

Abstract
Raf Kinase inhibitory protein (RKIP) is a well-established metastasis suppressor that is frequently downregulated in aggressive cancers. The impact of RKIP and its phosphorylated form on disease-free survival (DFS) and other clinicopathological parameters in breast cancer is yet to be discovered. To this end, we examined RKIP expression in 3 independent breast cancer cohorts. At the Protein level, loss or reduced total RKIP expression was associated with large-sized tumors characterized by high proliferative index, high-grade and diminished estrogen (ER) and progesterone receptor expression. Loss or diminution of RKIP expression was significantly associated with shorter DFS in all cohorts. Moreover, the complete loss of p-RKIP was an independent prognostic factor using multivariate analysis in operable invasive ductal breast cancer. We show for the first time that ER, partly, drives RKIP expression through MTA3-Snail axis. Consistent with this finding, we found that, at the mRNA level, RKIP expression varied significantly across the different molecular subtypes of breast cancer with the Luminal (ER+) subtype expressing high levels of RKIP and the more aggressive Claudin-low (ER-) subtype, which depicted the highest epithelial to mesenchymal transition (EMT) registered the lowest RKIP expression levels. In conclusion, loss of expression/diminution of RKIP or its phosphorylated form is associated with poor diseases-free survival in breast cancer. Determining the expression of RKIP and p-RKIP adds significant prognostic value to the management and subtyping of this disease.
Keywords: RKIP, PEBP1, ERK, estrogen receptor, aggressive cancer, breast cancer, Luminal, claudin-low, ERBB2, basal, prognosis, disease-free survival
Introduction
Breast cancer, and in particular invasive ductal carcinoma, remains the most common cancer inflicting women in the Western world. Treatment of advanced breast cancer remains challenging, and its heterogeneity, both at the cellular and molecular levels, continues to be at the realm of this challenge. Genomic and expression profiling have established five major breast cancer intrinsic subtypes and a normal breast cancer-like group that appear to have significant impact on prognosis and treatment [1-4].
It is becoming increasingly evident that the activation of the Raf/mitogen-activated and extracellular signal-regulated kinase (MEK)/extracellular signal-regulated kinase (ERK) cascade (RAS-Raf-MEK-ERK) plays key role in breast cancer progression and resistance to hormonal and chemotherapeutic drugs [5], and that the deregulation of the Raf-MEK-ERK protein expression, rather than kinase mutations, is more critical in breast cancer progression, especially in triple negative breast tumors [6-8] Given that RKIP is the only known physiological inhibitor of the Raf-MEK-ERK pathway [9], detailed RKIP expression profiling in breast cancer may be of significant clinical value.
RKIP is a master modulator of pivotal intracellular signaling pathways. RKIP binds either to Raf-1 or MEK and exerts its inhibitory effect by interfering with Raf-1 phosphorylation at residues S338 and T340/341 [10,11]. This process subdues downstream ERK kinase signaling [12].
The PKC isoenzymes -α, -βI, -βII, -γ, and atypical PKCξ—mediate the phosphorylation of RKIP at residue S153 [13], shifting RKIP from Raf-1 to G protein-coupled receptor kinase 2 (GRK2), therefore blocking its inhibitory activity on the G-protein coupled receptors (GPCR) signaling cascade resulting in its activation [14].
Loss or silencing of metastasis suppressor genes play key roles in modulating key processes involved in cellular growth, EMT, invasion and metastasis. RKIP silencing has been shown to destabilize GSK3β and to activate its downstream targets, which culminates in the stabilization of cyclin D1; cyclin D1 induces cell cycle progression, and the expression of β-catenin, SNAIL and SLUG, which are molecules known to promote EMT, cellular invasion and metastasis [15,16]. Current data suggest that RKIP is a cardinal metastasis suppressor that is subdued in almost all cancer types especially in more aggressive and metastatic ones [17]. However, regarding breast cancer, there is obvious lack of information in the current literature pertaining to the effects of RKIP expression and its phosphorylated form on patients’ survival and their association with clinicopathological characteristics. In this paper, we report a new role for RKIP in breast cancer prognosis, as well as a new model depicting how estrogen receptor induces the expression of RKIP through MTA3. The present data suggest that reduced expression of total RKIP and its phosphorylated form have significant influence on shorter Disease-free survival in breast cancer patients. More interestingly, was that RKIP expression level varied across the different subtypes of breast cancer with Luminal A breast cancer subtype (a subtype that largely express estrogen receptor), registered the highest RKIP expression, while Claudin-low (that show high EMT and estrogen receptor negativity), had the lowest RKIP expression. Therefore, our paper illuminates, for the first time, important and novel niches pertaining to the role RKIP and its phosphorylated form in breast cancer pathophysiology.
Materials and methods
Patients used for total RKIP expression
Patients presenting between 1995 and 1998 with primary invasive breast cancer at the Royal and Western Infirmaries and Stobhill Hospital, Glasgow were studied in the first cohort (n=421). Each tumor was printed in triplicate on the tissue microarray. Clinicopathological data were determined in most cases (Table 1). The Research Ethics Committees of North Glasgow University Hospitals and Kuwait University approved the use of human tissue in this study.
Table 1.
Clinicopathological characteristics of the breast cancer cohort. N=421
| Clinicopathological characteristics | Number of patients (%) | Number of missing patients |
|---|---|---|
| Age (≤50/>50 years) | 115 (27)/306 (73) | 0 |
| Size (≤20/21-50/>50 mm) | 240 (57)/168 (40)/11 (3) | 2 |
| Grade (I/II/III) | 84 (20)/180 (43)/155 (37) | 2 |
| Nodal status (negative/positive) | 233 (56)/182 (44) | 6 |
| Oestrogen receptor status (ER-/ER+) | 128 (31)/289 (69) | 4 |
| Progesterone receptor status (PR-/PR+) | 208 (50)/206 (50) | 7 |
| HER2 (HER2-/HER2+) | 348 (84)/68 (16) | 5 |
| Ki67 (≤15/>15) | 310 (75)/102 (25) | 9 |
| TUNEL (≤0.33/>0.33) | 229 (57)/174 (43) | 18 |
| Necrosis (absent/present) | 192 (47)/217 (53) | 12 |
| Chemotherapy (no/yes) | 247 (59)/172 (41) | 2 |
| Radiotherapy (no/yes) | 237 (57)/182 (43) | 2 |
| Endocrine therapy (no/yes) | 97 (23)/317 (77) | 7 |
Immunohistochemistry
ER and PR status was determined by immunohistochemistry in the CPA-accredited diagnostic pathology laboratory, Glasgow Southern General, with the inclusion of appropriate positive and negative controls. Dako ER-α antibody (clone 6F11 ⁄ 2, mouse anti-human, 1:50; Dako, Glostrup, Denmark), Leica PR antibody (clone R 636, mouse anti-human, 1:400). Immunohistochemistry for Epitomics’ monoclonal RKIP antibody (clone EPR2875Y, rabbit anti-human, 1:500; Epitomics, CA, USA) and Epitomics’ monoclonal p-RKIP antibody (clone, EP2845Y, rabbit anti-human, 1:250; Epitomics, CA, USA) were performed manually with DAKO Envision detection system in Glasgow Western Infirmary. The monoclonal anti-RKIP and anti-p-RKIP antibodies specificity was confirmed using immunohistochemistry and western blotting (Figure 1).
