

Abstract
Mutations of the MEN1 gene, encoding the tumor suppressor menin, predispose individuals to the cancer syndrome multiple endocrine neoplasia type 1, characterized by the development of tumors of the endocrine pancreas and anterior pituitary and parathyroid glands. We have targeted the murine Men1 gene by using Cre recombinase-loxP technology to develop both total and tissue-specific knockouts of the gene. Conditional homozygous inactivation of the Men1 gene in the pituitary gland and endocrine pancreas bypasses the embryonic lethality associated with a constitutional Men1−/− genotype and leads to β-cell hyperplasia in less than 4 months and insulinomas and prolactinomas starting at 9 months. The pituitary gland and pancreas develop normally in the conditional absence of menin, but loss of this transcriptional cofactor is sufficient to cause β-cell hyperplasia in some islets; however, such loss is not sufficient to initiate pituitary gland tumorigenesis, suggesting that additional genetic events are necessary for the latter.
The MEN1 tumor suppressor gene encodes the transcriptional cofactor menin, and individuals with germ line-inactivating mutations of this gene are predisposed to the cancer syndrome multiple endocrine neoplasia type 1 (MEN 1) (6, 21). Mutation carriers develop hyperplasia and tumors primarily of the endocrine pancreas and parathyroid and anterior pituitary glands. Somatic mutations of the MEN1 gene are commonly found in sporadic parathyroid tumors (13) and pancreatic tumors (35) but less frequently in pituitary tumors (36).
Highly conserved orthologs of the MEN1 gene have been found in a variety of organisms, including molluscs (31), Drosophila (11, 26), zebra fish (19, 25), rats (18), and mice (29). However, the 610-amino-acid human menin sequence provides few clues to the function of the protein, since it shows no homology to any other protein and has no identifiable functional motifs, except for two functionally independent, C-terminal nuclear localization signals (12). The protein has a predominantly nuclear distribution through all stages of the cell cycle (16, 32). Several menin-interacting partners have been identified and include the transcription factors JunD (1), Smad3 (17), NF-κB (14), and Pem (22), suggesting a role for menin in regulating gene expression. Additionally, a role for menin in DNA repair has been implicated through its interaction with the 32-kDa subunit (RPA2) of replication protein A (30), as well as FANCD2, a protein defective in some patients with Fanconi anemia (15).
Menin has been shown to repress the transcriptional activity of JunD, and several mutations clustered around exon 3 interfere with its binding to JunD (1). Further, suppression of AP-1-mediated transcription appears to result from menin reducing the induction of c-Fos expression (34) and inhibiting c-Jun N-terminal kinase-mediated phosphorylation of both c-Jun and JunD (9).
Menin is also necessary for the transcription of transforming growth factor β-responsive genes, an activity mediated through a direct interaction of menin and Smad3 (17). While menin does not influence Smad3-Smad4 dimerization or nuclear localization, it enhances Smad3 DNA binding and Smad3-mediated transcriptional activation. The finding that menin is a key regulator of growth-inhibitory signals arising from the activation of the transforming growth factor β pathway strongly supports its role as a tumor suppressor.
To directly assess the tumor suppressor function of menin in vivo, Crabtree and coworkers generated Men1 knockout mice by deleting exons 3 to 8 of the gene (7). From 9 months of age, heterozygous constitutional knockout mice (i.e., Men1+/− mice) developed hyperplasia and tumors of the parathyroid and anterior pituitary glands and pancreatic islets, the three principal tissues affected in MEN 1. Other tumors normally associated with MEN 1, such as adrenocortical carcinomas, pheochromocytomas, gastric neuroendocrine tumors, and thyroid adenomas, were also observed, indicating that this mouse model very closely mimics the human syndrome. Another murine model of MEN 1 has been developed by Bertolino et al. (3); the mice display a very similar tumor spectrum but with the addition of a high frequency of Leydig cell tumors in male mice and ovary sex cord stromal tumors in female mice. Men1-null mice die in utero at 10.5 to 12.5 days postcoitus (dpc), but the underlying cause of embryonic lethality has not been established (2, 7). The embryos are developmentally delayed and display a variety of abnormalities, including craniofacial defects, cardiac hypotrophy, edema, hemorrhage, and failure of neural tube closure.
To determine whether menin is essential for normal development of the endocrine pancreas and to circumvent the embryonic lethality of the Men1−/− genotype, we sought to produce lines of mice in which the Men1 gene was deleted in a tissue-specific manner in pancreatic β cells. To generate both constitutional and tissue-specific knockouts of the Men1 gene, exon 2 was selected for conditional deletion by the Cre recombinase-loxP system (28). When Cre recombinase is expressed in cells carrying a loxP-flanked (floxed) allele, DNA between the loxP recognition sites is deleted, generating a Men1−/− genotype restricted to the target tissue. Cre-mediated deletion of this exon removes the initiating methionine and approximately 20% of the coding sequence and results in a complete loss of menin production, as determined by Western blot analysis (5). Mice with a constitutive deletion of exon 2 of the Men1 gene were generated by in vitro Cre treatment of embryonic stem cells carrying the floxed allele prior to the establishment of chimeras (5). Additionally, mice in which conditional inactivation of the Men1 gene was specifically targeted to pancreatic β cells were generated by crossing mice homozygous for the floxed allele (Men1loxP/loxP) with mice expressing a Cre transgene under the control of the rat insulin II gene promoter [TgN(Ins2cre)25Mgn, hereafter referred to as Rip-cre+] (27).
