila models [24, 25, 30]. There are also numerous rodent models of diseases like Alzheimer’s Disease [33], Huntington’s Disease [34], Parkinson’s Disease [35], retinitis pigmentosa [47], retinopathies [51] and glaucoma [58, 60]. However, as has been recently suggested, there are additional model systems that may contribute a broader perspective and potentially yield new answers to old questions [84].
Mammalian photoreceptors require a tremendous amount of energy in order to maintain their normal functions and contain a high density of mitochondria, even when compared with neurons [85]. Therefore the proper functioning of mitochondria, the chief energy source in photoreceptors, is critical for normal retinal health. Though there is work being done to reduce the amount of oxidative stress in these milieus, there are not currently any leading contenders for ways to optimize mitochondrial health so that they are better able to survive in these challenging environments. We propose the use of the 13-lined ground squirrel (Spermophilus tridecemlineatus) as a model that we believe can provide further insight and open additional research avenues.
Ground squirrels are obligate hibernators and endure extreme metabolic challenges during hibernation [86]. Nonetheless, these animals manage to survive this period without accruing any permanent cellular damage secondary to either the global decrease in temperature and metabolic rate or the reperfusion that occurs with awakening from hibernation [40, 87]. In addition, because these animals are cone-dominant, they are well positioned to function as an animal model for diseases like AMD and cone dystrophies.
By studying these animals and the mitochondria in their retinas, one can attempt to identify the strategies adopted by photoreceptors in hibernating animals to cope with metabolic stress, a state that occurs often in retinal disease. In preliminary, unpublished work, we are seeing evidence of clear changes in the mitochondrial structure and function of the hibernating retinas. For these reasons, we believe that the 13-lined ground squirrel is an interesting animal deserving of additional study.
6. Summary
The process of autophagy is a well studied and well-recognized component of the normal cell functioning required to maintain a healthful state. Without autophagy, organisms can not survive [88]. Even deficiencies in autophagic functions are harmful and can lead to the accumulation of toxic substances, thereby accelerating the already deleterious processes of aging and disease. As a major subtype of autophagy, mitophagy plays an important role in cell homeostasis. This is particularly true in tissues with high metabolic needs, such as the brain and the retina. Unsuprisingly then, defects in mitophagy are known to play a role in many neurologic diseases, including Parkinson’s Disease [15, 89]. Autophagy and its effects on cellular and mitochondrial function have significant connections with the pathobiology of retinal diseases including AMD, diabetic retinopathy, optic atrophy and glaucoma. As such, therapeutic strategies that have a large impact on autophagy and mitochrondrial function may be effective in disease prevention and treatment.
The appropriate balance of mitochondrial fusion and fission is also essential for maintaining cellular health. While the rates of fusion and fission may vary, it is crucial that both be functional and responsive to the requirements of the tissue and organism. These dynamic processes are fundamentally linked with mitophagy, as they allow for the targeting of specific mitochondria for recycling and autophagy. Mitochondrial fusion and fission are therefore vital cellular activities.
A clear understanding of the mechanisms underlying mitochondrial adaptations to avoid retinal damage during episodes of acute stress can be instructive in generating strategies for disease treatment and prevention. For example, the systems that allow mitochondria in hibernating animals to adapt successfully to markedly altered metabolic states during hibernation may be also be helpful in the therapeutic rescue of cone cells in retinitis pigmentosa which experience metabolic derangements that resemble starvation [47]. Understanding mitochondrial function and how it can be modulated by autophagy may shed new light on disease mechanisms and open promising avenues to new therapeutic strategies.
