

Abstract
Purpose
Radiotherapy with concomitant and adjuvant temozolomide is the standard of care for newly diagnosed glioblastoma (GBM). O6-methylguanine-DNA methyltransferase (MGMT) methylation status may be an important determinant of treatment response. Dose-dense (DD) temozolomide results in prolonged depletion of MGMT in blood mononuclear cells and possibly in tumor. This trial tested whether DD temozolomide improves overall survival (OS) or progression-free survival (PFS) in patients with newly diagnosed GBM.
Patients and Methods
This phase III trial enrolled patients older than age 18 years with a Karnofsky performance score of ≥ 60 with adequate tissue. Stratification included clinical factors and tumor MGMT methylation status. Patients were randomly assigned to standard temozolomide (arm 1) or DD temozolomide (arm 2) for 6 to 12 cycles. The primary end point was OS. Secondary analyses evaluated the impact of MGMT status.
Results
A total of 833 patients were randomly assigned to either arm 1 or arm 2 (1,173 registered). No statistically significant difference was observed between arms for median OS (16.6 v 14.9 months, respectively; hazard ratio [HR], 1.03; P = .63) or median PFS (5.5 v 6.7 months; HR, 0.87; P = .06). Efficacy did not differ by methylation status. MGMT methylation was associated with improved OS (21.2 v 14 months; HR, 1.74; P < .001), PFS (8.7 v 5.7 months; HR, 1.63; P < .001), and response (P = .012). There was increased grade ≥ 3 toxicity in arm 2 (34% v 53%; P < .001), mostly lymphopenia and fatigue.
Conclusion
This study did not demonstrate improved efficacy for DD temozolomide for newly diagnosed GBM, regardless of methylation status. However, it did confirm the prognostic significance of MGMT methylation. Feasibility of large-scale accrual, prospective tumor collection, and molecular stratification was demonstrated.
INTRODUCTION
Glioblastoma (GBM) is the most common primary malignant brain tumor in adults. The recent phase III randomized clinical trial performed by the European Organisation for Research and Treatment of Cancer (EORTC) and the National Cancer Institute of Canada (NCIC) established the current standard of care for newly diagnosed GBM, which involves surgery or regional radiotherapy with concomitant temozolomide daily during the radiation therapy.1 Following completion of radiotherapy, patients undergo six cycles of single-agent temozolomide (5 days of a 28-day cycle). However, even with this treatment, the outcome for most patients with GBM remains grim, with a 2-year survival rate of only 27%.
Tumor specimens from a subset of patients enrolled onto the EORTC/NCIC clinical trial were evaluated for epigenetic changes in the promoter region of the O6-methylguanine-DNA methyltransferase (MGMT) DNA repair gene.2 The MGMT enzyme has been established as a major mechanism of resistance to alkylating agents, such as temozolomide.3 In this study of 203 tumor samples, epigenetic silencing by methylation of the promoter region of the MGMT gene was associated with statistically significant improvement in overall survival (OS) that was most marked in patients receiving the combined radiotherapy and temozolomide regimen. These findings suggest that modulation of MGMT enzymatic activity may increase sensitivity and response to temozolomide regimens.
Exposure to radiation has been shown to upregulate MGMT, whereas prolonged exposure to alkylating agents has been shown to deplete intracellular MGMT in peripheral blood mononuclear cells.4,5 Dose-dense (DD) schedules of temozolomide, designed to reduce MGMT levels and exhaust activity, have been tested in patients with recurrent or progressive GBM (reviewed in Wick et al6). Nonrandomized studies that tested a variety of treatment schedules reported treatment efficacy that compares favorably with the established temozolomide single-agent schedule without an apparent increase in treatment-related toxicity.7,8 The impact of two temozolomide schedules on MGMT activity were compared by using peripheral blood mononuclear cells as a surrogate for tumor.5 Both the alternating 7-days-on, 7-days-off and the 21-of-28-days schedules were shown to deplete MGMT activity. However, the latter schedule resulted in more prolonged and lower MGMT activity.