MATERIALS AND METHODS
Targeting vector.
The Men1 targeting construct, in which exon 2 was targeted for deletion by Cre recombinase and hence flanked by loxP sites, was described previously (5).
Mice.
Chimeras generated from floxed Men1 exon 2 embryonic stem cells were mated with C57BL/6J females, and pups were genotyped by PCR analysis as described previously (5). Men1+/loxP offspring were crossed with Rip-cre+ mice (27), and the offspring were interbred with Men1loxP/loxP mice to generate conditional Men1 knockout mice (Men1loxP/loxP Rip-cre+) and littermate controls (Men1loxP/loxP Rip-cre−).
Mice expressing the Rip-cre transgene were crossed with the Z/AP Cre-expressing reporter strain [Tg(ACTB-Bgeo/ALPP)1Lbe] (where Z represents lacZ and AP represents alkaline phosphatase) (23). Dually transgenic staged embryos (i.e., Z/AP+ Rip-cre+), adult offspring, and littermate controls (Z/AP+ Rip-cre−) were generated from mating of the respective singly transgenic mice.
All mice in the present study were treated in accordance with National Health and Medical Research Council guidelines for the care of experimental animals.
Genotyping.
Genomic DNA was extracted from a tail tip biopsy (or yolk sac) by overnight digestion at 55°C in 0.7 ml (0.4 ml) of a solution containing 50 mM Tris-HCl (pH 8.0), 20 mM EDTA (pH 8.0), 2.0% sodium dodecyl sulfate, and 0.5 mg of freshly added proteinase K/ml. Following salting out of proteins and cellular debris with 0.25 ml of saturated NaCl, DNA was precipitated with 0.7 ml of isopropanol. DNA pellets were washed with 70% ethanol, air dried, and resuspended in 50 μl of Tris-EDTA (pH 8.0) containing 1 μl of RNase A (10 mg/ml).
PCR analysis was used to detect an approximately 500-bp PCR product of the Rip-cre transgene with 2 μl of a 1:30 dilution of tail tip DNA in a 25-μl reaction mixture containing the supplied reaction buffer (Fisher Biotech), 1.5 mM MgCl2, 2 mM deoxynucleoside triphosphate mixture, 1.4 U of F1 Taq polymerase (Fisher Biotech), and 10 pmol each of primers Cre 3 (5′ CAC CCT GTT ACG TAT AGC 3′) and Cre 4 (5′ CTA ATC GCC ATC TTC CAG 3′). PCR cycling conditions were an initial denaturation step of 3 min at 94°C; 32 cycles of 30 s at 94°C, 30 s at 55°C, and 1 min at 72°C; and a final extension cycle of 5 min at 72°C. Some remaining tail tissue was used for the detection of Z/AP transgene status by staining with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) as outlined by Lobe et al. (23).
Preparation of tissue and embryo samples and analysis of Rip-cre transgene expression.
Protocols for tissue and embryo preparation and staining for lacZ and alkaline phosphatase (AP) activities were those outlined by Lobe et al. (23) but with the following modifications. Embryos were stained for AP alone by using the whole-mount protocol, sectioned, and counterstained with nuclear fast red (NFR) for 30 to 60 s. Dual staining was assessed with adult tissues; following nitroblue tetrazolium (NBT)-5-bromo-4-chloro-3-indolylphosphate (BCIP) staining, frozen sections were washed with phosphate-buffered saline (PBS), counterstained with NFR, and dehydrated through an ethanol-xylene series.
Immunohistochemical analysis.
Formalin-fixed, paraffin-embedded sections were used to detect hormone production in tumors. Sections were deparaffinized and hydrated by passage through xylene and a graded ethanol series. For pituitary tissue, endogenous peroxidase activity was quenched by incubating sections with 3% aqueous hydrogen peroxide for 5 min and then washing with PBS (pH 7.6). Samples were incubated with rabbit anti-human prolactin polyclonal primary antibody (1:1,200 dilution; Dako A0569) or rabbit anti-human adrenocorticotropin (ACTH) polyclonal primary antibody (1:3,500 dilution; Dako A0571) for 30 min. After being washed with PBS, slides were incubated with a biotin-conjugated anti-mouse or anti-rabbit immunoglobulin secondary antibody for 10 min, washed with PBS, incubated with peroxidase-conjugated streptavidin for 10 min, and washed with PBS. The chromogenic reaction was carried out with 3′,3′-diaminobenzidine (Dako K0675), yielding a brown product. Slides were washed with water and then counterstained with hematoxylin. All incubations were performed at room temperature, and all PBS washes were performed for 5 min.
Similarly, pancreatic tissues were stained for insulin but with the following modifications. The primary antibody used was a guinea pig anti-swine insulin polyclonal antibody (1:200 dilution; Dako A0564; 30 min), and the chromogen used was 9-amino-3-ethylcarbazole (Dako K3461). All washes were performed with 50 mM Tris-HCl (pH 7.6)- 0.05% Tween 20.
