

Abstract
Reactive oxygen species (ROS) are critical molecules produced as a consequence of aerobic respiration. It is essential for cells to control the production and activity of such molecules in order to protect the genome and regulate cellular processes such as stress response and apoptosis. Mitochondria are the major source of ROS within the cell, and as a result, numerous proteins have evolved to prevent or repair oxidative damage in this organelle. The recently discovered OXR1 gene family represents a set of conserved eukaryotic genes. Previous studies of the yeast OXR1 gene indicate that it functions to protect cells from oxidative damage. In this report, we show that human and yeast OXR1 genes are induced by heat and oxidative stress and that their proteins localize to the mitochondria and function to protect against oxidative damage. We also demonstrate that mitochondrial localization is required for Oxr1 protein to prevent oxidative damage.
It is critical for the aerobically respiring cell to defend against oxidative damage to cellular macromolecules in order to carry out metabolic activities and faithfully maintain the genome. Recently, much research has focused on the importance of reactive oxygen species (ROS) in various cellular processes, including DNA damage and repair (10), redox regulation of protein activity (18, 24), signal transduction (5, 11), ageing (30), and apoptosis (15, 21, 31).
There are numerous sources of ROS. Chemical agents such as menadione and paraquat produce mainly superoxide radicals (9), while hydrogen peroxide can be converted to the hydroxyl radical by the Fenton reaction (23). Ionizing radiation such as X rays and gamma rays damages cells primarily through reactive oxygen intermediates formed by electrolysis of water (4). However, the major source of ROS within the cell is the mitochondria (26). Electron transport activities occurring at the inner membrane of mitochondria have been shown to produce ROS at significant levels; as much as 1 to 5% of the O2 consumed by respiring cells is converted to ROS (7). As a result, cells express numerous antioxidant defenses that protect mitochondria, including Mn SOD, thioredoxins, glutathione, and DNA repair enzymes (2, 29, 39, 41). Recent investigations have also highlighted a role for antioxidant activity in the regulation of the mitochondrial apoptosis pathway. Inhibition of the electron transport chain, resulting in increased ROS production, has been shown to result in increased apoptosis (25, 40). Conversely, mutations in mitochondrial antioxidant functions (Trx-2, Mn SOD) have also been demonstrated to increase apoptosis through the mitochondrion-dependent pathway (32, 43). The important relationship between oxidative damage prevention and disease is illustrated by mice deficient in the mitochondrial apoptosis-inducing factor, which display increased oxidative stress and cell death in neuronal cells (22). It is becoming increasingly apparent that identification and characterization of ROS-regulating proteins in the mitochondria will be crucial in understanding how cells avoid oxidative injury and control apoptosis.
Recently we have identified a novel human gene, OXR1, on the basis of its ability to suppress oxidative DNA damage in Escherichia coli (42). OXR1 is an evolutionarily conserved gene, as homologues are present in many eukaryotic organisms from yeast to humans. To date, there is little known about its function. Deletion of the OXR1 gene in Saccharomyces cerevisiae (scOXR1) results in sensitivity to hydrogen peroxide damage (42). This suggests that the OXR1 gene product may play a particularly important and unique role, as many other mutations in individual genes that prevent or repair oxidative damage do not result in oxidation sensitivity phenotypes (3, 20). Drosophila melanogaster expresses seven isoforms of OXR1 (L82A to -G), and a mutant with the entire locus deleted is lethal as a result of a defect in eclosion (hatching from the pupal case) (37). The mouse homologue of OXR1, C7, was identified in a screen for genes induced upon cell attachment to extracellular matrix (14). The eukaryotic OXR1 genes encode proteins of various sizes, although they all contain a conserved ∼300-amino-acid C-terminal domain. As this domain corresponds to the entire S. cerevisiae Oxr1 protein, it likely represents a unique functional domain and possesses the proposed oxidation protection function of the scOxr1 and human Oxr1 (hOxr1) proteins.
In this report, we further characterize the expression of the scOXR1 and hOXR1 genes, as well as the cellular localization of their respective proteins. We provide evidence that the scOXR1 and hOXR1 genes exhibit a stress response in both yeast and human cells. We also present the first evidence that the hOxr1 protein provides protection from oxidative damage in a eukaryotic cell.
MATERIALS AND METHODS
Yeast strains, plasmids, and media.