To test the hypothesis that prolonged exposure to temozolomide improves survival in patients with newly diagnosed GBM, a randomized phase III clinical trial comparing the standard adjuvant temozolomide treatment (days 1 through 5 of a 28-day cycle) with the DD schedule (days 1 through 21 of a 28-day cycle) was initiated as a collaborative effort involving the Radiation Therapy Oncology Group (RTOG), the EORTC, and the North Central Cancer Therapy Group (NCCTG).
PATIENTS AND METHODS
Patients
Patients were eligible for the study if they were older than age 18 years with a newly diagnosed, histologically confirmed GBM (WHO grade 4 astrocytoma; Fig 1). Additional eligibility criteria included a Karnofsky performance score of at least 60 and adequate hematologic, renal, and hepatic function (defined as an absolute neutrophil count ≥ 1,500/mL, platelet count ≥ 100,000/mL, serum creatinine ≤ 1.7 mg/dL, serum blood urea nitrogen ≤ 25 mg/dL, serum total bilirubin ≤ 2.0 mg/dL, and serum ALT and serum AST ≤ 3× the upper limit for the laboratory). Patients taking corticosteroids had to be taking a stable or decreasing dose for the 5 days before study registration. Submission of a tumor tissue block with a minimum of 1 cm2 of tumor by day 14 of radiotherapy was a requirement. Before patient accrual, all patients were required to provide informed consent after study approval by an institutional review board or ethics committee.
Fig 1.

CONSORT diagram outlining accrual and eligibility. GBM, glioblastoma; IMRT, intensity-modulated radiotherapy.
Study Design and Treatment
The study schema is provided in Figure 2. Patients were required to be registered before the initiation of the concomitant radiotherapy and chemotherapy. Radiotherapy consisted of fractionated, conformal radiation given at a daily dose of 2 Gy. Treatment was delivered 5 days a week for a total of 6 weeks to a total dose of 60 Gy. Two radiotherapy protocols were allowed. In North America (RTOG, NCCTG), an initial volume consisting of enhancement, postoperative cavity, plus surrounding edema (or fluid-attenuated inversion-recovery [FLAIR] abnormality defined by magnetic resonance imaging [MRI]) and a 2-cm margin received 46 Gy in 23 fractions followed by a boost of 14 Gy in seven fractions to the area of enhancement plus the cavity and a 2.5-cm margin. In European (EORTC) centers, a single planning volume was used to deliver 60 Gy in 30 fractions to the area of enhancement and the cavity with a 2- to 3-cm margin. Temozolomide at a dose of 75 mg/m2 was started along with the radiotherapy and was continued on a daily basis until completion of radiation treatment, with a maximum of 49 doses. During the concomitant radiotherapy and temozolomide treatment, prophylaxis against Pneumocystis jiroveci pneumonia was required. Antiemetic prophylaxis was recommended at initiation of the concomitant radiotherapy and chemotherapy regimen.
Fig 2.

Protocol schema. MGMT, O6-methylguanine-DNA methyltransferase; RPA, recursive partitioning analysis; RT, radiotherapy; TMZ, temozolomide.
Patients were randomly assigned after completion of the concomitant radiotherapy and chemotherapy treatment to either standard or DD temozolomide in a permuted block design by using the method described by Zelen.9 Stratification factors included the RTOG recursive partitioning analysis (RPA) class (a compilation of clinical prognostic factors including age, performance status, extent of tumor resection, and neurologic function10), MGMT gene promoter methylation status, and radiotherapy plan (RTOG or EORTC). MGMT gene promoter methylation status was determined by using a quantitative methylation-specific polymerase chain reaction assay as described11 and performed centrally by Oncomethylome Science.
Adjuvant temozolomide treatment was initiated 4 weeks after completion of radiotherapy. Patients on the standard treatment arm received temozolomide at a starting dose of 150 mg/m2 for 5 consecutive days of a 28-day cycle, and temozolomide was increased for subsequent cycles to 200 mg/m2 if no treatment-related adverse events greater than grade 2 were noted. Treatment was planned for six cycles with the potential to extend treatment to a total of 12 cycles if treatment was well tolerated and there was evidence of continued benefit defined as either continued tumor response based on serial MRI, progressive improvement in the patient's performance status or neurologic function, or a decreasing requirement for corticosteroids. Patients randomly assigned to the DD treatment arm received an initial dose of 75 mg/m2 for 21 consecutive days of a 28-day cycle, which was increased for subsequent cycles to 100 mg/m2 if no treatment-related adverse events greater than grade 2 were noted. As with the standard dose arm, six cycles were planned with the potential to extend to a total of 12 cycles if the previously described criteria for benefit were met. Antiemetic therapy using a 5-hydroxytrytamine antagonist was strongly recommended for all patients. P. jiroveci pneumonia prophylaxis was recommended for patients with CD4 counts less than 200/mL.