Proliferation assays.
Assessment of the proliferation status of pancreatic islets with and without expression of the Cre transgene was carried out by using CDC47 (MCM7) immunohistochemical analysis. Sections of paraformaldehyde-fixed, paraffin-embedded pancreatic tissues were incubated overnight with a 1:200 dilution of biotinylated anti-human CDC47/MCM7/Ab-2 (NeoMarkers; MS-862), which specifically localizes to the nuclei of proliferating cells. This step was followed by streptavidin-peroxidase (Jackson Laboratories) and 3′,3′-diaminobenzidine detection. Sections were lightly counterstained with Mayer's hematoxylin. A proliferation index was calculated by counting proliferating cells (brown nuclei) and nonproliferating cells (blue nuclei) in at least five islets from each of three Cre+ and three littermate Cre− animals.
Detection of Cre-mediated deletion of exon 2.
For detecting conversion of the floxed allele to the null allele, genomic DNA was isolated from formalin-fixed tumor and normal tissue samples. Samples were coarsely chopped, suspended in 200 μl of extraction buffer (50 mM Tris-HCl [pH 8.5], 0.5% [vol/vol] Tween 20, 0.4 mg of proteinase K/ml), and incubated at 55°C for 3 to 4 h; this process was followed by overnight digestion at 37°C. Proteins were pelleted by the addition of 0.25 volume of saturated NaCl solution and centrifugation at 1,500 × g for 15 min. DNA was precipitated by the addition of 0.10 volume of 3 M sodium acetate, 2.5 volumes of absolute ethanol, and glycogen to 20 μg/ml, and samples were placed at −20°C for 2 to 4 h. After centrifugation at 16,000 × g for 20 min at 4°C, pellets were washed with 70% ethanol, air dried, and resuspended in 20 μl of Tris-EDTA.
DNA (1 μl) from either tumor or normal tissue samples was used in a multiplex genotyping PCR with a 25-μl reaction mixture containing the supplied reaction buffer (Fisher Biotech), 1.5 mM MgCl2, 2 mM deoxynucleoside triphosphate mixture, 1.4 U of F1 Taq polymerase, and 10 pmol each of primers 968F (5′ TTG GGA CTT GTG GGA GGC TG 3′), 207R (5′ AAG GAC AGG AGC ACC AGG TC 3′), and 2218R (5′ CTC ACA AGA GGC CTC AGA TGC 3′). PCR cycling conditions were an initial denaturation step of 3 min at 95°C; 2 cycles of 1 min at 95°C, 1 min at 68°C, and 1 min at 72°C; 2 cycles of 1 min at 95°C, 1 min at 66°C, and 1 min at 72°C; 24 cycles of 1 min at 95°C, 1 min at 64°C, and 1 min at 72°C; and a final extension cycle of 5 min at 72°C. PCR products were separated and visualized on a 2% agarose gel to detect both a 516-bp floxed exon 2 product (primers 968F and 207R) and a 500-bp deleted exon 2 band (primers 968F and 2218R).
RESULTS AND DISCUSSION
To assess the activity of the Rip-cre transgene and determine its tissue specificity, both during embryogenesis and in adulthood, Rip-cre+ mice were crossed with the Z/AP reporter strain (23). These mice ubiquitously express a floxed lacZ gene in the absence of the Cre enzyme. In Cre-expressing cells, the enzyme excises this portion of the transgene, allowing the expression of the downstream human AP reporter gene and resulting in a switch from blue staining (X-Gal staining for lacZ activity) to purple staining (NBT-BCIP staining for AP activity). While Postic et al. (27) have shown that the Rip-cre transgene is strongly expressed in β cells of the pancreas, we (this study) and others (8, 10) have determined that Cre expression driven by the Rip promoter is not restricted solely to β cells but is found in a variety of other tissues of neuroendocrine lineage. When the Z/AP reporter strain is used, Rip-cre transgene expression (AP staining) is detected as early as 8.5 dpc in Rathke's pouch (Fig. 1a), which develops to become the anterior pituitary gland; by 11.5 dpc, it is also found in the diencephalon and floor plate of the neural tube (Fig. 1b to d, f, and g). Some cells stain in a region of the gut that corresponds to the position of early pancreatic cell precursors (Fig. 1f). AP staining in adult tissues is detected in pancreatic islets (Fig. 2b) and both the anterior and the intermediate lobes of the pituitary gland (Fig. 2f). Low levels are also found in a few regions of the epididymis and thyroid gland and scattered throughout the cerebral hemispheres of the brain (data not shown).
FIG. 1.

Cre recombinase expression in Z/AP+/Rip-cre+ embryos. (a and b) AP whole-mount staining of Z/AP+/Rip-cre+ embryos (right) and Z/AP+/Rip-cre− littermates (left). (a) Embryo at 8.5 dpc showing Cre expression (purple AP staining) in the region of Rathke's pouch (arrow). (b) Embryo at 11.5 dpc showing scattered staining of cells in the lower dorsal region of the embryo and the vagal (X) and accessory (XI) cranial nerve trunk (arrow). (c to g) Sections from 11.5-dpc embryos stained with AP as whole mounts (as above). Sections were counterstained with NFR. (c and d) Z/AP+/Rip-cre+ embryos showing AP staining (magnification, ×40). (e) Z/AP+/Rip-cre− littermate control. (f and g) Z/AP+/Rip-cre+ embryos (magnification, ×100) showing AP staining in the pituitary primordium, i.e., Rathke's pouch (rp), the diencephalon (di), the floor plate of the neural tube (nt), and scattered cells which may correspond to pancreatic precursor cells (pa). Liver (li), heart (he), and other tissues are not stained by AP.