S. cerevisiae strain N1-4 (42) was used for wild-type controls. Strains N74 and N76 (containing plasmids pMV656 and pMV657, respectively) were derived from oxr1Δ::URA3 strain N1-9 (see Table 1). The mitochondrial targeting sequence (MTS) of SOD2 was fused to hOXR1 by PCR with the primer 5′ AAGGATCCATGTTCGCGAAAACAGCAGCTGCTAATTTAACCAAGAAGGGTGGTTTGTCATTGCTCTCCCAAAGGGAAAATATTCAAC 3′. The product was inserted into the expression vector pMV611 to generate pMV656. pMV657 was constructed in the same way, except that the upstream hOXR1 primer lacked the SOD2 MTS sequence. Strains N34 and N39 were derived from strain N1-4 by integration into the OXR1 locus of a PCR product containing a C-terminal green fluorescent protein (GFP) or hemagglutinin (HA) tag fused to the 3′ end of the OXR1 open reading frame and flanked by 40 bp of chromosomal sequence upstream and downstream of the OXR1 open reading frame, in accordance with published procedures (28). Correct integration into the chromosome was confirmed by PCR analysis with a internal primer corresponding to OXR1 and an external primer corresponding to the selectable kanamycin resistance marker. Also, strains were tested phenotypically for wild-type sensitivity to hydrogen peroxide to confirm OXR1 function. The plasmid template for the HA tag was a gift from M. Longtine (Oklahoma State University, Stillwater). Yeast peptone dextrose and synthetic minimal dextrose media were prepared as described by Adams et al. (1).
TABLE 1.
Yeast strains and plasmids used in this study
Cell lines and culture.
HeLa cells were cultured in Dulbecco's modified eagle medium (GIBCO BRL) supplemented with 10% fetal bovine serum (Sigma), 1 mg of l-glutamine (Gibco) per ml and penicillin-streptomycin (Gibco) at 37°C in a 5% CO2 incubator.
Immunofluorescence microscopy.
Yeast immunofluorescence microscopy was carried out as previously described (1), with mouse anti-HA monoclonal antibody 16B12 (Covance) at a 1:1,000 dilution and secondary anti-mouse AlexaFluor 568 antibody (Molecular Probes) at a 1:500 dilution. For HeLa cell immunofluorescence microscopy, cells grown on 12-mm-diameter poly-d-lysine-coated glass coverslips (Becton Dickinson) were washed five times in phosphate-buffered saline (PBS), fixed in 2.5% paraformaldehyde, washed once in PBS, and permeabilized with 0.1% Triton X-100. Cells were incubated with a 1:200 dilution of either rabbit anti-C7C (0.78-mg/ml stock) or anti-C7 M (0.25-mg/ml stock) antibodies, which were gifts from E. Engvall (Burnham Institute, La Jolla, Calif.). Antibodies were produced and affinity purified with the C7C or C7M domain peptides as described by Fischer et al. (14). Cells were then washed three times in PBS and incubated with anti-rabbit AlexaFluor 488 secondary antibody (Molecular Probes) at a 1:200 dilution, followed by visualization by fluorescence microscopy. For mitochondrial labeling, cells were incubated with 100 nM MitoTracker Red (Molecular Probes) for 15 min prior to fixation.
Protein extracts and immunoblotting.
Yeast and HeLa cells were pelleted, washed once in PBS, resuspended in sodium dodecyl sulfate (SDS) protein loading buffer, and immediately boiled for 10 min. Proteins were then separated by SDS-polyacrylamide gel electrophoresis and immunoblotted with mouse anti-HA monoclonal antibody 16B12 (Covance) and a horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (Amersham Life Science). Where shown, mouse anti-α-tubulin antibody (Lab Vision) was used to probe blots as a loading control.
RNA isolation and Northern blotting.
Total yeast RNA was extracted by the acid phenol method as described by Sambrook et al. (36). Approximately 20 μg of total RNA per sample was separated on a 1% formaldehyde gel and transferred to a nylon membrane (Hybond) by capillary transfer. PCR products of the scOXR1 open reading frame or the SCR1 open reading frame were used as templates in random primed labeling reactions to generate 32P-labeled probes. Hybridization was carried out at 42°C overnight. Band intensities were scanned on a Personal Densitometer SI (Molecular Dynamics) and quantified with Molecular Analyst software (Bio-Rad).
RESULTS
Mitochondrial localization of scOxr1 protein
Sequence analysis of the scOXR1 open reading frame revealed a putative mitochondrial localization signal at the N terminus of the protein (PSORTII; http://psort.nibb.ac.jp). We therefore sought to confirm this prediction by the use of a fluorescently tagged scOxr1 protein. The GFP open reading frame was fused in frame with the 3′ end of the OXR1 gene, and this fusion construct was integrated by homologous recombination, replacing the endogenous OXR1 gene on chromosome XVI. This allows expression of an Oxr1-GFP fusion protein from the endogenous OXR1 promoter in the normal OXR1 chromosomal context. Figure 1 shows the localization of Oxr1-GFP to discrete cytoplasmic compartments which correspond with the extranuclear 4′,6′-diamidino-2-phenylindole (DAPI)-stained regions (compare panels A and B), indicating that Oxr1-GFP and mitochondrial DNA reside in the same compartment. To further support this, yeast cells were stained with the mitochondrion-specific probe MitoTracker (Molecular Probes). As shown in Fig. 1C, the regions stained with MitoTracker clearly overlap those identified by Oxr1-GFP fluorescence. Very little, if any, protein is associated with the nucleus (large, DAPI-stained region), indicating that scOxr1 protein resides almost exclusively in the mitochondria.