Patient Evaluation and Follow-Up
Baseline evaluation included a neurologic evaluation, complete blood counts, blood chemistries including renal and hepatic function, a serum pregnancy test (as appropriate), and tumor imaging with either MRI scans (preferred) or computed tomography. A subset of patients were enrolled onto the Net Clinical Benefits study to evaluate the impact of tumor and treatment. They underwent baseline as well as longitudinal testing with the MD Anderson Symptom Inventory for Brain Tumors (MDASI-BT); a neurocognitive function battery; and the EORTC QLQ30/BN20 (the EORTC general quality-of-life and brain cancer–specific quality-of-life questionnaire).12–14 During radiotherapy, patients were assessed weekly for adverse events and underwent weekly complete blood counts and monthly blood chemistries. During the adjuvant phase of treatment, patients in the standard adjuvant treatment arm had blood counts and blood chemistries measured on days 21 and 28 of each cycle; patients in the DD treatment arm had blood testing on days 14, 21, and 28 of each cycle. A repeat tumor imaging study was performed approximately 4 weeks after completion of radiotherapy and before initiation of cycle 4 (and 7 and 10, if given). Patients who completed adjuvant treatment underwent tumor imaging every 3 months until tumor progression. Assessment of response was made by using serial measures of the product of the two largest cross-sectional diameters, and progression was defined as an increase in tumor size by at least 25% or development of a new lesion.15 In light of the recognition of early radiation reactions emulating tumor progression, investigators were encouraged not to declare tumor progression within the first 12 weeks after completion of radiation unless there was either a new lesion or the patient had neurologic worsening.16 Toxicities were graded according to the National Cancer Institute's Common Terminology Criteria for Adverse Events version 3.0.
Statistical Considerations
The primary end point was OS. Secondary end points included progression-free survival (PFS) and toxicities associated with each treatment. The hypothesis was that there would be at least 20% reduction in hazard rate for the DD temozolomide arm, corresponding to a median survival time of 17.5 months. A one-sided log-rank test at a significance level of 0.025 would have 80% power to detect this survival difference with a sample size of 750 randomly assigned patients (647 deaths were required for the final analysis). With 750 analyzable patients, the study had at least 90% power to detect a 2-month increase in PFS for patients treated with DD temozolomide.
The statistical analysis is based on the modified intent-to-treat principle (including all of the eligible and randomly assigned patients, regardless of treatment receipt). OS and PFS were estimated by using the Kaplan-Meier method, and differences between treatment groups were tested by using the log-rank test.17,18 The Cox proportional hazards model was used to estimate the treatment hazard ratios (HRs) associated with each end point while adjusting for stratification factors.19 OS was defined as the interval from random assignment to death as a result of any cause or the last follow-up date on which the patient was reported alive. PFS was defined as the interval from random assignment to progression or death or being censored at the last clinical or radiology assessment date on which the patient was reported alive without progression. For all end points except the OS comparison between two treatments for the study, a P value of less than .05 (two-sided) was defined as the significance threshold. All of the analyses were performed by using SAS version 9 (SAS Institute, Cary, NC).
RESULTS
Patient Demographics
The study opened for accrual in January 2006 and closed to accrual in June 2008 with a total of 1,173 patients entered (Fig 1). Among registered patients, 1,125 were eligible for concurrent radiotherapy and temozolomide and 48 (4%) were ineligible. A total of 833 patients (74% of those enrolled) were randomly assigned for the adjuvant stage of treatment. Inadequate tissue (n = 144) and early disease progression (n = 48) were the most frequent reasons for not being randomly assigned to one of the two study arms. The characteristics of the two treatment groups were well balanced at baseline (Table 1). Tumor tissue blocks were submitted from 1,141 (97%) of the enrolled patients. All randomly assigned patients had centrally confirmed GBM. MGMT gene promoter methylation status determination was available for 91% of samples.