Figure 1.

Specificity of human RKIP and p-RKIP monoclonal antibodies. RKIP immunohistochemistry performed on formalin-fixed paraffin-embedded human brain tissue (A) and p-RKIP detected in human liver (B). Note the intense cytoplasmic and nuclear staining. Western blotting detection of 23kDa RKIP in 4 different cell lines (C) and p-RKIP (S153) in HeLa cells before – and after + treatment with calyculin A, the pp1 and pp2 phosphatases’ inhibitor (D). Courtesy of Epitomics.
Slide scanning and scoring
Stained slides were scanned at an objective magnification of 40 × using a Hamamatsu NanoZoomer scanner (Hamamatsu, Welwyn Garden City, UK). Visualization and automated assessment were carried out with the Slidepath Tissue IA system version 1.0 (Slidepath, Dublin, Ireland). Staining intensities for ER-α, PR and total RKIP for all cores were assessed visually using the weighted histoscore (H-score) quantitative method as previously described [18,19]. The H-score method takes into account both the area and intensity of staining to generate values between 0-300 using the following formula: Σ (1 × % cells staining weakly positive) + (2 × % cells staining moderately positive) + (3 × % cells staining strongly positive).
Seventy-six cores were independently scored by two observers blinded to patients’ data for RKIP and p-RKIP (L.B and J.E). L.B. then scored all slides for analysis.
Quartile assignment of total and p-RKIP H-scores
For statistical comparisons, p-RKIP cytoplasmic expression was categorized into 4 quartiles depending on the obtained H-score values; the first quartile, contained tumors, which H-score values were <5, the second quartile p-RKIP expression was determined between 5-30, the third between 31-54 and the fourth quartile >54. Nuclear expression data were similarly categorized. For total RKIP the values of the H-Scores quartiles are shown in Figure 2.
Figure 2.
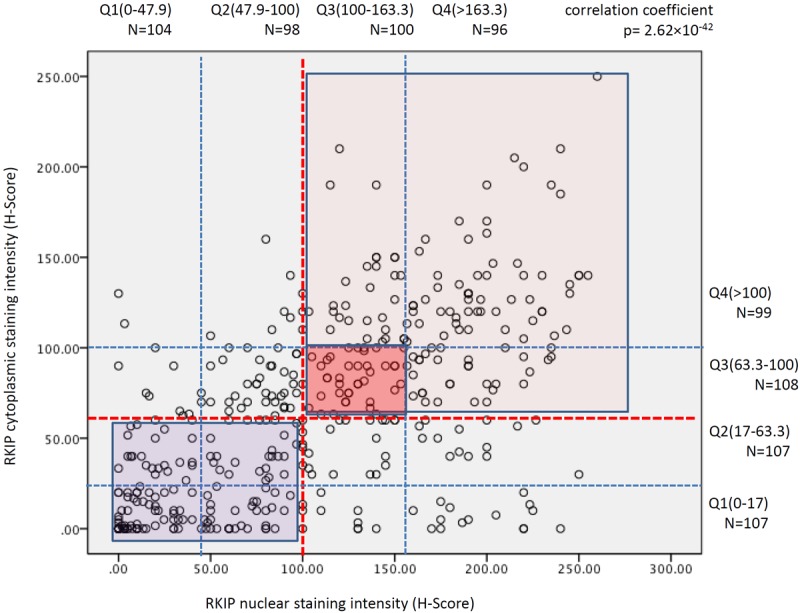
Correlation between nuclear and cytoplasmic RKIP expression in breast cancer. Red dashed lines indicate the median H-Score staining values for RKIP expression and the thin blue dashed lines demarcate the quartiles shown above for cytoplasmic and to the right for nuclear expression. Dark blue rectangle indicate samples with diminished or loss of RKIP expression in the cytoplasm and nuclei, dark pink square demarcates samples with intermediate expression and pink square shows those samples that expressed RKIP strongly in their nuclei and cytoplasm.
Tissue culture, in vitro transfection assays and western blotting
The preparation of rabbit anti-RKIP antibody was reported previously [9]. Mouse monoclonal antibodies specific for α-tubulin and actin antibody were purchased from Sigma. Rabbit snail and e-cadherin polyclonal antibodies were from Abcam and Cell Signaling Technology, respectively. Rabbit MTA3 and ERα polyclonal antibodies were generous gifts from Drs. Paul Wade and Manohar Ratnam, respectively.
T47D, MCF7 and MDA-MB231 cells were cultured in Dulbecco’s modified Eagle’s medium with 10% FBS. Cells were grown in a humidified tissue culture incubator at 37°C in 5% CO2.
3 × 105 cells were transfected with a total of 2 μg of plasmid DNA using LipofectAMINE Plus Reagent (Invitrogen). Forty to 48 h after transfection, cells were collected for immunoblot analyses.
Total cell extracts were prepared as described previously [12], and protein concentrations of lysates were determined using the Bradford assay kit (Bio-Rad). Proteins (10-50 μg) were separated by SDS-PAGE and electrophoretically transferred from the gel to immobilin membranes (millipores). Proteins recognized by the antibodies were detected by enhanced chemiluminescence reagents (Pierce) with the BioRad ChemiDoc EQ system.
Preparation of knockdown cells
To silence expression of estrogen receptor-α (ERα) in MCF7 cells, we infected cells with retroviruses encoding a ERα-specific shRNA or a scramble siRNA as control in the retroviral shRNA expression vectors kindly provided by Dr. Brian Rowen at the Tulane University. The retroviruses were prepared as previously described [20].