FIG. 1.

Localization of scOxr1 protein in yeast. Fluorescence microscopy was carried out on strain N34 expressing C-terminally GFP-tagged Oxr1 protein. Cells were preloaded with 100 μM MitoTracker Green probe for 30 min prior to collection and mounting in DAPI-containing mounting buffer. Panels: A, DAPI-stained cells; B, MitoTracker; C, Oxr1p-GFP. Immunofluorescence microscopy of strain N-39 expressing Oxr1p-HA was conducted. Panels: D, DAPI; E, anti-HA immunostaining.
We also confirmed the localization of Oxr1 protein by the use of an epitope-tagged Oxr1 protein and immunofluorescence techniques. A C-terminally HA-tagged form of Oxr1 protein was generated and expressed from the endogenous OXR1 locus, as was done for Oxr1-GFP. Immunofluorescence was then performed to determine the localization of the HA-tagged protein. Figure 1D and E show the colocalization of mitochondrial DNA and the Oxr1-HA protein. These results are in agreement with the Oxr1-GFP studies and strongly suggest that scOxr1 protein is associated with the yeast mitochondria.
Expression of scOXR1 is induced by heat and oxidative stress.
Global transcription profiling experiments with yeast have demonstrated that OXR1 is one of a subset of genes induced by stress conditions, particularly those conditions associated with an increase in oxidative stress (6, 17). To confirm that OXR1 expression is stress inducible, we subjected yeast cells to both heat and oxidative stress conditions, and the levels of OXR1 transcripts were monitored by Northern blotting. Figure 2A shows the results of a Northern blot assay of total yeast RNA from untreated cells and from those subjected to heat stress at 37°C. The OXR1 transcript levels increased within the first 15 min of treatment and returned to unstressed levels by 45 min. These data closely resemble those obtained with a probe to the classical yeast heat shock gene HSP12 (data not shown) (35). The heat stress-inducible expression of OXR1 is also apparent at the translational level. As shown in Fig. 2B, heat stress causes accumulation of the Oxr1-HA protein, as determined by immunofluorescence immunomicros copy. These results suggest that expression of OXR1 is regulated at least in part by growth temperature. We also asked if OXR1 expression could be induced by oxidative stress. Figure 2A shows the effect of exposure to 0.5 mM H2O2 on the OXR1 transcript level as monitored by Northern blotting. As with heat stress, there was a rapid increase in the OXR1 transcript level during the first 15 min of treatment, although transcript levels remained elevated throughout the time course of the experiment. Together with the heat stress data and previously published microarray data, these results strongly suggest that OXR1 is a stress-induced gene in S. cerevisiae.
FIG. 2.

Heat and oxidative stress induction of scOXR1. (A) Yeast cells were grown at 22°C to log phase and shifted to 37°C for the times shown (left). Alternatively, cells were grown at 30°C and treated with 0.5 mM H2O2 for the times indicated (right). At each time point, cells were harvested and total RNA was analyzed by Northern blotting. Fold induction values are given for each time point relative to the zero-time control. (B) Yeast cells were grown at 22°C to log phase and incubation was continued at 22°C, or cells were shifted to 37°C and incubation was continued for 1 h. Cells were then fixed and stained with anti-HA antibody to visualize Oxr1p-HA. Left side, DAPI staining; right side, anti-HA staining.
The hOxr1 protein is localized to mitochondria in human cells.
Previous work with the mouse homolog of hOxr1, C7, has generated antibodies to two domains of the C7 protein. An antibody to domain II of C7 was used to demonstrate nucleolar localization of C7 in several rodent cell lines (14). A second antibody specific for the C-terminal domain (domain III) of C7 and called C7C was also generated. This domain is highly homologous to the corresponding region of the hOxr1 protein (∼90% identity) and is also homologous to scOxr1 protein. (We reasoned that mouse antibody C7C would provide a useful tool for examining the localization of the hOxr1 protein in the human HeLa cell line, since it recognizes hOxr1 expressed in bacteria [data not shown].) We conducted immunofluorescence experiments with HeLa cells and the C7C antibody, and as shown in Fig. 3, the hOxr1 protein is localized to a specific cytoplasmic compartment, enriched around the nuclear periphery, and also found in long, tubular projections extending from the perinuclear region to the tips of the adherent cell. The observed staining is strikingly similar to that of the mitochondria in HeLa cells (8). We therefore stained the cells with MitoTracker prior to immunofluorescence microscopy in order to establish colocalization. Figure 3 clearly shows that MitoTracker stains the same cellular compartment as the Oxr1 antibody, indicating that in HeLa cells, hOxr1 is associated with the mitochondria. As is the case in yeast, little if any Oxr1 protein is detected in the nucleus. Similar localization results were obtained in experiments with two additional mammalian cell lines, human Hep2 cells and monkey COS cells (data not shown).