Acknowledgement
We thank Dr. Wai Wong for critical reading of the review.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Letters. 1993;333(1–2):169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 2.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komatsu M. Liver autophagy: physiology and pathology. J Biochem. 2012;152(1):5–15. doi: 10.1093/jb/mvs059. [DOI] [PubMed] [Google Scholar]
- 4.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lum JJD, J. R, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–438. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 6.Rabinowitz JD, White E. Autophagy and Metabolism. Science. 2010;330(6009):1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plomp PJ, et al. Energy dependence of different steps in the autophagic-lysosomal pathway. Journal of Biological Chemistry. 1989;264(12):6699–6704. [PubMed] [Google Scholar]
- 8.Boya P, et al. Inhibition of Macroautophagy Triggers Apoptosis. Molecular and Cellular Biology. 2005;25(3):1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111(9):1208–21. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoshino A, et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. Journal of Molecular and Cellular Cardiology. 2012;52(1):175–184. doi: 10.1016/j.yjmcc.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Narendra DP, et al. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010;8(1):e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greene JC, et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proceedings of the National Academy of Sciences. 2003;100(7):4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narendra D, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of Cell Biology. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nisoli E, Carruba MO. Nitric oxide and mitochondrial biogenesis. Journal of Cell Science. 2006;119(14):2855–2862. doi: 10.1242/jcs.03062. [DOI] [PubMed] [Google Scholar]
- 15.Youle RJK, M. Mitochondrial fission in apoptosis. Nature Reviews. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 16.Brunk CF. Mitochondrial proliferation during myogenesis. Experimental Cell Research. 1981;136(2):305–309. doi: 10.1016/0014-4827(81)90008-2. [DOI] [PubMed] [Google Scholar]
- 17.Vorobjev IA, Zorov DB. Diazepam inhibits cell respiration and induces fragmentation of mitochondrial reticulum. FEBS Letters. 1983;163(2):311–314. doi: 10.1016/0014-5793(83)80842-4. [DOI] [PubMed] [Google Scholar]
- 18.Alberts B JA, Lewis J, et al. Molecular Biology of the Cell. 5th ed Garland Science; New York: 2008. [Google Scholar]
- 19.He Y, Tombran-Tink J. In: Mitochondrial Decay and Impairment of Antioxidant Defenses in Aging RPE Cells, in Retinal Degenerative Diseases. Anderson RE, Hollyfield JG, LaVail MM, editors. Springer; New York: 2010. pp. 165–183. [DOI] [PubMed] [Google Scholar]
- 20.Rossignol R, et al. Energy Substrate Modulates Mitochondrial Structure and Oxidative Capacity in Cancer Cells. Cancer Research. 2004;64(3):985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 21.Rossignol R, Gilkerson R, Aggeler R, et al. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985Y993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 22.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11(12):872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 23.Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27(2):433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poole AC, et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proceedings of the National Academy of Sciences. 2008;105(5):1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng H, et al. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proceedings of the National Academy of Sciences. 2008;105(38):14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steketee MB, et al. Mitochondrial Dynamics Regulate Growth Cone Motility, Guidance, and Neurite Growth Rate in Perinatal Retinal Ganglion Cells In Vitro. Investigative Ophthalmology & Visual Science. 2012;53(11):7402–7411. doi: 10.1167/iovs.12-10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kjer B, et al. Dominant optic atrophy mapped to chromosome 3q region .2. Clinical and epidemiological aspects. Acta Ophthalmologica Scandinavica. 1996;74(1):3–7. doi: 10.1111/j.1600-0420.1996.tb00672.x. [DOI] [PubMed] [Google Scholar]