Data preprocessing of Affymetrix microarray gene expression
Microarray gene expression data of human breast cancer on Affymetrix U133A or U133Plus2 platforms were downloaded from Array Express and Gene Expression Omnibus (GEO). For this study, we included all of publicly available dataset at the time the analysis was initiated. This panel of data comprises 3,992 human breast tumor samples from 26 cohorts, including E-TABM-158 (n=130), GSE11121 (n=200), GSE12276 (n=204), GSE1456 (n=159), GSE1561 (n=49), GSE19615 (n=115), GSE20181 (n=176), GSE2034 (n=286), GSE21653 (n=266), GSE23177 (n=116), GSE23593 (n=50), GSE23988 (n=61), GSE25066 (n=508), GSE26639 (n=226), GSE31519 (n=67), GSE3494 (n=251), GSE3744 (n=47), GSE4922 (n=40), GSE5327 (n=58), GSE5460 (n=127), GSE5764 (n=10), GSE6532 (n=414), GSE6596 (n=24), GSE7390 (n=198), GSE9195 (n=77), and HESS cohort (n=133) [21]. Robust Multichip Average (RMA) normalization was performed on each dataset. The normalized data was combined and subsequently standardized using ComBat [22] to remove batch effect.
Similarly for breast cancer cell line panel, it was established using two large dataset on Affymetrix U133A or U133plus2 platform: GSE15026 (n=30 samples corresponding to 19 cell lines) and E-TABM-157 (n=51 samples corresponding to 51 cell lines). The data was subjected to the same pre-processing as the clinical samples. The RMA-normalized and ComBat-standardized breast cancer cell line panel consists of 81 samples corresponding to 70 cell lines (Table 2).
Table 2.
All subtyped breast cancer cell lines
| Index | Cell Line | Cohort | Subtype |
|---|---|---|---|
| 1 | 600MPE | E-TABM-157 | Luminal |
| 2 | AU565 | E-TABM-157 | Luminal |
| 3 | BT20 | E-TABM-157 | Basal-A |
| 4 | BT474 | E-TABM-157 | Luminal |
| 5 | BT483 | E-TABM-157 | Luminal |
| 6 | BT549 | E-TABM-157 | Basal-B/Claudin-Low |
| 7 | CAMA1 | E-TABM-157 | Luminal |
| 8 | HBL100 | E-TABM-157 | Basal-B/Claudin-Low |
| 9 | HCC1007 | E-TABM-157 | Luminal |
| 10 | HCC1143 | E-TABM-157 | Basal-A |
| 11 | HCC1187 | E-TABM-157 | Basal-A |
| 12 | HCC1428 | E-TABM-157 | Luminal |
| 13 | HCC1500 | E-TABM-157 | Basal-B/Claudin-Low |
| 14 | HCC1569 | E-TABM-157 | Basal-A |
| 15 | HCC1937 | E-TABM-157 | Basal-A |
| 16 | HCC1954 | E-TABM-157 | Basal-A |
| 17 | HCC202 | E-TABM-157 | Luminal |
| 18 | HCC2157 | E-TABM-157 | Basal-A |
| 19 | HCC2185 | E-TABM-157 | Luminal |
| 20 | HCC3153 | E-TABM-157 | Basal-A |
| 21 | HCC38 | E-TABM-157 | Basal-B/Claudin-Low |
| 22 | HCC70 | E-TABM-157 | Basal-A |
| 23 | HS578T | E-TABM-157 | Basal-B/Claudin-Low |
| 24 | LY2 | E-TABM-157 | Luminal |
| 25 | MCF10A | E-TABM-157 | Basal-B/Claudin-Low |
| 26 | MCF12A | E-TABM-157 | Basal-B/Claudin-Low |
| 27 | MCF7 | E-TABM-157 | Luminal |
| 28 | MDAMB134VI | E-TABM-157 | Luminal |
| 29 | MDAMB157 | E-TABM-157 | Basal-B/Claudin-Low |
| 30 | MDAMB175VII | E-TABM-157 | Luminal |
| 31 | MDAMB231 | E-TABM-157 | Basal-B/Claudin-Low |
| 32 | MDAMB361 | E-TABM-157 | Luminal |
| 33 | MDAMB415 | E-TABM-157 | Basal-B/Claudin-Low |
| 34 | MDAMB435 | E-TABM-157 | Basal-B/Claudin-Low |
| 35 | MDAMB436 | E-TABM-157 | Luminal |
| 36 | MDAMB453 | E-TABM-157 | Basal-A |
| 37 | MDAMB468 | E-TABM-157 | Basal-A |
| 38 | SKBR3 | E-TABM-157 | Luminal |
| 39 | SUM1315MO2 | E-TABM-157 | Basal-B/Claudin-Low |
| 40 | SUM149PT | E-TABM-157 | Basal-B/Claudin-Low |
| 41 | SUM159PT | E-TABM-157 | Basal-B/Claudin-Low |
| 42 | SUM185PE | E-TABM-157 | Luminal |
| 43 | SUM190PT | E-TABM-157 | Basal-A |
| 44 | SUM225 | E-TABM-157 | Basal-A |
| 45 | SUM44PE | E-TABM-157 | Luminal |
| 46 | SUM52PE | E-TABM-157 | Luminal |
| 47 | T47D | E-TABM-157 | Luminal |
| 48 | UACC812 | E-TABM-157 | Luminal |
| 49 | ZR751 | E-TABM-157 | Luminal |
| 50 | ZR7530 | E-TABM-157 | Luminal |
| 51 | ZR75B | E-TABM-157 | Luminal |
| 52 | BT20(1) | GSE15026 | Basal-A |
| 53 | BT20(2) | GSE15026 | Basal-A |
| 54 | BT474(1) | GSE15026 | Luminal |
| 55 | BT474(2) | GSE15026 | Luminal |
| 56 | BT483(1) | GSE15026 | Luminal |
| 57 | BT483(2) | GSE15026 | Luminal |
| 58 | BT549(1) | GSE15026 | Basal-B/Claudin-Low |
| 59 | BT549(2) | GSE15026 | Basal-B/Claudin-Low |
| 60 | CAMA1(2) | GSE15026 | Luminal |
| 61 | HCC1143(2) | GSE15026 | Basal-A |
| 62 | HCC1428(2) | GSE15026 | Luminal |
| 63 | HCC1806(2) | GSE15026 | --- |
| 64 | HCC38(2) | GSE15026 | Basal-B/Claudin-Low |
| 65 | HS578T(2) | GSE15026 | Basal-B/Claudin-Low |
| 66 | MCF7(1) | GSE15026 | Luminal |
| 67 | MCF7(2) | GSE15026 | Luminal |
| 68 | MDAMB157(2) | GSE15026 | Basal-B/Claudin-Low |
| 69 | MDAMB231(1) | GSE15026 | Basal-B/Claudin-Low |
| 70 | MDAMB231(2) | GSE15026 | Basal-B/Claudin-Low |
| 71 | MDAMB361(1) | GSE15026 | Luminal |
| 72 | MDAMB361(2) | GSE15026 | Luminal |
| 73 | MDAMB435(1) | GSE15026 | Basal-B/Claudin-Low |
| 74 | MDAMB435S(2) | GSE15026 | Basal-B/Claudin-Low |
| 75 | MDAMB453(2) | GSE15026 | Basal-A |
| 76 | SKBR3(1) | GSE15026 | Luminal |
| 77 | SKBR3(2) | GSE15026 | Luminal |
| 78 | T47D(1) | GSE15026 | Luminal |
| 79 | T47D(2) | GSE15026 | Luminal |
| 80 | ZR75(1) | GSE15026 | --- |
| 81 | ZR751(2) | GSE15026 | Luminal |
Identification of breast cancer subtypes
To obtain the molecular subtype of breast cancer for the clinical samples, we employed Single sample Gene Set Enrichment Analysis (ssGSEA) [23] to compute for each sample, enrichment scores of breast cancer subtype signature (Basal, Claudin-low, Luminal-A, Luminal-B, ERBB2, or Normal-like) [24]. Each clinical sample was then assigned as the subtype it has the highest enrichment. On the other hand, subtype of breast cancer cell lines was taken from Neve et al. [25] based on the cell line name. Basal-B subtype in the original paper [25] was labeled as Claudin-low in this study.