FIG. 2.

Detection of Cre-mediated excision. Pancreatic and pituitary tissues (magnification, ×400) from Z/AP+/Rip-cre+ (a, b, e, and f) and Z/AP+/Rip-cre− (c, d, g, and h) mice were examined. Tissues stained with X-Gal and AP are indicated by blue staining and purple staining, respectively. Hematoxylin and eosin staining of parallel sections for all tissues is shown in panels a, c, e, and g. (b) Islet cells showing mosaic expression of the lacZ reporter gene. Cre excision is indicated by AP staining. X-Gal staining is present in surrounding acinar cells. (d) Pancreatic section showing expression of the lacZ reporter transgene only. is, islet cells; ac, acinar cells. (f) Pituitary section showing extensive Cre activity (AP staining) in the pars distalis (p.d.) and pars intermedia (p.i.). Note that only lacZ expression is faintly detected in the pars nervosa (p.n.). (h) Pituitary section counterstained with NFR and showing expression of the lacZ reporter transgene only.
To assess endocrine organ development and tumorigenesis in Men1loxP/loxP/Rip-cre+ mice, a cohort of animals that were approximately 12 months old (n = 21; median age, 12 months; range, 4 to 16 months) was examined by full necropsy. All of the mice had developed prominent hyperplasia of multiple islets (Fig. 3a), and seven animals also had developed islet cell tumors (Fig. 3e and f). Generally, tumor development occurred earlier and was more severe (larger tumors and/or a larger number of foci) in Men1loxP/loxP/Rip-cre− mice than in our line of Men1+/− mice, which had developed a tumor spectrum very similar to that reported by others (3, 7) (data not shown). Hyperplastic islets and islet cell tumors were stained positively for insulin in all six samples examined (Fig. 3c, g, and h).
FIG. 3.

Pancreatic β-cell hyperplasia and insulinoma in Men1loxP/loxP/Rip-cre+ mice. (a to d) β-Cell hyperplasia in a 4-month-old Men1loxP/loxP/Rip-cre+ mouse (a and c) compared to a Men1loxP/loxP/Rip-cre− littermate (b and d). Staining was done with hematoxylin and eosin (H&E) (a and b; magnification, ×20) and with a horseradish peroxidase-conjugated mouse anti-insulin antibody (c and d; magnification, ×20) (brown). (e to h) Pancreatic islet cell tumor and islet cell hyperplasia in a Men1loxP/loxP/Rip-cre+ mouse. Staining was done with H&E (e and f; magnifications, ×20 and ×200, respectively). Immunoperoxidase staining (brown) of islet cell lesions was done with a mouse anti-insulin antibody (g and h; magnifications, ×20 and ×200, respectively).
After we had submitted the results of this study for publication, two other groups reported β-cell-specific deletion of the Men1 gene in mice (4, 8). The timing of islet hyperplasia and insulinoma formation observed in our mice mirrors their findings. Both of the other studies showed that insulinoma development corresponded to an increase in serum insulin levels and a concomitant decrease in blood glucose concentrations. Although we did not measure these parameters in our mice, we would expect similar changes to have occurred as a direct consequence of the observed β-cell hyperplasia.
Fifteen of the Men1loxP/loxP/Rip-cre+ mice (5 males and 10 females) developed adenomas of the anterior pituitary gland (Fig. 4a and b). All six pituitary adenomas examined were stained positively for prolactin (Fig. 4c and d) and were negative for growth hormone, ACTH, thyroid-stimulating hormone, follicle-stimulating hormone, and luteinizing hormone. The predominance of prolactinomas in these mice is consistent with the human MEN 1 syndrome, in which these are the most commonly seen pituitary lesions. None of the Men1loxP/loxP/Rip-cre− littermate controls examined (n = 22; median age, 17 months; range, 4 to 25 months) had pancreatic or pituitary abnormalities.
FIG. 4.

Prolactinoma and hyperplasia of the pars intermedia in the pituitary glands of Men1loxP/loxP/Rip-cre+ mice. (a and b) Adenoma (ad) of anterior pituitary gland (pars distalis) stained with hematoxylin and eosin (magnifications, ×20 [a] and ×400 [b]). (c and d) Immunoperoxidase staining (brown) of adenoma cells with a rabbit antiprolactin polyclonal antibody (magnifications, ×20 [c] and ×400 [d]). (e and f) Immunoperoxidase staining of the pars intermedia with a rabbit anti-ACTH polyclonal antibody in a Men1loxP/loxP/Rip-cre+ mouse (e) and a Men1loxP/loxP/Rip-cre− littermate (f) (magnification for both panels, ×20).