Table 1.
Patient Characteristics
| Characteristic | Standard Dose Temozolomide (arm 1) |
Dose-Dense Temozolomide (arm 2) |
||
|---|---|---|---|---|
| No. | % | No. | % | |
| Age, years | ||||
| < 50 | 112 | 27 | 111 | 26 |
| ≥ 50 | 299 | 73 | 311 | 74 |
| Sex | ||||
| Male | 239 | 58 | 237 | 56 |
| Female | 172 | 42 | 185 | 44 |
| Race/ethnicity | ||||
| White | 319 | 78 | 328 | 78 |
| Nonwhite | 13 | 3 | 15 | 3 |
| Unknown | 79 | 19 | 79 | 19 |
| KPS | ||||
| 60-80 | 138 | 34 | 146 | 35 |
| 90-100 | 273 | 66 | 276 | 65 |
| Surgery | ||||
| Biopsy | 14 | 3 | 13 | 3 |
| Partial resection | 167 | 41 | 188 | 45 |
| Total resection | 230 | 56 | 221 | 52 |
| Radiation | ||||
| RTOG/NCCTG | 337 | 82 | 349 | 83 |
| EORTC | 74 | 18 | 73 | 17 |
| Neurologic function | ||||
| No symptoms | 140 | 34 | 147 | 35 |
| Minor symptoms | 185 | 45 | 196 | 45 |
| Moderate symptoms | 84 | 20 | 75 | 18 |
| Severe symptoms | 2 | 1 | 4 | 1 |
| MGMT status | ||||
| Methylated | 122 | 30 | 123 | 29 |
| Unmethylated | 254 | 62 | 263 | 62 |
| Unknown (invalid, indeterminate) | 35 | 8 | 36 | 9 |
| RPA class | ||||
| III | 85 | 21 | 86 | 20 |
| IV | 251 | 61 | 259 | 61 |
| V | 75 | 18 | 77 | 19 |
Abbreviations: EORTC, European Organisation for Research and Treatment of Cancer; KPS, Karnofsky performance status; MGMT, O6-methylguanine-DNA methyltransferase; NCCTG, North Central Cancer Therapy Group; RPA, recursive partitioning analysis; RTOG, Radiation Therapy Oncology Group.
Treatment Administration
At the completion of radiotherapy, patients were randomly assigned to receive either standard dose (n = 411) or DD temozolomide (n = 422). For the standard dose arm, the median number of cycles was three, and 155 patients (37%) received at least six cycles of temozolomide, whereas for the DD arm, the median number of cycles was four, and 181 patients (43%) received at least six cycles of treatment. More than six cycles of adjuvant treatment were given to 117 and 122 patients on the standard and DD arms, respectively. The full 12 cycles of adjuvant treatment were administered to 57 and 59 patients on the standard and DD arms, respectively. Tumor progression or death prompted treatment cessation in 255 patients (62%) on the standard dose arm and 211 patients (50%) on the DD arm. Toxicity or intercurrent illness resulted in treatment cessation in 49 patients (12%) on the standard dose arm and 94 patients (22%) on the DD arm. The median total dose of temozolomide administered was 7,500 mg in the standard dose arm and 17,010 mg in the DD arm.
Treatment Outcomes
At the time of final study analysis (using data through January 6, 2011), 219 patients (20%) were still alive, with a median follow-up time of 31.9 months. The median OS and PFS (from the time of registration) were 16 months (95% CI, 15.2 to 16.7 months) and 7.5 months (95% CI, 7.1 to 8.0 months), respectively.