Epithelial-mesenchymal transition signature
An Epithelial-Mesenchymal Transition (EMT) signature was developed by comparing expression profiles of CDH1 with CDH2 expressing ovarian carcinomas cell lines using Binary Regression method [26]. The BinReg ovarian cancer EMT signature was then applied to predict the EMT status of breast cancer tumors or cell lines. Subsequently, the top 5% (~100 breast cancer tumor samples or ~10 breast cancer cell lines) with the highest probabilities for epithelial or mesenchymal phenotype were used to obtain the epithelial or mesenchymal specific gene list for the breast cancer tumor or cell line (EMT signature) using Significance Analysis of Microarray (SAM) q-value=0 and Receiver Operating Curve (ROC) value of 0.85.
To obtain EMT score of a breast cancer clinical sample or cell line, the enrichment scores of the breast cancer tumor-specific or cell line-specific epithelial and mesenchymal gene list were computed using single sample GSEA [23]. EMT score is defined as the normalized subtraction of the mesenchymal from epithelial enrichment score. A higher or lower EMT score indicate that a breast cancer tumor/cell line exhibits a more mesenchymal or epithelial phenotype respectively.
Statistical analysis
For immunohistochemistry and clinicopathological correlations were assessed with using the chi-squared or Fisher’s exact tests for trend as appropriate. DFS indicates the time period between the operation date and disease reappearance in the form of local or distant metastatic recurrence. Differences in DFS were tested using the Log-rank test in univariate analysis and displayed using Kaplan-Meier curves. Cox proportional-hazards model was used to calculate the hazard ratio. Analysis was performed with SPSS version 17 (SPSS, Chicago, IL, USA). For Affymetrix expression data, differences in the means/medians values were evaluated using Mann-Whitney test and Spearman correlations were computed using Matlab®. This study complied with the Reporting recommendations for tumor marker prognostic studies (REMARK) criteria [27]. A level of P≤0.05 was considered to be significant.
Results
Clinicopathological correlations of total RKIP expression in invasive ductal breast cancer
421 primary invasive ductal breast carcinomas in triplicate tissue microarrays were utilized in the assessment of total and 153-phosphorylated form of RKIP (p-RKIP) expression and their correlation with known clinicopathological parameters. The clinicopathological characteristics of the cohort are shown in Table 1.
Total RKIP was expressed in the cytoplasm and/or in nuclei of breast cancer cells. Consequently, the cytoplasmic or nuclear staining was scored independently of each other and was categorized into 4 quartiles (Figures 2 and 3). There was a statistically significant and direct correlation between nuclear and cytoplasmic expression of total RKIP (Figure 2).
Figure 3.
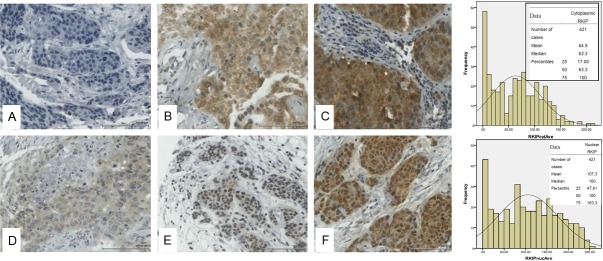
Representative immunohistochemical staining for RKIP in breast cancer tissue microarray. Top panel shows cytoplasmic RKIP expression while lower panel shows nuclear RKIP expression. The bar charts depict the frequencies against the H-Scores obtained in 421 cases of breast cancer samples with the means, medians and percentiles shown in the inset. A and D show negative, B and E intermediate and C and F strong RKIP expression in cytoplasm and nuclei respectively.
Our data show that loss or reduced total nuclear RKIP expression (Table 3) or combined nuclear and cytoplasmic staining (Table 4) was significantly associated with large-sized tumors having high Ki-67 index, high tumor grade, and the presence of extensive necrosis. A reciprocal trend between loss or diminished total RKIP expression and lymph nodes metastasis was evident in this cohort. However, the association was not statistically significant (Table 3). Using univariate analysis, patients whose tumors showed loss or diminished total nuclear RKIP expression (quartile 1; Q1) had significantly shorter DFS compared to high expressors (quartile 4; Q4) (Table 3 and Figure 4A). Similar clinicopathological correlative scores were obtained when the expression patterns were categorized into Q1 (negative-low), Q2-3 (intermediate) or Q4 (high) total RKIP expression in both the nuclei and the cytoplasm (Table 4 and Figure 4B). In multivariate analysis, the presence of lymph node metastasis, necrosis, high Ki-67 proliferative index and the absence of progesterone receptors, but not total RKIP expression, were independent prognostic factors (Table 5).
Table 3.