Surprisingly, in neither of the other models of Rip-cre transgene-mediated conditional deletion of the Men1 gene were pituitary lesions reported (4, 8), in stark contrast to our own observations, where the majority of animals developed prolactinomas. It is difficult to envisage that these differences could have arisen through the alternative regions of the Men1 gene that were targeted for deletion in each of the studies (exons 3 to 8 [8], exon 3 [4], and exon 2 [this study]). It is more likely that the variations in phenotype were due to subtle differences in the founder line carrying the Rip-cre transgene, giving rise to subtle differences in the tissue distribution of Cre expression.
While Z/AP analysis in our mice indicated that the Rip-cre transgene was expressed in the intermediate lobe of the pituitary gland, which in mice produces ACTH (24), only one intermediate lobe adenoma was observed in Men1loxP/loxP/Rip-cre+ mice. However, two other animals did show enlargement of this tissue, confirmed by ACTH immunostaining in one (Fig. 4e and f). No intermediate lobe abnormalities were observed in Men1loxP/loxP/Rip-cre− animals. This finding is consistent with the observed expression of the Rip-cre transgene in the pars intermedia (Fig. 2f) and suggests that deletion of the Men1 gene alone in this part of the pituitary gland has a more limited, although appreciable, effect in promoting neoplasia. This finding contrasts markedly with the effect of deletion of the Rb1 gene in this tissue, since up to 100% of Rb1+/− mice in some studies developed intermediate lobe tumors (33), and is indicative of a general action of menin as a tumor suppressor, even in tissues not normally associated with MEN 1.
Histological changes similar to the changes normally observed during late pregnancy were observed in the mammary tissue of two virgin female mice with pituitary adenomas. These are likely to have been caused by endocrine action of tumor-derived prolactin on the gland rather than to have been a consequence of deletion of the Men1 gene in the mammary tissue. Similarly, another incidental finding was observed in the lung tissue from a female mouse: several pulmonary blood vessels showed marked thickening of the smooth muscle layer and narrowing of the lumen suggestive of pulmonary hypertension. The vascular smooth muscle proliferation is likely to have been induced by factors secreted by the pancreatic islet cell tumor in this animal. A similar phenomenon occurs in carcinoid syndrome, in which molecules (especially serotonin) produced by intestinal neuroendocrine tumor cells cause endocardial fibrosis of the right side of the heart and lead to systemic symptoms, such as flushing and diarrhea.
Histological analysis of tissues at an earlier age (i.e., 4 months) showed normal pituitary development. In pancreatic tissue, mild to moderate islet cell hyperplasia was detected in some islets, although the majority appeared normal. These observations suggest that both of these tissues develop normally in the absence of menin. They also indicate that a complete lack of menin is sufficient to initiate hyperplasia of β cells but not the cells of the anterior pituitary gland. This conclusion is supported by the finding of a much lower frequency of MEN1 gene mutations in pituitary tumors (36) than in tumors of the endocrine pancreas (20, 35) and suggests that other genetic events are necessary to initiate pituitary tumorigenesis, even in patients with MEN 1.
To determine whether the early islet cell hyperplasia was associated with increased proliferation in β cells lacking menin, we counted MCM7-stained proliferating nuclei in islets from mice with and without conditional deletion of the Men1 gene. In a total of 28 wild-type islets from three mice, the median proliferation index was 0 (range, 0 to 3). This index was significantly lower (the P value, determined by the Mann-Whitney U test, was <0.0003) than the median of 2.5 (range, 0 to 22) found in 25 islets of comparable sizes from three Men1loxP/loxP/Rip-cre+ mice. The latter was also significantly lower (P value, <0.0007) than the median of 48.8 (range, 22 to 51) found in three small pancreatic adenomas in mice with β-cell-specific deletion of the Men1 gene. Representative examples of islets and tumors from the mice are shown in Fig. 5.
FIG. 5.

Comparison of proliferation rates in islets or tumors from wild-type and Men1loxP/loxP/Rip-cre+ mice. Anti-MCM7 was used to stain proliferating nuclei in a normal nonproliferating islet from a wild-type mouse (a); a small, moderately proliferating islet from a Men1loxP/loxP; Rip-cre+ mouse (b); and a highly proliferating tumor from a Men1loxP/loxP/Rip-cre+ mouse (c). Scale bars, 50 μm.
We also attempted to determine whether the observed β-cell hyperplasia also might have been due in part to a lower degree of apoptosis in Men1-null cells; however, we were unable to assess this possibility, since all cells in all islets from both wild-type and Men1loxP/loxP/Rip-cre+ mice showed positive staining in an assay for terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL). This phenomenon was restricted to the islets, since only an occasional TUNEL-positive cell was seen in the surrounding exocrine tissues of all animals (data not shown). This finding in pancreatic islets was observed in tissues that were placed in fixative within 15 min of necropsy and most likely was due to the rapid onset of autolysis specifically associated with these tissues.
To confirm that Cre-mediated deletion of exon 2 had occurred in tumor cells but not in cells not expressing Cre, multiplex PCR analysis was performed with representative samples of pancreatic and pituitary lesions to detect conversion of the floxed Men1 allele to the allele with exon 2 deleted. Only the band with exon 2 deleted was detected in tumor cells, while the floxed Men1 allele remained intact in tissues not expressing Cre (Fig. 6).
FIG. 6.