For the primary analyses of this study, OS and PFS were measured from the time of random assignment. For reference, summary outcome data are provided from study registration in Table 2. There were 652 deaths among the 833 randomly assigned patients, with 22% of patients still alive. The median follow-up from random assignment was 31.5 months. The median OS was 16.6 months (95% CI, 14.9 to 18.0 months) for patients on the standard dose arm and 14.9 months (95% CI, 13.7 to 16.5 months) for patients on the DD arm (Fig 3A). The P value from the one-sided log-rank test was .63 with an associated HR of 1.03 (95% CI, 0.88 to 1.20) for DD versus standard dose. The protocol-specified six cycles of maintenance therapy were completed in 336 patients, but 75 (22%) stopped because of progression or toxicity. Only 22 (7%) stopped at treatment completion, and 239 (71%) received additional cycles. The median OSs for those 22 and 239 patients are 24.9 months (95% CI, 19.2 to 36.2 months) and 30.2 months (95% CI, 25.5 to 35.4 months), respectively.
Table 2.
Outcomes Data From Study Registration
| Patient Group | No. of Patients | OS (months) | PFS (months) | 2-Year Survival Rate (%) |
|---|---|---|---|---|
| All eligible | 1,120 | 16.0 | 7.5 | |
| All randomly assigned | 833 | 17.7 | 8.2 | |
| Arm 1 | 411 | 18.9 | 7.5 | 34.2 |
| Arm 2 | 422 | 16.8 | 8.8 | 33.9 |
| MGMT | ||||
| Methylated | 245 | 23.2 | 10.5 | 47.3 |
| Unmethylated | 517 | 16.0 | 7.8 | 25.4 |
| Methylated | ||||
| Arm 1 | 122 | 23.5 | 8.8 | |
| Arm 2 | 123 | 21.9 | 11.7 | |
| Unmethylated | ||||
| Arm 1 | 254 | 16.6 | 7.1 | |
| Arm 2 | 263 | 15.4 | 8.2 |
Abbreviations: MGMT, O6-methylguanine-DNA methyltransferase; OS, overall survival; PFS, progression-free survival.
Fig 3.

Outcomes comparing standard and dose-dense temozolomide therapy. (A, B) Overall and progression-free survival for all randomly assigned patients. (C, D) Overall and progression-free survival for patients with O6-methylguanine-DNA methyltransferase (MGMT) unmethylated tumors. (E, F) Overall and progression-free survival for patients with MGMT methylated tumors. HR, hazard ratio.
At the time of analysis, 753 patients (91%) had experienced tumor progression or had died. The median PFS was 5.5 months (95% CI, 4.7 to 6.1 months) for the standard dose arm and 6.7 months (95% CI, 6.2 to 7.7 months) for the DD arm, yielding a two-sided P value of .06 with an associated HR of 0.87 (95% CI, 0.75 to 1.00; Fig 3B).
MGMT methylation status was available for 762 of the randomly assigned patients. In this cohort, there were 595 deaths and disease progression occurred in an additional 93 patients. The median OS was 21.2 months (95% CI, 17.9 to 24.8 months) and 14.0 months (95% CI, 12.9 to 14.7 months) for tumors with methylated and unmethylated MGMT gene promoter, respectively. The HR (methylated v unmethylated) was 0.58 (95% CI, 0.48 to 0.69; P < .001; Fig 4A). Similarly, the median PFS was 8.7 months (95% CI, 6.6 to 11.2 months) and 5.7 months (95% CI, 5.1 to 6.1 months), respectively, for tumors with methylated and unmethylated MGMT gene promoter with an HR (methylated v unmethylated) of 0.61 (95% CI, 0.52 to 0.73; P < .001; Fig 4B).
Fig 4.
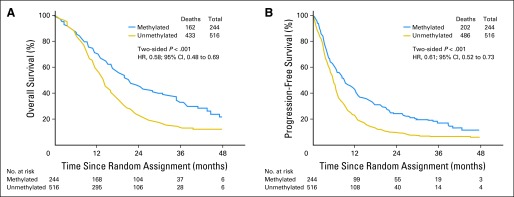
Outcomes based on tumor O6-methylguanine-DNA methyltransferase (MGMT) methylation status. (A) Overall survival. (B) Progression-free survival. HR, hazard ratio.