Correlations between nuclear RKIP expression and the patients’ clinicopathological characteristics
| Clinico-pathological characteristics | Patients (n=421) | RKIP expression H-Score quartile | p value | |||
|---|---|---|---|---|---|---|
|
| ||||||
| 1 (n=107) | 2 (n=104) | 3 (n=106) | 4 (n=104) | |||
| Age (≤50/>50 years) | 115/306 | 25/82 | 31/73 | 33/73 | 26/78 | 0.736 |
| Size (≤20/21-50/>50 mm) | 240/168/11 m2 | 53/51/3 | 52/48/2 m2 | 59/44/3 | 76/25/3 | 0.002 |
| Grade (I/II/III) | 84/180/155 m2 | 11/34/61 m1 | 15/50/39 | 21/54/31 | 37/42/24 m1 | <0.001 (7.98 × 10-5) |
| Nodal status (negative/positive) | 233/182 m6 | 59/47 m1 | 54/49 m1 | 53/52 m1 | 67/34 m3 | 0.101 |
| Oestrogen receptor status (ER-/ER+) | 128/289 m4 | 53/52 m2 | 34/69 m1 | 23/83 | 18/85 m1 | <0.001 (4.68 × 10-5) |
| Progesterone receptor status (PR-/PR+) | 208/206 m7 | 73/32 m2 | 49/54 m1 | 45/59 m2 | 41/61 m2 | <0.001 (2.51 × 10-5) |
| HER2 (HER2-/HER2+) | 348/68 m5 | 79/26 m2 | 82/21 m1 | 93/12 m1 | 94/9 m1 | <0.001 (4.40 × 10-5) |
| Ki67 (≤15/>15) | 310/102 m9 | 68/37 m2 | 74/30 | 84/20 m2 | 84/15 m5 | <0.001 (2.50 × 10-4) |
| TUNEL (≤0.33/>0.33) | 229/174 m18 | 63/40 m4 | 58/45 m1 | 58/44 m4 | 50/45 m9 | 0.262 |
| Necrosis (absent/positive) | 192/217 m12 | 41/65 m1 | 39/60 m5 | 46/57 m3 | 66/35 m3 | <0.001 (2.58 × 10-4) |
| Chemotherapy (no/yes) | 247/172 m2 | 53/53 m1 | 48/55 m1 | 65/41 | 81/23 | <0.001 (5.25 × 10-6) |
| Radiotherapy (no/yes) | 237/182 m2 | 69/37 m1 | 50/53 m1 | 63/43 | 55/49 | 0.080 |
| Endocrine therapy (no/yes) | 97/317 m7 | 37/69 m1 | 21/81 m2 | 21/82 m3 | 18/85 m1 | 0.013 |
| DFS (months) | n=104 m3 | n=98 m6 | n=100 m6 | n=96 m8 | Overall 0.074 | |
| Mean (95% CI) | 82.4 (76.3-88.5) | 89.7 (83.4-96.0) | 88.7 (83.0-94.4) | 96.0 (90.4-101.6) | Quartile 1 vs 4 (p=0.01) | |
m (number of patients) denotes missing cases.
Table 4.
Clinicopathological characteristics correlated with categorical low, intermediate and high RKIP expression both in the nucleus and cytoplasm
| Clinico-pathological characteristics | Patients (n=310) | RKIP expression | p value | ||
|---|---|---|---|---|---|
|
| |||||
| Low RKIP nuclear & cytoplasmic expression (n=158) | Intermediate RKIP nuclear & cytoplasmic expression (n=41) | High RKIP nuclear & cytoplasmic expression (n=111) | |||
| Age (≤50/>50 years) | 77/233 | 39/119 | 11/30 | 27/84 | 0.994 |
| Size (≤20/21-50/>50 mm) | 177/122/10 m1 | 81/71/5 m1 | 21/18/2 | 75/33/3 | 0.031 |
| Grade (I/II/III) | 69/130/109 m2 | 24/63/70 m1 | 7/19/15 | 38/48/24 m1 | <0.001 (2.9 × 10-4) |
| Nodal status (negative/positive) | 172/134 m4 | 81/75 m2 | 24/17 | 67/42 m2 | 0.290 |
| Oestrogen receptor status (ER-/ER+) | 89/217 m4 | 61/94 m3 | 6/35 | 22/88 m1 | <0.001 (1.7 × 10-4) |
| Progesterone receptor status (PR-/PR+) | 151/154 m5 | 92/63 m3 | 14/27 | 45/64 m2 | 0.001 |
| HER2 (HER2-/HER2+) | 257/49 m4 | 122/33 m3 | 35/6 | 100/10 m1 | 0.008 |
| Ki67 (≤15/>15) | 224/79 m7 | 104/52 m2 | 28/12 m1 | 92/15 m4 | 0.001 |
| TUNEL (≤0.33/>0.33) | 167/132 m11 | 93/62 m3 | 24/17 | 50/53 m8 | 0.089 |
| Necrosis (absent/present) | 155/147 m8 | 70/83 m5 | 20/21 | 65/43 m3 | 0.067 |
| Chemotherapy (no/yes) | 194/115 m1 | 85/72 m1 | 28/13 | 81/30 | 0.005 |
| Radiotherapy (no/yes) | 176/133 m1 | 94/63 m1 | 27/14 | 55/56 | 0.113 |
| Endocrine therapy (no/yes) | 74/233 m3 | 44/113 m1 | 5/35 m1 | 25/85 m1 | 0.002 |
| DFS (months) | n=152 m6 | n=39 m2 | n=101 m10 | Group 1 vs. Group 2 p=0.023* | |
| Mean (95% CI) | 83.7 (78.6-88.8) | 81.9 (76.2-87.6) | 96.6 (91.6-101.6) | ||
m (number of patients) denotes missing cases.
p value for disease free survival pooled over strata (for the whole graph) is 0.072.
Figure 4.
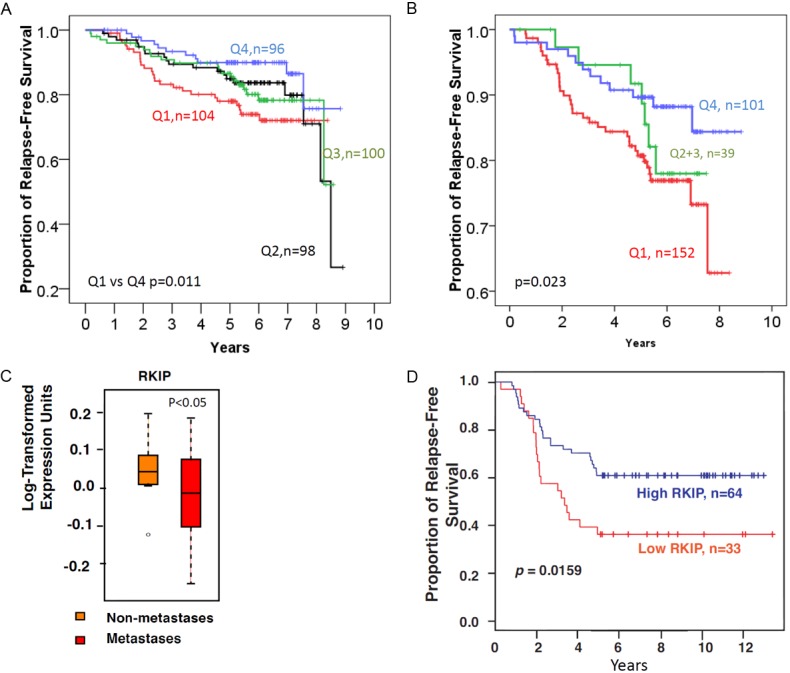
Loss or diminished total RKIP expression is associated with reduced disease-free survival in breast cancer. A: Total RKIP expression in nuclei divided into 4 quartile using the weighted H-Score. B: Total RKIP expression scores combined for both the nuclei and cytoplasm. C: Box-plot showing that Loss or diminished RKIP expression at the mRNA level is associated with metastatic breast cancer. D: Low mRNA (≤median) RKIP expression is associated with reduced disease-free survival in breast cancer compared to high RKIP expression (>median). The Log-Rank test was used to compare the statistical difference in disease-free survival.