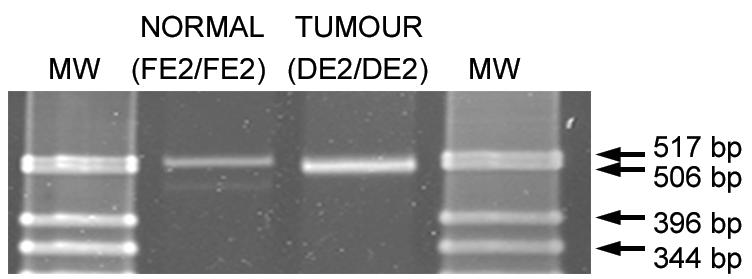
Detection of Cre-mediated deletion of exon 2 of the Men1 gene in tumors from Men1loxP/loxP/Rip-cre+ mice. Multiplex PCR products were run on a 3% agarose gel and stained with Vista Green, which revealed the presence of homozygous floxed Men1 exon 2 (FE2/FE2) and deleted exon 2 (DE2/DE2) alleles. Lanes: MW; molecular size markers (GIBCO-BRL 1-kb ladder); NORMAL, liver tissue; TUMOUR, pituitary adenoma.
In summary, the generation of Men1loxP/loxP/Rip-cre+ mice has succeeded in bypassing the embryonic lethality of the total knockout while inactivating the Men1 gene in parts of the pituitary gland and β cells of the pancreas. This model recapitulates part of the human MEN 1 syndrome and indicates that menin is not essential for the normal development of either the pancreas or the pituitary gland. As predicted, when both copies of the gene are inactivated during embryogenesis, the onset of tumorigenesis occurs significantly earlier than in Men1+/− mice, where tumors occur only after somatic loss of the wild-type allele. The development of hyperplasia at such a young age suggests that at least in mice, the loss of both copies of the Men1 gene is sufficient for the development of endocrine pancreatic hyperplasia. In contrast, only ∼60% of the mice had pituitary tumors by 12 months of age. These findings imply divergent pathways for tumor development in these primary MEN 1 target tissues, suggesting that other events, such as loss or gain of function of additional mutations and/or altered cell signaling through hormonal imbalances, are required for the development of pituitary lesions. Men1loxP/loxP/Rip-cre+ mice provide a unique opportunity to examine the relationship between the onset of pancreatic lesions and the onset of pituitary lesions in the MEN 1 syndrome and will allow further analysis of the different genetic pathways underlying the development of these tumor types.
Acknowledgments
We are grateful to Melanie Trivett and Frank Feleppa for immunohistological analysis.
This work was supported by the National Health and Medical Research Council of Australia and the Queensland Cancer Fund.
REFERENCES
- 1.Agarwal, S. K., S. C. Guru, C. Heppner, M. R. Erdos, R. M. Collins, S. Y. Park, S. Saggar, S. C. Chandrasekharappa, F. S. Collins, A. M. Spiegel, S. J. Marx, and A. L. Burns. 1999. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell 96:143-152. [DOI] [PubMed] [Google Scholar]
- 2.Bertolino, P., I. Radovanovic, H. Casse, A. Aguzzi, Z. Q. Wang, and C. X. Zhang. 2003. Genetic ablation of the tumor suppressor menin causes lethality at mid-gestation with defects in multiple organs. Mech. Dev. 120:549-560. [DOI] [PubMed] [Google Scholar]
- 3.Bertolino, P., W. M. Tong, D. Galendo, Z. Q. Wang, and C. X. Zhang. 2003. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 17:1880-1892. [DOI] [PubMed] [Google Scholar]
- 4.Bertolino, P., W. M. Tong, P. L. Herrera, H. Casse, C. X. Zhang, and Z. Q. Wang. 2003. Pancreatic beta-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Res. 63:4836-4841. [PubMed] [Google Scholar]
- 5.Biondi, C., M. Gartside, I. Tonks, C. Paterson, N. K. Hayward, and G. F. Kay. 2002. Targeting and conditional inactivation of the murine Men1 locus using the Cre recombinase:loxP system. Genesis 32:150-151. [DOI] [PubMed] [Google Scholar]
- 6.Chandrasekharappa, S. C., S. C. Guru, P. Manickam, S. E. Olufemi, F. S. Collins, M. R. Emmert-Buck, L. V. Debelenko, Z. Zhuang, I. A. Lubensky, L. A. Liotta, J. S. Crabtree, Y. Wang, B. A. Roe, J. Weisemann, M. S. Boguski, S. K. Agarwal, M. B. Kester, Y. S. Kim, C. Heppner, Q. Dong, A. M. Spiegel, A. L. Burns, and S. J. Marx. 1997. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 276:404-407. [DOI] [PubMed] [Google Scholar]