For patients with tumors that were designated as MGMT unmethylated (n = 517), there were 433 deaths and 53 additional patients with tumor progression. The median OS was 14.6 months (95% CI, 13.2 to 16.5 months) for standard dose and 13.3 months (95% CI, 12.3 to 14.3 months) for the DD arm (HR, 0.99; 95% CI, 0.82 to 1.19; P = .44; Fig 3C). Median PFS was 5.1 months (95% CI, 4.3 to 5.7 months) for the standard arm and 6.0 months (95% CI, 5.5 to 6.5 months) for the DD arm (HR, 0.88; 95% CI, 0.73 to 1.05; P = .15; Fig 3D). For patients with MGMT methylated tumors (n = 245), there were 162 deaths and an additional 40 patients with tumor progression. The median OS was 21.4 months (95% CI, 17.6 to 29.0 months) for the standard arm and 20.2 months (95% CI, 15.4 to 25.1 months) for the DD arm (HR, 1.19; 95% CI, 0.87 to 1.62; P = .86; Fig 3E). Median PFS was 6.5 months (95% CI, 4.1 to 9.6 months) for the standard arm and 10.1 months (95% CI, 7.9 to 12.4 months) for the DD arm (HR, 0.87; 95% CI, 0.66 to 1.15; P = .33; Fig 3F).
Cox proportional hazard modeling for OS and PFS included recursive partitioning analysis (RPA) class (III, IV, V), tumor MGMT status (methylated, unmethylated), and radiation type (European v US). For both the OS and PFS end points, RPA class and MGMT status remained in the model. After adjusting for these factors, the OS HR for DD versus standard treatment was 1.06 (95% CI, 0.90 to 1.24; P = .51) and the PFS HR for DD versus standard treatment was 0.89 (95% CI, 0.77 to 1.03; P = .12). Neither a global test for interaction of treatment with covariates of MGMT methylation status, RPA class, or radiation type nor any pairwise tests of interaction were significant.
Safety and Toxicity
Adverse events were evaluated separately during the concurrent chemotherapy and radiotherapy and the adjuvant treatment phase. Grade 3 or 4 lymphopenia was the most common toxicity occurring in 12% of patients but without significant opportunistic infections. Serious neutropenia and thrombocytopenia occurred in 3.6% and 6.8%, respectively, of all patients undergoing radiation. There was one treatment-related death as a result of neutropenia.
During the adjuvant phase of treatment, grade 3 to 5 adverse events were more prevalent with the DD (n = 194 of 369) compared with standard dose treatment (n = 120 of 351). However, most of the difference was because of a higher number of patients with lymphopenia (107 v 51) and fatigue (33 v 12). Overall, neutropenia and thrombocytopenia were infrequent and were similar between the two arms (Table 3).
Table 3.
Serious Toxicities Associated with Standard Dose and Dose-Dense Temozolomide Treatment
| Category | Grade 3 |
Grade 4 |
||||||
|---|---|---|---|---|---|---|---|---|
| Standard Dose |
Dose Dense |
Standard Dose |
Dose Dense |
|||||
| No. | % | No. | % | No. | % | No. | % | |
| Anemia | 4 | 1 | 4 | 1 | 0 | 0 | ||
| Leukopenia | 13 | 4 | 33 | 9 | 7 | 2 | 3 | 1 |
| Neutropenia | 16 | 5 | 29 | 8 | 8 | 2 | 7 | 2 |
| Lymphopenia | 41 | 12 | 76 | 20 | 10 | 3 | 31 | 9 |
| Thrombocytopenia | 20 | 6 | 18 | 5 | 13 | 4 | 8 | 2 |
| Fatigue | 12 | 3 | 33 | 9 | 0 | 0 | ||
| Nausea/vomiting | 5 | 1 | 8 | 2 | 0 | 0 | ||
DISCUSSION
The landmark study performed by the EORTC and NCIC provided the first Level 1 evidence for the clinical benefit of chemotherapy for patients with newly diagnosed GBM.1 MGMT has been proposed as a major mechanism of resistance to alkylating agents and, therefore, tumor cells with low levels of MGMT expression were expected to have a better response to these drugs.4 A companion correlative subset study to the EORTC trial demonstrated a relationship between the tumor promoter region methylation status of the MGMT gene and outcome.2 Tumor cells with high levels of MGMT protein are thought to be resistant to alklyating agents, and it would follow that successful depletion of intracellular MGMT would be expected to enhance treatment response.