- 28.Votruba M, Moore AT, Bhattacharya SS. Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy. Journal of Medical Genetics. 1998;35(10):793–800. doi: 10.1136/jmg.35.10.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies V, Votruba M. Focus on molecules: the OPA1 protein. Exp Eye Res. 2006;83(5):1003–4. doi: 10.1016/j.exer.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 30.Dorn GW, et al. MARF and Opa1 Control Mitochondrial and Cardiac Function in Drosophila. Circulation Research. 2011;108(1):12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan DC. Mitochondrial Fusion and Fission in Mammals. Annual Review of Cell and Developmental Biology. 2006;22(1):79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 32.Mink JW, Blumenschine RJ, Adams DB. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 1981;241(3):R203–R212. doi: 10.1152/ajpregu.1981.241.3.R203. [DOI] [PubMed] [Google Scholar]
- 33.McManus MJ, Murphy MP, Franklin JL. The Mitochondria-Targeted Antioxidant MitoQ Prevents Loss of Spatial Memory Retention and Early Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. The Journal of Neuroscience. 2011;31(44):15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xun Z, et al. Targeting of XJB-5-131 to Mitochondria Suppresses Oxidative DNA Damage and Motor Decline in a Mouse Model of Huntington’s Disease. Cell Reports. 2012;2(5):1137–1142. doi: 10.1016/j.celrep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mudò G, et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cellular and Molecular Life Sciences. 2012;69(7):1153–1165. doi: 10.1007/s00018-011-0850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardy J. Genetic Analysis of Pathways to Parkinson Disease. Neuron. 2010;68(2):201–206. doi: 10.1016/j.neuron.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–30. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Human Molecular Genetics. 2002;11(9):1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 39.Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 40.Remé CEY, R. W. The effects of hibernation on cone visual cells in the ground squirrel. Invest. Ophthalmol. Visual Sci. 1977;16(9):815–840. [PubMed] [Google Scholar]
- 41.Remé CE. Autophagy in visual cells and pigment epithelium. Invest Ophthalmol Vis Sci. 1977;16(9):807–814. [PubMed] [Google Scholar]
- 42.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. The Journal of Cell Biology. 2005;169(3):425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laughlin SB, de Ruyter van Steveninck RR, Anderson JC. The metabolic cost of neural information. Nat Neurosci. 1998;1(1):36–41. doi: 10.1038/236. [DOI] [PubMed] [Google Scholar]
- 44.Ames A.r. Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: a commentary based on studies on retina. Can J Physiol Pharmacol. 1992;70:S158–164. doi: 10.1139/y92-257. [DOI] [PubMed] [Google Scholar]
- 45.Roy ST, K., Tien T, Barrette KF. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Diabetic Retinopathy: A Mechanistic Insight for High Glucose-Induced Retinal Cell Death. Curr Clin Pharmacol. 2012 doi: 10.2174/1574884711308040003. [DOI] [PubMed] [Google Scholar]
- 46.Ambati J, Fowler Benjamin J. Mechanisms of Age-Related Macular Degeneration. Neuron. 2012;75(1):26–39. doi: 10.1016/j.neuron.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009;12(1):44–52. doi: 10.1038/nn.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barot M, Gokulgandhi MR, Mitra AK. Mitochondrial dysfunction in retinal diseases. Curr Eye Res. 2011;36(12):1069–77. doi: 10.3109/02713683.2011.607536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitter S, et al. In: Autophagy in the Retina: A Potential Role in Age-Related Macular Degeneration, in Retinal Degenerative Diseases. LaVail MM, et al., editors. Springer; US: 2012. pp. 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jarrett SG, Lewin AS, Boulton ME. The Importance of Mitochondria in Age-Related and Inherited Eye Disorders. Ophthalmic Research. 2010;44(3):179–190. doi: 10.1159/000316480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maeda A, et al. Retinopathy in Mice Induced by Disrupted All-trans-retinal Clearance. Journal of Biological Chemistry. 