- 7.Crabtree, J. S., P. C. Scacheri, J. M. Ward, L. Garrett-Beal, M. R. Emmert-Buck, K. A. Edgemon, D. Lorang, S. K. Libutti, S. C. Chandrasekharappa, S. J. Marx, A. M. Spiegel, and F. S. Collins. 2001. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. USA 98:1118-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crabtree, J. S., P. C. Scacheri, J. M. Ward, S. R. McNally, G. P. Swain, C. Montagna, J. H. Hager, D. Hanahan, H. Edlund, M. A. Magnuson, L. Garrett-Beal, A. L. Burns, T. Ried, S. C. Chandrasekharappa, S. J. Marx, A. M. Spiegel, and F. S. Collins. 2003. Of mice and MEN1: insulinomas in a conditional mouse knockout. Mol. Cell. Biol. 23:6075-6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gallo, A., C. Cuozzo, I. Esposito, M. Maggiolini, D. Bonofiglio, A. Vivacqua, M. Garramone, C. Weiss, D. Bohmann, and A. M. Musti. 2002. Menin uncouples Elk-1, JunD and c-Jun phosphorylation from MAP kinase activation. Oncogene 21:6434-6445. [DOI] [PubMed] [Google Scholar]
- 10.Gannon, M., C. Shiota, C. Postic, C. V. Wright, and M. Magnuson. 2000. Analysis of the Cre-mediated recombination driven by rat insulin promoter in embryonic and adult mouse pancreas. Genesis 26:139-142. [DOI] [PubMed] [Google Scholar]
- 11.Guru, S. C., N. B. Prasad, E. J. Shin, K. Hemavathy, J. Lu, Y. T. Ip, S. K. Agarwal, S. J. Marx, A. M. Spiegel, F. S. Collins, B. Oliver, and S. C. Chandrasekharappa. 2001. Characterization of a MEN1 ortholog from Drosophila melanogaster. Gene 263:31-38. [DOI] [PubMed] [Google Scholar]
- 12.Guru, S. C., P. K. Goldsmith, A. L. Burns, S. J. Marx, A. M. Spiegel, F. S. Collins, and S. C. Chandrasekharappa. 1998. Menin, the product of the MEN1 gene, is a nuclear protein. Proc. Natl. Acad. Sci. USA 95:1630-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heppner, C., M. B. Kester, S. K. Agarwal, L. V. Debelenko, M. R. Emmert-Buck, S. C. Guru, P. Manickam, S. E. Olufemi, M. C. Skarulis, J. L. Doppman, R. H. Alexander, Y. S. Kim, S. K. Saggar, I. A. Lubensky, Z. Zhuang, L. A. Liotta, S. C. Chandrasekharappa, F. S. Collins, A. M. Spiegel, A. L. Burns, and S. J. Marx. 1997. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat. Genet. 16:375-378. [DOI] [PubMed] [Google Scholar]
- 14.Heppner, C., K. Y. Bilimoria, S. K. Agarwal, M. Kester, L. J. Whitty, S. C. Guru, S. C. Chandrasekharappa, F. S. Collins, A. M. Spiegel, S. J. Marx, and A. L. Burns. 2001. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene 20:4917-4925. [DOI] [PubMed] [Google Scholar]
- 15.Jin, S., H. Mao, R. W. Schnepp, S. M. Sykes, A. C. Silva, A. D. D'Andrea, and X. Hua. 2003. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 63:4204-4210. [PubMed] [Google Scholar]
- 16.Kaji, H., L. Canaff, D. Goltzman, and G. N. Hendy. 1999. Cell cycle regulation of menin expression. Cancer Res. 59:5097-5101. [PubMed] [Google Scholar]
- 17.Kaji, H., L. Canaff, J. J. Lebrun, D. Goltzman, and G. N. Hendy. 2001. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc. Natl. Acad. Sci. USA 98:3837-3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karges, W., S. Maier, A. Wissmann, H. Dralle, H. M. Dosch, and B. O. Boehm. 1999. Primary structure, gene expression and chromosomal mapping of rodent homologs of the MEN1 tumor suppressor gene. Biochim. Biophys. Acta 1446:286-294. [DOI] [PubMed] [Google Scholar]
- 19.Khodaei, S., K. P. O'Brien, J. Dumanski, F. K. Wong, and G. Weber. 1999. Characterization of the MEN1 ortholog in zebrafish. Biochem. Biophys. Res. Commun. 264:404-408. [DOI] [PubMed] [Google Scholar]
- 20.Ki Wong, F., J. Burgess, M. Nordenskjold, C. Larsson, and B. Tean Teh. 2000. Multiple endocrine neoplasia type 1. Semin. Cancer Biol. 10:299-312. [DOI] [PubMed] [Google Scholar]
- 21.Lemmens, I., W. J. Van de Ven, K. Kas, C. X. Zhang, S. Giraud, V. Wautot, N. Buisson, K. De Witte, J. Salandre, G. Lenoir, M. Pugeat, A. Calender, F. Parente, D. Quincey, P. Gaudray, M. J. De Wit, C. J. Lips, J. W. Hoppener, S. Khodaei, A. L. Grant, G. Weber, S. Kytola, B. T. Teh, F. Farnebo, and R. V. Thakker. 1997. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum. Mol. Genet. 6:1177-1183. [DOI] [PubMed] [Google Scholar]
- 22.Lemmens, I. H., L. Forsberg, A. A. Pannett, E. Meyen, F. Piehl, J. J. Turner, W. J. Van de Ven, R. V. Thakker, C. Larsson, and K. Kas. 2001. Menin interacts directly with the homeobox-containing protein Pem. Biochem. Biophys. Res. Commun. 286:426-431. [DOI] [PubMed] [Google Scholar]