The study reported here sought to determine whether prolonged exposure to an alkylating agent with known activity in GBM would improve patient outcomes as measured by OS or PFS. The DD schedule was designed to deplete cellular MGMT and restore sensitivity to temozolomide.5 Alternatively, in tumor cells with no or low MGMT expression, the DD schedule was projected to improve response by an enhancement of tumor cell exposure, as has been demonstrated with other cancer regimens, particularly in breast cancer.20 Unfortunately, no therapeutic benefits were detected for the DD regimen. A recent study that evaluated temozolomide dosing regimens in chemotherapy-naive patients with recurrent high-grade gliomas did not demonstrate improved survival for the DD temozolomide regimen using the same 21-of-28-days schedule.21 The investigators postulated that peak temozolomide concentrations, rather than prolonged exposure, may be most important for treatment efficacy. Furthermore, comparison of the OS results with those in prior studies is limited by differences in eligibility (age, need for tumor resection) and loss of patients before random assignment.
Although a therapeutic advantage for DD temozolomide was not established, the study provides important information and precedence regarding the design and feasibility of large-scale, international, collaborative cancer clinical trials. The high rate of tumor tissue acquisition (97%) from the eligible patients provided the opportunity to confirm the prognostic significance of MGMT gene promoter methylation and this tissue repository has provided samples for advanced multiplatform molecular analyses. Patient outcomes instruments, including tests of neurocognitive function, symptom burden, and quality of life were completed by a subset of patients. These analyses, reported separately, provide an unprecedented opportunity to examine the comparative impact of the study treatments on patient outcomes. This study design was successfully implemented for a recently completed international phase III study in patients with newly diagnosed GBM.
Supplementary Material
Acknowledgment
We thank Kathryn Okrent, Barbara Kaiser, Sandrine Geinoz, and Wilma Hoffman (deceased) for protocol development and management; Denise Manfredi, Nick Barror, Tom Budiharto, Oscar Matzinger, Paul Fenton, Edwin Aird, and Rachel Bar-Dermona for radiation therapy quality assurance; and Robert Janzer (deceased) and Benoit Lhermitte for pathology review.
Footnotes
See accompanying article on page 4076
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information: NCT00304031.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Mark R. Gilbert, Genentech (C), EMD Serono (C), Merck (C); Roger Stupp, Merck (C), Merck Serono (C), Genentech (C); Monika E. Hegi, Merck (C), EMD Serono (C), MDxHealth (C); Jeffrey S. Wefel, Genentech (C); Deborah T. Blumenthal, Merck (C); Brigitta G. Baumert, Schering-Plough (C); Minesh P. Mehta, Merck (C), Roche (C), Novocure (C) Stock Ownership: Minesh P. Mehta, Accuray, Pharmacyclics Honoraria: Mark R. Gilbert, Merck, Genentech, Novartis; Roger Stupp, Merck, Genentech; Monika E. Hegi, Merck, EMD Serono, MDxHealth; Terri S. Armstrong, Merck; Minesh P. Mehta, Merck Research Funding: Mark R. Gilbert, GlaxoSmithKline, Genentech; Monika E. Hegi, MDxHealth; Terri S. Armstrong, Merck, Genentech; Jeffrey S. Wefel, Merck Expert Testimony: None Patents: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Mark R. Gilbert, Meihua Wang, Kenneth D. Aldape, Roger Stupp, Kurt A. Jaeckle, Terri S. Armstrong, Jeffrey S. Wefel, Minhee Won, Arnab Chakravarti, Walter J. Curran Jr, Minesh P. Mehta
Administrative support: Roger Stupp
Provision of study materials or patients: Mark R. Gilbert, Roger Stupp, Deborah T. Blumenthal, Christopher J. Schultz, Sara Erridge, Tzahala Tzuk-Shina, Minesh P. Mehta