FIG. 3.

Localization of hOXR1 protein in HeLa cells. HeLa cells were preloaded with 100 μM MitoTracker Red probe, fixed, and immunostained with rabbit anti-C7C antibody and anti-rabbit AlexFluor 488 secondary antibody. Panels: A, anti-C7C staining; B, MitoTracker; C, merged images of panels A and B.
hOxr1 protein is induced by heat and oxidative stress in HeLa cells.
Given the similarities between scOxr1 and hOxr1 in terms of protein homology and localization, we asked if hOXR1 expression is also induced by stress conditions in human cells. First, we subjected HeLa cells to a 1 mM dose of H2O2 for 15 min and allowed 1 h for recovery in fresh growth medium. hOxr1 protein was then visualized by immunofluorescence microscopy, as shown in Fig. 4A. The staining of Oxr1 protein is more intense in peroxide-treated cells than in untreated cells, and the protein accumulation appears to be localized primarily to the mitochondria, with some diffuse staining in the cytoplasm (compare Oxr1 staining with MitoTracker staining). There is no visible difference in the intensity of MitoTracker staining between peroxide-treated and untreated cells, indicating that the difference seen in Oxr1 staining is due to protein levels and not mitochondrial content. To support the immunofluorescence microscopy results, we also monitored hOxr1 protein levels during stress by Western blotting (Fig. 4B). In unstressed HeLa cells, the C7C antibody recognizes a 37.5-kDa protein, which is consistent with the predicted size (41.6 kDa) of the hOXR1 gene product originally isolated (42). Following 1 mM H2O2 treatment, Oxr1 protein accumulated within the first hour after treatment, and by 4 h the level of protein reached a maximum and began to decrease by 5 h. We also observed the accumulation of a higher-molecular-weight protein recognized by the C7C antibody at 58.8 kDa, which presumably reflects a larger splice variant that includes additional upstream exons detected by DNA sequence analysis of chromosome 8. We also asked if heat stress induces expression of hOxr1 protein, as it does for scOxr1 protein. Figure 4C shows a Western blot of HeLa cells extracts following 30 min of heat stress at 42°C. hOxr1 protein accumulates in a manner very similar to that in peroxide-treated cells, with a maximum level occurring by 2.5 h and reduction apparent by the 4.5-h mark. The immunofluorescence results of peroxide-treated HeLa cells, together with the Western blots of peroxide- and heat-treated cells, indicate that hOxr1 protein expression is induced by heat and oxidative stress in human cells.
FIG. 4.

(A) H2O2-induced accumulation of hOxr1 protein in HeLa cells. Cells were loaded with 100 μM MitoTracker Red for 15 min and then treated with 1 mM H2O2 for 15 min or left untreated and allowed to recover in fresh medium for 1 h prior to fixation and staining with anti-C7C antibody. (B) HeLa cells were treated with 1 mM H2O2 for 15 min and allowed to recover in fresh medium for the times indicated. Cells were harvested, and crude protein extracts were separated by SDS-polyacrylamide gel electrophoresis and immunoblotted with anti-C7C antibody. Samples were also stained with Coomassie as a loading control. (C) Cells were heat stressed at 42°C for 30 min and allowed to recover at 37°C for the times indicated. Western blot assays were conducted as in panel B. Blots were probed with anti-α-tubulin antibody as a loading control. U, unstressed.
Multiple transcripts of hOXR1 are expressed in a tissue-specific manner.
To further characterize hOXR1 expression, we determined the abundance of hOXR1 message in multiple human tissues by Northern blotting. A blot of poly(A)+ RNA from several human tissues was hybridized with a hOXR1 cDNA probe corresponding to the entire C-terminal OXR1 homology domain, and the results are shown in Fig. 5. Two transcripts, 2.9 and 4.9 kb, are observed in nearly all of the tissues, with the exception of brain tissue, which expresses a unique 5.1-kb transcript in addition to the 2.9-kb transcript. Also, the relative abundances of the transcripts differ in the tissues examined, with the 4.9-kb transcript being most abundant in placenta, lung, liver, kidney, and pancreas tissues. In heart and skeletal muscle tissues, the smaller transcript appears to be as abundant as the 4.9-kb transcript. From this finding, we conclude that hOXR1 is expressed as two transcripts whose relative abundances differ among various human tissues.
FIG. 5.
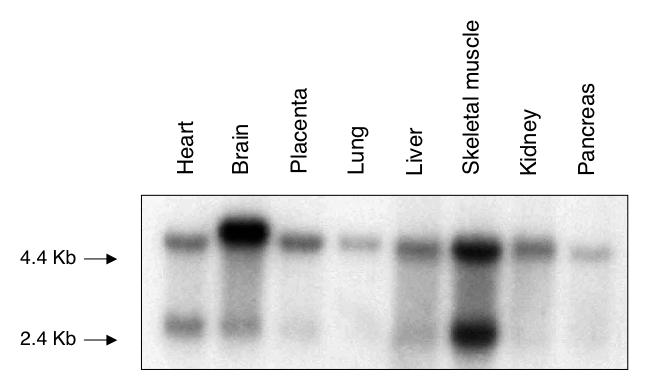
Multiple-tissue Northern blot assay of hOxr1. A blot of poly(A)+ RNA from several human tissues (Clontech) was probed with 32P-labeled hOXR1 cDNA.