2008;283(39):26684–26693. doi: 10.1074/jbc.M804505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, et al. Autophagy Protects the Retina from Light-induced Degeneration. Journal of Biological Chemistry. 2013;288(11):7506–7518. doi: 10.1074/jbc.M112.439935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jarrett SG, L.H., Godley BF, Boulton ME. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog Retin Eye Res. 2008;27:596–607. doi: 10.1016/j.preteyeres.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 54.Altomare E, et al. Oxidative protein damage in human diabetic eye: evidence of a retinal participation. Eur J Clin Invest. 1997;27(2):141–7. doi: 10.1046/j.1365-2362.1997.780629.x. [DOI] [PubMed] [Google Scholar]
- 55.Santos JM, et al. Mitochondrial biogenesis and the development of diabetic retinopathy. Free Radical Biology and Medicine. 2011;51(10):1849–1860. doi: 10.1016/j.freeradbiomed.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santos JM, Tewari S, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic Biol Med. 2012;53(9):1729–37. doi: 10.1016/j.freeradbiomed.2012.08.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ju W-K, et al. Elevated Hydrostatic Pressure Triggers Mitochondrial Fission and Decreases Cellular ATP in Differentiated RGC-5 Cells. Investigative Ophthalmology & Visual Science. 2007;48(5):2145–2151. doi: 10.1167/iovs.06-0573. [DOI] [PubMed] [Google Scholar]
- 58.Ju WK, et al. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest Ophthalmol Vis Sci. 2008;49(11):4903–11. doi: 10.1167/iovs.07-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ju WK, K., Duong-Polk KX, Lindsey JD, Ellisman MH, Weinreb RN. Increased optic atrophy type 1 expression protects retinal ganglion cells in a mouse model of glaucoma. Molecular Vision. 2010;16:1331–1342. [PMC free article] [PubMed] [Google Scholar]
- 60.Munemasa Y, et al. Modulation of mitochondria in the axon and soma of retinal ganglion cells in a rat glaucoma model. Journal of Neurochemistry. 2010;115(6):1508–1519. doi: 10.1111/j.1471-4159.2010.07057.x. [DOI] [PubMed] [Google Scholar]
- 61.Ali M, et al. Null Mutations in LTBP2 Cause Primary Congenital Glaucoma. The American Journal of Human Genetics. 2009;84(5):664–671. doi: 10.1016/j.ajhg.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoilov I, Akarsu AN, Sarfarazi M. Identification of Three Different Truncating Mutations in Cytochrome P4501B1 (CYP1B1) as the Principal Cause of Primary Congenital Glaucoma (Buphthalmos) in Families Linked to the GLC3A Locus on Chromosome 2p21. Human Molecular Genetics. 1997;6(4):641–647. doi: 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- 63.Vithana EN, et al. Genome-wide association analyses identify three new susceptibility loci for primary angle closure glaucoma. Nat Genet. 2012;44(10):1142–1146. doi: 10.1038/ng.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charliat GJ, D., Blanchard F. Genetic risk factor in primary open-angle glaucoma. Ophtalmic Epidemiology. 1994;1:131–138. doi: 10.3109/09286589409047221. [DOI] [PubMed] [Google Scholar]
- 65.Abu-Amero KK, Morales J, Bosley TM. Mitochondrial Abnormalities in Patients with Primary Open-Angle Glaucoma. Investigative Ophthalmology & Visual Science. 2006;47(6):2533–2541. doi: 10.1167/iovs.05-1639. [DOI] [PubMed] [Google Scholar]
- 66.Kong YXG, et al. Increase in mitochondrial DNA mutations impairs retinal function and renders the retina vulnerable to injury. Aging Cell. 2011;10(4):572–583. doi: 10.1111/j.1474-9726.2011.00690.x. [DOI] [PubMed] [Google Scholar]
- 67.Gruber J, et al. Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnology Advances. (0) doi: 10.1016/j.biotechadv.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 68.Sheu S-S, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2006;1762(2):256–265. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 69.Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010;12(9):842–846. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- 70.Rajawat YS, Hilioti Z, Bossis I. Aging: Central role for autophagy and the lysosomal degradative system. Ageing Research Reviews. 2009;8(3):199–213. doi: 10.1016/j.arr.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 71.Rubinsztein David C., Mariño G, Kroemer G. Autophagy and Aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 72.Schutt F, et al. Moderately reduced ATP levels promote oxidative stress and debilitate autophagic and phagocytic capacities in human RPE cells. Invest Ophthalmol Vis Sci. 2012;53(9):5354–61. doi: 10.1167/iovs.12-9845. [DOI] [PubMed] [Google Scholar]