Collection and assembly of data: Mark R. Gilbert, Roger Stupp, Monika E. Hegi, Jeffrey S. Wefel, Deborah T. Blumenthal, Anita Mahajan, Kristen I. Hopkins, Tzahala Tzuk-Shina, Arnab Chakravarti, Walter J. Curran Jr
Data analysis and interpretation: Mark R. Gilbert, Meihua Wang, Kenneth D. Aldape, Roger Stupp, Kurt A. Jaeckle, Terri S. Armstrong, Jeffrey S. Wefel, Minhee Won, Anita Mahajan, Christopher J. Schultz, Sara Erridge, Brigitta G. Baumert, Paul D. Brown, Arnab Chakravarti, Walter J. Curran Jr, Minesh P. Mehta
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 3.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 4.Liu L, Gerson SL. Targeted modulation of MGMT: Clinical implications. Clin Cancer Res. 2006;12:328–331. doi: 10.1158/1078-0432.CCR-05-2543. [DOI] [PubMed] [Google Scholar]
- 5.Tolcher AW, Gerson SL, Denis L, et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br J Cancer. 2003;88:1004–1011. doi: 10.1038/sj.bjc.6600827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wick W, Platten M, Weller M. New (alternative) temozolomide regimens for the treatment of glioma. Neuro Oncol. 2009;11:69–79. doi: 10.1215/15228517-2008-078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wick A, Felsberg J, Steinbach JP, et al. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J Clin Oncol. 2007;25:3357–3361. doi: 10.1200/JCO.2007.10.7722. [DOI] [PubMed] [Google Scholar]
- 8.Tosoni A, Franceschi E, Ermani M, et al. Temozolomide three weeks on and one week off as first line therapy for patients with recurrent or progressive low grade gliomas. J Neurooncol. 2008;89:179–185. doi: 10.1007/s11060-008-9600-y. [DOI] [PubMed] [Google Scholar]
- 9.Zelen M. The randomization and stratification of patients to clinical trials. J Chronic Dis. 1974;27:365–375. doi: 10.1016/0021-9681(74)90015-0. [DOI] [PubMed] [Google Scholar]
- 10.Curran WJ, Jr, Scott CB, Horton J, et al. Recursive partitioning analysis of prognostic factors in three Radiation Therapy Oncology Group malignant glioma trials. J Natl Cancer Inst. 1993;85:704–710. doi: 10.1093/jnci/85.9.704. [DOI] [PubMed] [Google Scholar]
- 11.Vlassenbroeck I, Califice S, Diserens AC, et al. Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J Mol Diagn. 2008;10:332–337. doi: 10.2353/jmoldx.2008.070169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armstrong TS, Mendoza T, Gning I, et al. Validation of the M.D. Anderson Symptom Inventory Brain Tumor Module (MDASI-BT) J Neurooncol. 2006;80:27–35. doi: 10.1007/s11060-006-9135-z. [DOI] [PubMed] [Google Scholar]
- 13.Osoba D, Aaronson NK, Muller M, et al. The development and psychometric validation of a brain cancer quality-of-life questionnaire for use in combination with general cancer-specific questionnaires. Qual Life Res. 1996;5:139–150. doi: 10.1007/BF00435979. [DOI] [PubMed] [Google Scholar]
- 14.Wefel JS, Cloughesy T, Zazzali JL, et al. Neurocognitive function in patients with recurrent glioblastoma treated with bevacizumab. Neuro Oncol. 2011;13:660–668. doi: 10.1093/neuonc/nor024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macdonald DR, Cascino TL, Schold SC, Jr, et al. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 16.Sanghera P, Perry J, Sahgal A, et al. Pseudoprogression following chemoradiotherapy for glioblastoma multiforme. Can J Neurol Sci. 2010;37:36–42. doi: 10.1017/s0317167100009628. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 18.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966;50:163–170. [PubMed] [Google Scholar]
- 19.Cox DR. Regression models and life tables. J R Stat Soc B. 1972;34:187–220. [Google Scholar]
- 20.Citron ML, Berry DA, Cirrincione C, et al. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: First report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol. 2003;21:1431–1439. doi: 10.1200/JCO.2003.09.081. [DOI] [PubMed] [Google Scholar]
- 21.Brada M, Stenning S, Gabe R, et al. Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. J Clin Oncol. 2010;28:4601–4608. doi: 10.1200/JCO.2009.27.1932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.