Mitochondrion-targeted hOxr1 can complement the peroxide sensitivity of a yeast oxr1Δ mutant strain.
Previous work has shown that the yeast oxr1Δ mutant is approximately 10-fold more sensitive to hydrogen peroxide lethality than is the wild-type strain (42). In order to determine if the hOxr1 protein is capable of complementing the peroxide sensitivity of the yeast oxr1Δ mutant, we expressed mitochondrion-targeted and untargeted hOxr1 proteins in the yeast oxr1Δ mutant background and tested resistance to hydrogen peroxide. To target hOxr1 protein to the yeast mitochondria, the MTS from yeast Sod2 protein was fused to the N terminus of the hOxr1 protein. This targeting signal has been used previously to direct ectopically expressed proteins into the yeast mitochondria (13). The protein was tagged at its C terminus with the 13-Myc epitope tag for detection by immunofluorescence microscopy and Western blotting. The untargeted hOxr1-myc protein lacked the Sod2 protein MTS. Figure 6A shows the hydrogen peroxide survival curves of the wild-type, oxr1Δ, and oxr1Δ strains expressing either the mitochondrion-targeted hOXR1 (mt-hOxr1-myc) or the untargeted hOxr1 (phOxr1-myc) gene from the constitutive GPD (glyceraldehyde-phosphate dehydrogenase) promoter. The mt-hOxr1-myc-expressing strain shows wild-type resistance to hydrogen peroxide, while the untargeted hOxr1-myc-expressing strain is as sensitive as the oxr1Δ mutant strain. Even though the untargeted hOxr1-myc protein is more highly expressed than the mt-hOxr1-myc protein (Fig. 6B), it does not confer peroxide resistance on the strain in which it is expressed, indicating that mitochondrial localization is required for protection from oxidation by Oxr1 protein. We also confirmed the mitochondrial localization of the mt-hOxr1-myc protein by immunofluorescence microscopy as shown in Fig. 6C. The anti-myc antibody staining colocalizes with the MitoTracker probe. In contrast, the untargeted hOxr1-myc protein displays a more diffuse staining pattern, with a significant amount of signal present in the nucleus and little colocalization with MitoTracker. These results indicate that hOxr1 can functionally complement the hydrogen peroxide sensitivity of a yeast oxr1Δ mutant strain and that mitochondrial localization is a requirement for function.
FIG. 6.

Mitochondrion-targeted hOxr1 protein complements the peroxide sensitivity of an oxr1Δ::URA3 mutant S. cerevisiae strain. (A) Yeast strains were grown to an optical density at 600 nm of 0.6 and treated with the indicated doses of H2O2, and dilutions were plated on synthetic minimal dextrose medium plates without leucine. Colonies were counted after 3 days of growth at 30°C. Strains bearing plasmids expressing either the mitochondrion-targeted (pmt-hOXR1-myc) or the untargeted (phOXR1-myc) form of hOXR1 are compared with wild-type (WT) and vector control (oxr1Δ::URA3) strains. The data shown are representative of three independent experiments. conc., concentration. (B) Protein extracts from oxr1Δ::URA3 strains expressing myc-tagged hOxr1 protein fused to the MTS (mt-hOXR1-myc) or untargeted (hOxr1-myc) were immunoblotted with anti-myc monoclonal antibody 9E10. (C) Yeast cells were loaded with 100 μM MitoTracker Green probe and immunostained for hOxr1-myc proteins with anti-myc 9E10 antibody and AlexaFluor 568 secondary antibody. Cells expressing targeted and untargeted hOxr1 proteins are labeled as in panel B.
DISCUSSION
The recently discovered hOXR1 gene represents a family of well-conserved eukaryotic genes whose function is proposed to include resistance to oxidative damage (42). In this report, we demonstrate that the regulation of gene expression, protein localization, and function of Oxr1 is also conserved from yeast to humans.
Numerous proteins are localized to the mitochondria to counteract the deleterious effects of ROS, including glutathione peroxidase, thioredoxin, SOD, and multiple DNA repair enzymes (2, 29, 39, 41). Despite the seeming overabundance of these oxidative damage resistance functions, the yeast oxr1Δ mutant remains sensitive to oxidative damage, indicating an important role for this gene in protecting cells from oxidative damage (42). Consistent with this idea are the results of several microarray experiments addressing stress-induced gene expression in yeast. The scOXR1 gene has been shown to be induced under conditions of heat stress, stationary phase, and diauxic shift (6, 17). Interestingly, these same conditions have been reported to result in significant increases in ROS within the cell (12, 19, 27). Our findings corroborate the microarray data and expand these observations by showing that scOXR1 is part of a stress response pathway turned on under conditions of ROS production and provide further evidence that scOxr1 protein serves to protect yeast cells from oxidative damage. We also demonstrate that the scOxr1 protein can be functionally replaced by its human orthologue containing the Oxr1 homology domain.