- 73.Congdon NOC, B., Klaver CC, Klein R, Muñoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P. Eye Diseases Prevalence Research Group, Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- 74.Fisher SA, et al. Meta-analysis of genome scans of age-related macular degeneration. Human Molecular Genetics. 2005;14(15):2257–2264. doi: 10.1093/hmg/ddi230. [DOI] [PubMed] [Google Scholar]
- 75.Klein R, et al. The epidemiology of age-related macular degeneration. American Journal of Ophthalmology. 2004;137(3):486–495. doi: 10.1016/j.ajo.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 76.Seddon JM, Ajani UA, Mitchell BD. Familial aggregation of age-related maculopathy. American Journal of Ophthalmology. 1997;123(2):199–206. doi: 10.1016/s0002-9394(14)71036-0. [DOI] [PubMed] [Google Scholar]
- 77.Kanda A, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proceedings of the National Academy of Sciences. 2007;104(41):16227–16232. doi: 10.1073/pnas.0703933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheng Y, et al. Genetic and Functional Dissection of ARMS2 in Age-Related Macular Degeneration and Polypoidal Choroidal Vasculopathy. PLoS ONE. 2013;8(1):e53665. doi: 10.1371/journal.pone.0053665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nordgaard CL, et al. Proteomics of the Retinal Pigment Epithelium Reveals Altered Protein Expression at Progressive Stages of Age-Related Macular Degeneration. Investigative Ophthalmology & Visual Science. 2006;47(3):815–822. doi: 10.1167/iovs.05-0976. [DOI] [PubMed] [Google Scholar]
- 80.Feher J, et al. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiology of Aging. 2006;27(7):983–993. doi: 10.1016/j.neurobiolaging.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 81.Chen Y, et al. Autophagy protects the retina from light-induced degeneration. J Biol Chem. 2013;288(11):7506–7518. doi: 10.1074/jbc.M112.439935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Labrousse AM, Z.M., Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell. 1999;4:815–26. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- 83.Jagasia R, G.P., Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–60. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 84.Bolker J. There’s more to life than rats and flies. Nature. 2012;491:31–32. doi: 10.1038/491031a. [DOI] [PubMed] [Google Scholar]
- 85.Eakin R. In: Structure of Invertebrate Photoreceptors, in Photochemistry of Vision. Dartnall HA, editor. Springer; Berlin Heidelberg: 1972. pp. 625–684. [Google Scholar]
- 86.Vaughan DKG, A. R., Michalski ML, Seidling J, Schlink S. Capture, care, and captive breeding of 13-lined ground squirrels, Spermophilus tridecemlineatus. Lab Animal. 2006;35(4):33–40. doi: 10.1038/laban0406-33. [DOI] [PubMed] [Google Scholar]
- 87.Andrews MT. Advances in molecular biology of hibernation in mammals. Bioessays. 2007;29(5):431–40. doi: 10.1002/bies.20560. [DOI] [PubMed] [Google Scholar]
- 88.Seibenhener ML, et al. Sequestosome 1/p62 Is a Polyubiquitin Chain Binding Protein Involved in Ubiquitin Proteasome Degradation. Molecular and Cellular Biology. 2004;24(18):8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schapira AHV. Mitochondrial Pathology in Parkinson’s Disease. Mount Sinai Journal of Medicine. 2011;78:872–881. doi: 10.1002/msj.20303. [DOI] [PubMed] [Google Scholar]
- 90.Mizushima N, Komatsu M. Autophagy: Renovation of Cells and Tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 91.Blackstone C, Chang C-R. Mitochondria unite to survive. Nat Cell Biol. 2011;13(5):521–522. doi: 10.1038/ncb0511-521. [DOI] [PubMed] [Google Scholar]
- 92.Youle RJ, van der Bliek AM. Mitochondrial Fission, Fusion, and Stress. Science. 2012;337(6098):1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]