Table 5.
Multivariate Cox-regression analysis of RKIP expression and established prognostic markers in operable invasive ductal breast cancer
| Clinico-pathological characteristics | HR (95% C.I) | p value |
|---|---|---|
| Nodal metastasis | ||
| Negative | 1 | 0.002 |
| positive | 2.25 (1.3-3.8) | |
| Progesterone receptor status | ||
| PR- | 1 | 0.001 |
| PR+ | 0.39 (0.22-0.7) | |
| Necrosis | ||
| Negative/mild | 1 | 0.01 |
| Extensive | 2.27 (1.24-4.1) | |
| Ki-67 | ||
| ≤15 | 1 | 0.012 |
| >15 | 1.92 (1.15-3.2) | |
| HER2 | ||
| HER2- | 1 | 0.044 |
| HER2+ | 1.8 (1.04-3.1) |
Further experiments were conducted to examine the impact of RKIP loss or diminution on patients’ DFS at the mRNA level. We analyzed a publicly available breast cancer gene expression microarray data set from 115 women published previously [28]. Figure 4C shows that RKIP mRNA levels were significantly lower in metastatic compared to non-metastatic primary breast cancers. In this public set, DFS was available for 97 patients. As shown in Figure 4D, patients with low RKIP expressing breast cancer had significantly shorter DFS compared to those whose tumors expressed high RKIP mRNA levels. These data, independently, confirm the relationship between diminished RKIP expression and reduced DFS in breast cancer.
Absence of phosphorylated raf kinase inhibitory protein (pRKIP) is an independent predictor of bad prognosis in primary operable breast cancer
p-RKIP staining was informative in 373 cases. Of these 81.0% (302/373) expressed p-RKIP in the cytoplasm and 62.5% (233/373) expressed p-RKIP in the nucleus (Figure 5). There was a significant and direct correlation between p-RKIP cytoplasmic and nuclear staining (Figure 6A). For example, 74/373 (21%) were negative (quartile 1) for both cytoplasmic and nuclear expression (termed group 1), while 59/373 (15.8%) expressed p-RKIP strongly (quartile 4) in both the nuclei and cytoplasm (termed group 2). We focused our analysis on comparing these two polarized groups. p-RKIP expression levels were not significantly associated with any clinicopathological parameters, including the presence of lymph node metastasis (Table 6). However, in univariate analysis, the estimated DFS was 95.4 months (95% C.I of 89-101.7 months) for patients overexpressing (group 2 or Q4) p-RKIP, while DFS was 77.6 months (95% C.I of 70-80 months) for group 1 (or Q1) patients (Figure 6B). Multivariate analysis using a Cox-regression model, predicted a significantly reduced hazard ratio (HR) for disease recurrence in tumors overexpressing p-RKIP (HR= 0.625; 95% C.I 0.435-0.899), independent of tumor size, grade, presence of lymph node metastasis, ER, PR and Her-2 amplification status (Table 7).
Figure 5.
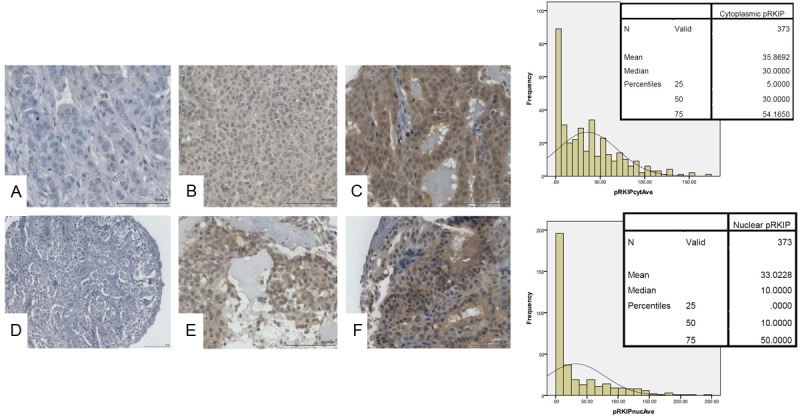
Representative immunohistochemical staining for p-RKIP in breast cancer tissue microarray. Top panel shows cytoplasmic p-RKIP expression while lower panel shows nuclear p-RKIP expression. The bar charts depict the frequencies against the H-Scores obtained in 373 cases of breast cancer samples with the means, medians and percentiles shown in the inset. A and D show negative, B and E intermediate and C and F strong p-RKIP expression in cytoplasm and nuclei respectively.
Figure 6.
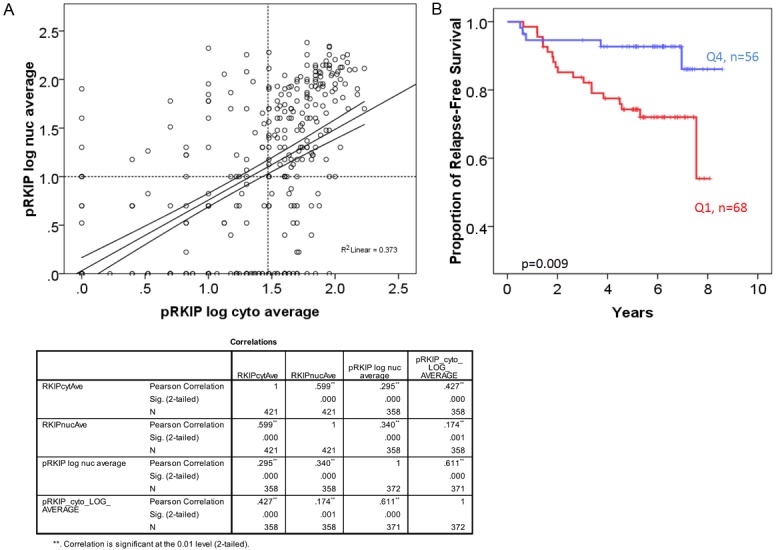
p-RKIP expression in breast cancer tissues. A: Pearson’s correlation between nuclear and cytoplasmic p-RKIP expression. B: Complete loss (Q1-negative) of 153 p-RKIP expression in both the nuclei and the cytoplasm is associated with reduced disease-free survival in breast cancer compared to very high expression (Q4). The Log-Rank test was used to compare the statistical difference in disease-free survival.
Table 6.