The mouse homologue of hOXR1, C7, was isolated by others in a screen for genes up regulated upon cell attachment to extracellular matrix. With the C7M antibody generated to domain II of C7, this protein was shown to localize to the nucleolus in several rodent cell lines (14). We have used the C7C antibody produced from this study and shown it to recognize specifically mitochondrial protein in HeLa cells (Fig. 3), as well as in Hep2 and COS cells. This is consistent with the Western blot data showing that the C7C antibody recognizes only one major protein in untreated HeLa cell extracts (Fig. 4B, untreated control lane). A second species is detectable after induction by oxidative or heat stress and also appears to be largely mitochondrial. No nucleolar staining is detectable, even after stress induction. Our results indicate that, in the cell lines tested, hOxr1 is associated with the mammalian mitochondria. That we observed no nucleolar staining with the C7C antibody suggests either that the nucleolar isoform of Oxr1 lacks the amino acid sequence recognized by C7C or that the antibody cannot access such sequences. Recent studies failed to detect nucleolar staining in human cells (Eva Engvall, personal communication) and are similar to our results with the C7C or C7M antibodies (Fig. 3 and 4 and data not shown). This suggests that the nucleolar staining in rodent cells is due to a species difference or is a species-related artifact. Mitochondrial localization of the Oxr1 homology domain is consistent with the finding that Oxr1 must be targeted to this cellular compartment for the antioxidant function of this domain in yeast.
As is the case with scOXR1, the hOXR1 gene is induced by stress conditions in human cells. The first evidence of stress-induced expression of the hOXR1 gene came from immunofluorescence experiments with HeLa cells after hydrogen peroxide treatment (Fig. 4A). During a 1-h recovery from oxidative stress, hOxr1 protein visibly accumulated in the mitochondria. We also saw a more intense signal in the cytoplasm, which may be due to leakage of hOxr1 protein from the mitochondria, incomplete importation of all of the protein into the mitochondria, or the expression of a distinct cytoplasmic isoform of hOxr1. The latter possibility is consistent with the Western blot results showing the appearance of multiple Oxr1 bands following peroxide treatment (Fig. 4B). This Western blot finding also confirms the oxidative stress-induced accumulation of the 37.5-kDa hOxr1 protein. As in yeast, heat stress has been shown to lead to increased ROS and induction of antioxidant functions in mammalian cells (38). We have shown that heat stress induces expression of hOxr1 protein in a manner very similar to that of oxidative stress (Fig. 4C). Although the induction of mitochondrial heat shock proteins by heat and oxidative stress is well known (16), there is little evidence of mitochondrial proteins outside of this well-conserved protein family induced by both heat and oxidative stress. Also, the most well-characterized mitochondrial heat shock proteins are chaperonins (33), and it is unclear what, if any, antioxidant activity they possess. hOxr1 may therefore represent one of a small set of proteins that are responsive to multiple stress conditions and provide protection against ROS in human mitochondria. In this respect, it is interesting that hOXR1 mRNA appears to be abundant in tissues with a relatively high respiration capacity (heart, skeletal muscle, brain; Fig. 5), where it would be advantageous to counteract mitochondrial ROS production.
It has been hypothesized that ROS play a role in mediating cell death in mammalian cells, particularly through the mitochondrial apoptosis pathway (15, 21, 31). Conditions that increase the amount of mitochondrial ROS production (for example, inhibition of the electron transport chain) lead to increased apoptosis. Conversely, depletion of mitochondrial antioxidant functions has also been shown to increase cell death by apoptosis (22). Regulation of the mitochondrial redox state has been shown to be important for resistance to oxidative stress in S. cerevisiae, as well as in mammals. Deletion of the mitochondrial thioredoxin reductase TRR2 in yeast causes increased sensitivity to hydrogen peroxide (34), while homozygous mutation of mitochondrial thioredoxin (Trx-2) in mice results in elevated apoptosis and embryonic lethality (32). We have found that targeting hOxr1 to the yeast mitochondria is necessary for complementing the hydrogen peroxide sensitivity of an oxr1Δ mutant (Fig. 6A). The hOxr1 protein containing an N-terminal MTS is targeted to the yeast mitochondria (Fig. 6C) and exhibits wild-type resistance to peroxide, particularly at the highest doses tested. A strain expressing an identical copy of hOXR1 lacking the MTS is as sensitive to peroxide as is the oxr1Δ mutant. These data suggest that the peroxide-induced lethality seen in yeast is mediated by a mitochondrial process, and mitochondrial localization of OXR1 function (either yeast or human) is required for wild-type resistance to peroxide damage. Furthermore, these results support the claim that the hOxr1 and scOxr1 proteins are functionally homologous. Our findings suggest that both hOxr1 and scOxr1 may be part of a mitochondrial stress response. Since hOxr1 is capable of providing yeast cells protection from oxidative damage when localized to the mitochondria, it is likely that it plays a similar role in oxidative stress resistance in human cells as well. It will be interesting to determine if hOxr1 is involved in the regulation of ROS production or detoxification and protection from oxidation-mediated apoptosis in human cells.