- 23.Lobe, C. G., K. E. Koop, W. Kreppner, H. Lomeli, M. Gertsenstein, and A. Nagy. 1999. Z/AP, a double reporter for Cre-mediated recombination. Dev. Biol. 208:281-292. [DOI] [PubMed] [Google Scholar]
- 24.Mahler, J. F., and M. R. Elwell. 2002. Pituitary gland, p. 491-507. In R. R. Maronpot (ed.), Pathology of the mouse, vol. 1. Cache River Press, Vienna, Ill. [Google Scholar]
- 25.Manickam, P., A. M. Vogel, S. K. Agarwal, T. Oda, A. M. Spiegel, S. J. Marx, F. S. Collins, B. M. Weinstein, and S. C. Chandrasekharappa. 2000. Isolation, characterization, expression and functional analysis of the zebrafish ortholog of MEN1. Mamm. Genome 11:448-454. [DOI] [PubMed] [Google Scholar]
- 26.Maruyama, K., T. Tsukada, M. Honda, N. Nara-Ashizawa, K. Noguchi, J. Cheng, N. Ohkura, K. Sasaki, and K. Yamaguchi. 2000. Complementary DNA structure and genomic organization of Drosophila menin. Mol. Cell. Endocrinol. 168:135-140. [DOI] [PubMed] [Google Scholar]
- 27.Postic, C., M. Shiota, K. D. Niswender, T. L. Jetton, Y. Chen, J. M. Moates, K. D. Shelton, J. Lindner, A. D. Cherrington, and M. A. Magnuson. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274:305-315. [DOI] [PubMed] [Google Scholar]
- 28.Sauer, B., and N. Henderson. 1988. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA 85:5166-5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart, C., F. Parente, F. Piehl, F. Farnebo, D. Quincey, G. Silins, L. Bergman, G. F. Carle, I. Lemmens, S. Grimmond, C. Z. Xian, S. Khodei, B. T. Teh, J. Lagercrantz, P. Siggers, A. Calender, V. Van de Vem, K. Kas, G. Weber, N. Hayward, P. Gaudray, and C. Larsson. 1998. Characterization of the mouse Men1 gene and its expression during development. Oncogene 17:2485-2493. [DOI] [PubMed] [Google Scholar]
- 30.Sukhodolets, K. E., A. B. Hickman, S. K. Agarwal, M. V. Sukhodolets, V. H. Obungu, E. A. Novotny, J. S. Crabtree, S. C. Chandrasekharappa, F. S. Collins, A. M. Spiegel, A. L. Burns, and S. J. Marx. 2003. The 32-kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol. Cell. Biol. 23:493-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Kesteren, R. E., N. I. Syed, D. W. Munno, Bouwman, Z. P. J., Feng, W. P. Geraerts, and A. B. Smit. 2001. Synapse formation between central neurons requires postsynaptic expression of the MEN1 tumor suppressor gene. J. Neurosci. 21:RC161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wautot, V., S. Khodaei, L. Frappart, N. Buisson, E. Baro, G. M. Lenoir, A. Calender, C. X. Zhang, and G. Weber. 2000. Expression analysis of endogenous menin, the product of the multiple endocrine neoplasia type 1 gene, in cell lines and human tissues. Int. J. Cancer 85:877-881. [DOI] [PubMed] [Google Scholar]
- 33.Williams, B. O., L. Remington, D. M. Albert, S. Mukai, R. T. Bronson, and T. Jacks. 1994. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat. Genet. 7:480-484. [DOI] [PubMed] [Google Scholar]
- 34.Yumita, Y. W., Ikeo, K. Yamauchi, A. Sakurai, and K. Hashizume. 2003. Suppression of insulin-induced AP-1 transactivation by menin accompanies inhibition of c-Fos induction. Int. J. Cancer 103:738-744. [DOI] [PubMed] [Google Scholar]
- 35.Zhuang, Z., A. O. Vortmeyer, S. Pack, S. Huang, T. A. Pham, C. Wang, W. S. Park, S. K. Agarwal, L. V. Debelenko, M. Kester, S. C. Guru, P. Manickam, S. E. Olufemi, F. Yu, C. Heppner, J. S. Crabtree, M. C. Skarulis, D. J. Venzon, M. R. Emmert-Buck, A. M. Spiegel, S. C. Chandrasekharappa, F. S. Collins, A. L. Burns, S. J. Marx, and I. A. Lubensky. 1997. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res. 57:4682-4686. [PubMed] [Google Scholar]
- 36.Zhuang, Z., S. Z. Ezzat, A. O. Vortmeyer, R. Weil, E. H. Oldfield, W. S. Park, S. Pack, S. Huang, S. K. Agarwal, S. C. Guru, P. Manickam, L. V. Debelenko, M. B. Kester, S. E. Olufemi, C. Heppner, J. S. Crabtree, A. L. Burns, A. M. Spiegel, S. J. Marx, S. C. Chandrasekharappa, F. S. Collins, M. R. Emmert-Buck, L. A. Liotta, S. L. Asa, and I. A. Lubensky. 1997. Somatic mutations of the MEN1 tumor suppressor gene in pituitary tumors. Cancer Res. 57:5446-5451. [PubMed] [Google Scholar]