Clinicopathological characteristics correlated with categorical low and high p-RKIP expression both in the nucleus and cytoplasm
| Clinico-pathological characteristics | Patients (n=133) | pRKIP expression | p value | |
|---|---|---|---|---|
|
| ||||
| pRKIP low in nucleus and cytoplasm (n=74) | pRKIP high in nucleusand cytoplasm (n=59) | |||
| Age (≤50/>50 years) | 34/99 | 19/55 | 15/44 | 0.974 |
| Size (≤20/21-50/>50 mm) | 78/49/6 | 47/25/2 | 31/24/4 | 0.140 |
| Grade (I/II/III) | 27/58/47 m1 | 11/33/30 | 16/25/17 m1 | 0.154 |
| Nodal status (negative/positive) | 68/63 m2 | 36/37 m1 | 32/26 m1 | 0.505 |
| Oestrogen receptor status (ER-/ER+) | 39/92 m2 | 24/50 | 15/42 m2 | 0.450 |
| Progesterone receptor status (PR-/PR+) | 71/60 m2 | 41/33 | 30/27 m2 | 0.753 |
| HER2 (HER2-/HER2+) | 115/17 m1 | 61/13 | 54/4 m1 | 0.070 |
| Ki67 (≤15/>15) | 96/33 m4 | 51/21 m2 | 45/12 m2 | 0.296 |
| TUNEL (≤0.33/>0.33) | 73/52 m8 | 42/28 m4 | 31/24 m4 | 0.683 |
| Necrosis (absent/present) | 69/62 m2 | 37/37 | 32/25 m2 | 0.485 |
| Chemotherapy (no/yes) | 78/55 | 41/33 | 37/22 | 0.395 |
| Radiotherapy (no/yes) | 74/59 | 42/32 | 32/27 | 0.771 |
| Endocrine therapy (no/yes) | 29/103 m1 | 16/57 m1 | 13/46 | 0.987 |
| DFS (months) | n=68 m6 | n=56 m3 | 0.009 | |
| Mean (95% CI) | 77.6 (70.1-85.1) | 95.4 (89.0-101.8) | ||
m (number of patients)=missing.
Table 7.
Multivariate Cox-regression analysis of p-RKIP expression and established prognostic markers in operable invasive ductal breast cancer
| Clinico-pathological characteristics | HR (95% C.I) | p value |
|---|---|---|
| p-RKIP expression | ||
| Quartile 1 (negative) | 1 | 0.01 |
| Quartile 4 (high expression) | 0.625 (0.4-0.89) | |
| Size (mm) | ||
| ≤50 | 1 | 0.003 |
| >50 | 3.1 (1.46-6.5) | |
| Nodal metastasis | ||
| Negative | 1 | 0.006 |
| positive | 4.23 (1.5-11.7) | |
| Oestrogen receptor status | ||
| ER- | 1 | 0.001 |
| ER+ | 0.23 (0.1-0.53) |
Estrogen receptor and RKIP are connected
Table 3 depicts a significant linear relationship between the percentage of breast cancer cases expressing estrogen receptor and their RKIP expression levels. Therefore, in relation to quartiles 1 to 4 of total nuclear RKIP expression, 48.5%, 66%, 78.3% and 82% of tumors expressed estrogen receptor respectively (p=4.68 × 10-5). A similar trend was also observed with progesterone receptor (Table 3). Furthermore, this association was confirmed at the mRNA level using Van’t Veer et al. data (Figure 7). Intriguingly, total RKIP dosage in breast cancer had the opposite linear trend with respect to Her-2/neu gene amplification (Tables 3 and 4). Our data suggest that ER may be involved in the transcriptional regulation of RKIP. We next tested this hypothesis in vitro.
Figure 7.
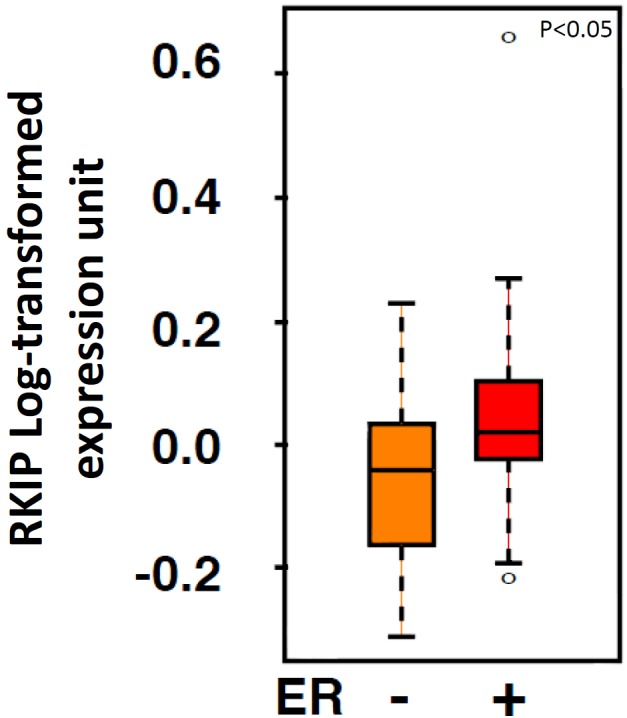
Box plot analysis depicting RKIP expression levels in relation to ER status in breast cancer samples obtained by reanalyzing published microarray data.
RKIP is a downstream target of the ER-MTA3-Snail transcription regulatory pathway
To investigate the molecular mechanism(s) connecting ER to RKIP expression, we determined the basal levels of ER, RKIP and Snail in 3 well-characterized breast cancer cell lines. MDA-MB231, which is ER negative, expressed high level of Snail, and consequently low RKIP and e-cadherin (Figure 8A). Conversely, ER expressing MCF7 and T47D cell lines expressed low levels of Snail protein resulting in higher RKIP and e-cadherin expression compared to the MDA-MB231 cell line. Previous work has established that MTA3, a component of the Mi-2/NuRD complex and an avid repressor of Snail is transcriptionally induced by ER [29]. Our data show that MTA3 protein level was lower in ER negative MB231 cell line. Further experiments were conducted to examine the impact of modulating ER expression on MTA-Snail-RKIP protein levels in these cells. Silencing of ER in MCF7 cells induced the expression of Snail and the concomitant reduction of RKIP and e-cadherin proteins (Figure 8B). Conversely, overexpressing ER or MTA3 in MDA-MB231 cell line reduced Snail expression with the subsequent induction of RKIP expression (Figure 8C and 8D respectively). A model of the ER-MTA-Snail-RKIP axis is shown in Figure 8E.
A.