Acknowledgments
This work was supported by grant GM56420 from the National Institutes of Health.
We thank E. Engvall for mouse C7 antibodies and M. Marinus and E. Engvall for critical reading of the manuscript.
REFERENCES
- 1.Adams, A., D. E. Gottschling, C. A. Kaiser, and T. Stearns. 1997. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
- 2.Arner, E. S., and A. Holmgren. 2000. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267:6102-6109. [DOI] [PubMed] [Google Scholar]
- 3.Avery, A. M., and S. V. Avery. 2001. Saccharomyces cerevisiae expresses three phospholipid hydroperoxide glutathione peroxidases. J. Biol. Chem. 276:33730-33735. [DOI] [PubMed] [Google Scholar]
- 4.Breen, A. P., and J. A. Murphy. 1995. Reactions of oxyl radicals with DNA. Free Radic. Biol. Med. 18:1033-1077. [DOI] [PubMed] [Google Scholar]
- 5.Carmody, R. J., and T. G. Cotter. 2001. Signalling apoptosis: a radical approach. Redox Rep. 6:77-90. [DOI] [PubMed] [Google Scholar]
- 6.Causton, H. C., B. Ren, S. S. Koh, C. T. Harbison, E. Kanin, E. G. Jennings, T. I. Lee, H. L. True, E. S. Lander, and R. A. Young. 2001. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 12:323-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chance, B., H. Sies, and A. Boveris. 1979. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 59:527-605. [DOI] [PubMed] [Google Scholar]
- 8.Collins, T. J., M. J. Berridge, P. Lipp, and M. D. Bootman. 2002. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21:1616-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Comporti, M. 1989. Three models of free radical-induced cell injury. Chem. Biol. Interact. 72:1-56. [DOI] [PubMed] [Google Scholar]
- 10.Cooke, M. S., M. D. Evans, M. Dizdaroglu, and J. Lunec. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17:1195-1214. [DOI] [PubMed] [Google Scholar]
- 11.Costa, V., and P. Moradas-Ferreira. 2001. Oxidative stress and signal transduction in Saccharomyces cerevisiae: insights into ageing, apoptosis and diseases. Mol. Aspects Med. 22:217-246. [DOI] [PubMed] [Google Scholar]
- 12.Davidson, J. F., B. Whyte, P. H. Bissinger, and R. H. Schiestl. 1996. Oxidative stress is involved in heat-induced cell death in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 93:5116-5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobson, A. W., Y. Xu, M. R. Kelley, S. P. LeDoux, and G. L. Wilson. 2000. Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J. Biol. Chem. 275:37518-37523. [DOI] [PubMed] [Google Scholar]
- 14.Fischer, H., X. U. Zhang, K. P. O'Brien, P. Kylsten, and E. Engvall. 2001. C7, a novel nucleolar protein, is the mouse homologue of the Drosophila late puff product L82 and an isoform of human OXR1. Biochem. Biophys. Res. Commun. 281:795-803. [DOI] [PubMed] [Google Scholar]
- 15.Fleury, C., B. Mignotte, and J. L. Vayssiere. 2002. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 84:131-141. [DOI] [PubMed] [Google Scholar]
- 16.Garrido, C., S. Gurbuxani, L. Ravagnan, and G. Kroemer. 2001. Heat shock proteins: endogenous modulators of apoptotic cell death. Biochem. Biophys. Res. Commun. 286:433-442. [DOI] [PubMed] [Google Scholar]
- 17.Gasch, A. P., and M. Werner-Washburne. 2002. The genomics of yeast responses to environmental stress and starvation. Funct. Integr. Genomics 2:181-192. [DOI] [PubMed] [Google Scholar]
- 18.Georgiou, G. 2002. How to flip the (redox) switch. Cell 111:607-610. [DOI] [PubMed] [Google Scholar]
- 19.Grant, C. M., S. Luikenhuis, A. Beckhouse, M. Soderbergh, and I. W. Dawes. 2000. Differential regulation of glutaredoxin gene expression in response to stress conditions in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 1490:33-42. [DOI] [PubMed] [Google Scholar]
- 20.Grant, C. M., G. Perrone, and I. W. Dawes. 1998. Glutathione and catalase provide overlapping defenses for protection against hydrogen peroxide in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 253:893-898. [DOI] [PubMed] [Google Scholar]
- 21.Kim, J. Y., and J. H. Park. 2003. ROS-dependent caspase-9 activation in hypoxic cell death. FEBS Lett. 549:94-98. [DOI] [PubMed] [Google Scholar]
- 22.Klein, J. A., C. M. Longo-Guess, M. P. Rossmann, K. L. Seburn, R. E. Hurd, W. N. Frankel, R. T. Bronson, and S. L. Ackerman. 2002. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419:367-374. [DOI] [PubMed] [Google Scholar]
- 23.Koppenol, W. H. 2001. The Haber-Weiss cycle—70 years later. Redox Rep. 6:229-234. [DOI] [PubMed] [Google Scholar]
- 24.Kuge, S., M. Arita, A. Murayama, K. Maeta, S. Izawa, Y. Inoue, and A. Nomoto. 2001. Regulation of the yeast Yap1p nuclear export signal is mediated by redox signal-induced reversible disulfide bond formation. Mol. Cell. Biol. 21:6139-6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li, N., K. Ragheb, G. Lawler, J. Sturgis, B. Rajwa, J. A. Melendez, and J. P. Robinson. 2003. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 278:8516-8525. [DOI] [PubMed] [Google Scholar]
- 26.Liu, Y., G. Fiskum, and D. Schubert. 2002. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80:780-787. [DOI] [PubMed] [Google Scholar]
- 27.Longo, V. D., E. B. Gralla, and J. S. Valentine. 1996. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J. Biol. Chem. 271:12275-12280. [DOI] [PubMed] [Google Scholar]
- 28.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953-961. [DOI] [PubMed] [Google Scholar]
- 29.Macmillan-Crow, L. A., and D. L. Cruthirds. 2001. Invited review: manganese superoxide dismutase in disease. Free Radic. Res. 34:325-336. [DOI] [PubMed] [Google Scholar]
- 30.Melov, S., J. Ravenscroft, S. Malik, M. S. Gill, D. W. Walker, P. E. Clayton, D. C. Wallace, B. Malfroy, S. R. Doctrow, and G. J. Lithgow. 2000. Extension of life-span with superoxide dismutase/catalase mimetics. Science 289:1567-1569. [DOI] [PubMed] [Google Scholar]
- 31.Newmeyer, D. D., and S. Ferguson-Miller. 2003. Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112:481-490. [DOI] [PubMed] [Google Scholar]
- 32.Nonn, L., R. R. Williams, R. P. Erickson, and G. Powis. 2003. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 23:916-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parcellier, A., S. Gurbuxani, E. Schmitt, E. Solary, and C. Garrido. 2003. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem. Biophys. Res. Commun. 304:505-512. [DOI] [PubMed] [Google Scholar]
- 34.Pedrajas, J. R., E. Kosmidou, A. Miranda-Vizuete, J. A. Gustafsson, A. P. Wright, and G. Spyrou. 1999. Identification and functional characterization of a novel mitochondrial thioredoxin system in Saccharomyces cerevisiae. J. Biol. Chem. 274:6366-6373. [DOI] [PubMed] [Google Scholar]
- 35.Praekelt, U. M., and P. A. Meacock. 1990. HSP12, a new small heat shock gene of Saccharomyces cerevisiae: analysis of structure, regulation and function. Mol. Gen. Genet. 223:97-106. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 37.Stowers, R. S., S. Russell, and D. Garza. 1999. The 82F late puff contains the L82 gene, an essential member of a novel gene family. Dev. Biol. 213:116-130. [DOI] [PubMed] [Google Scholar]
- 38.Subjeck, J. R., and T. T. Shyy. 1986. Stress protein systems of mammalian cells. Am. J. Physiol. 250:C1-C17. [DOI] [PubMed] [Google Scholar]
- 39.Takao, M., H. Aburatani, K. Kobayashi, and A. Yasui. 1998. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 26:2917-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taylor, E. R., F. Hurrell, R. J. Shannon, T. K. Lin, J. Hirst, and M. P. Murphy. 2003. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 278:19603-19610. [DOI] [PubMed] [Google Scholar]
- 41.Ueda, S., H. Masutani, H. Nakamura, T. Tanaka, M. Ueno, and J. Yodoi. 2002. Redox control of cell death. Antioxid. Redox Signal. 4:405-414. [DOI] [PubMed] [Google Scholar]
- 42.Volkert, M. R., N. A. Elliott, and D. E. Housman. 2000. Functional genomics reveals a family of eukaryotic oxidation protection genes. Proc. Natl. Acad. Sci. USA 97:14530-14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao, Y., T. D. Oberley, L. Chaiswing, S. M. Lin, C. J. Epstein, T. T. Huang, and D. St Clair. 2002. Manganese superoxide dismutase deficiency enhances cell turnover via tumor promoter-induced alterations in AP-1 and p53-mediated pathways in a skin cancer model. Oncogene 21:3836-3846. [DOI] [PubMed] [Google Scholar]