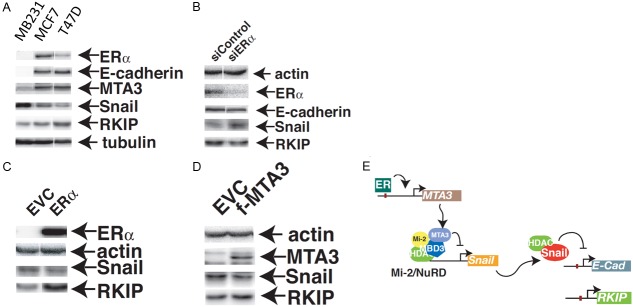
Representative western blot analysis of ERα, e-cadherin, MTA3, Snail, RKIP and Tubulin in metastatic, triple negative (MDA-MB231) and non-metastatic (MCF7, T47D) breast cancer cell lines. Tubulin expression was served as a loading control. (B) Protein expression of RKIP and Snail in MCF7 cells expressing ERα specific or scramble control siRNA. (C) Representative western blot analysis of Snail and RKIP expression in protein lysates derived from MDA-MB231 cells expressing the ERα (C) or MTA3 (D) proteins. EVC: Empty vector control. Actin expression was used as a loading control. The experiments have repeated at least three times with very similar results. (E) A representative model depicting the mechanism through which RKIP expression is induced by ER.
RKIP expression and breast cancer molecular subtypes
Based on our ER finding, we hypothesized that ER negative breast cancers, like the Basal-like tumors and especially Claudin-low tumors (that exhibit high EMT phenotype) should express lower levels of RKIP compared to ER positive tumors.
To test this hypothesis, we downloaded and reanalyzed microarray mRNA expression data of human breast cancer performed using Affymetrix U133A or U133Plus2 platforms. The data comprised of information from 3,992 human breast tumor samples from 26 cohorts and 81 samples corresponding to 70 different breast cancer cell lines (See materials and methods and Table 2). An EMT signature was developed to assess the EMT phenotypic status of a clinical sample or cell line (See Materials and Methods). The breast cancer subtype was predicted using signature from Prat et al., 2010 [24], and ssGSEA [23] (Materials and Methods).
In both cell lines (Figure 9A) and clinical samples (Figure 9B), Luminal A breast cancer subtype had the highest RKIP expression levels followed by Luminal B subtype. Claudin-low subtype had the lowest levels of RKIP expression, followed by Her2-enriched, Basal-like and near normal subtypes (Figure 9A and 9B). Intriguingly, an inverse relationship was attainable between RKIP levels and EMT scores in both the cell lines (Figure 9C) and the clinical samples (Figure 9D). Figure 9E shows that a direct linear and significant correlation exists between ER and RKIP positivity. ER positive breast cancer expressed significantly higher RKIP compared to ER negative tumors (Figure 9F). Consistent with the previous cohorts, low RKIP had significantly shorter DFS compared to high-RKIP expressors (Figure 9G).
Figure 9.
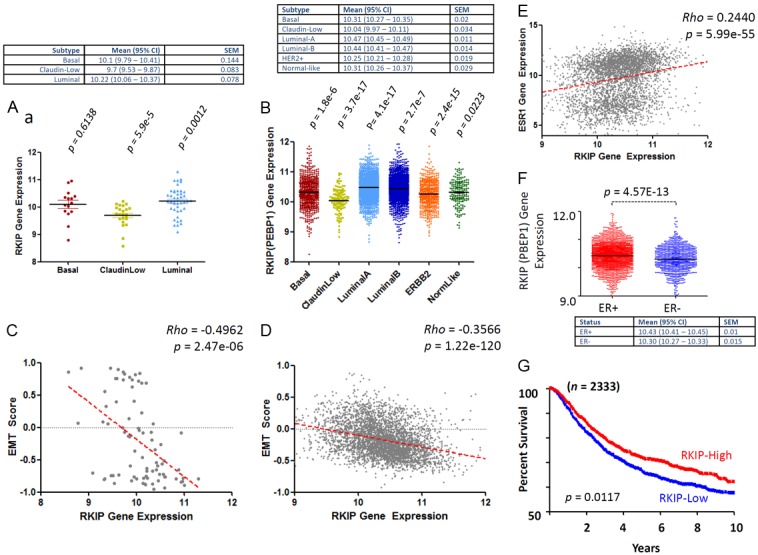
Association between RKIP expression, EMT, molecular subclasses, ER and DFS of breast cancer. Median RKIP expression levels obtained from 81 samples (Table 2) from 70 different breast cancer cell lines. (A) and 3992 clinical breast cancer cases (B) after appropriate molecular subclassification. (C and D) Show the inverse correlation between RKIP expression and EMT scores obtained in cell lines and clinical breast cancer samples respectively. (E) Direct and linear correlation between RKIP and ER expression in breast cancer samples. (F) Shows the median values and distribution of RKIP mRNA levels in ER+ and ER- breast cancer samples. (G) Shows Kaplan-Meier’s disease-free survival curves between high (>median) and low RKIP (≤median) expressing breast cancer samples. The p values compare each subclass with the rest of subclasses. Rho statistic was used to establish the linear correlation between RKIP expression and EMT. The Log-Rank test was used to compare the statistical difference in DFS for this cohort.
Discussion
Our data represent an important advance in understanding the role of RKIP in breast cancer progression. At the protein level, we show that the absence or diminution of RKIP, but not p-RKIP is associated with increased tumor size and proliferative index, high tumor grade, and increased necrosis in invasive ductal carcinoma. Our data are reminiscent of earlier reports depicting RKIP as a significant modulator of cellular growth and cell cycle kinetics [16,30]. In ovarian cancer, reduced RKIP expression has been associated with poor differentiation and increased cellular proliferation [31].
Hagan et al., were the first to study and report on RKIP expression in breast cancer using immunohistochemistry [32]. The authors observed no significant correlation between RKIP expression and histological type, tumor grade, size, or estrogen receptor status. However, they demonstrated that reduced or diminished RKIP expression was prevalent in lymph node metastases. Similarly, another study reported on the reduction of RKIP expression in breast cancer lymph node metastases but offered no indication on its influences on tumor size or proliferation [33]. These data are consistent with the metastasis suppression function of RKIP.
Using three independent cohorts, we show, by mean of univariate analysis, that RKIP expression loss or diminution is associated with reduced DFS in breast cancer. Obtaining highly congruent data from 3 independent studies undoubtedly support a genuine role for RKIP in breast cancer progression. We have previously identified RKIP as a useful prognostic marker, particularly in identifying patients with early-stage colorectal cancer at risk of metastatic relapse [34]. Moreover, loss or diminution of RKIP has been linked to poor survival in many cancer types (reviewed in Al-Mulla et al. [17]). Therefore, our data add breast cancer to the panoply of evidence linking RKIP expression loss to metastatic disease and poor survival. In addition, we show that the complete absence of p-RKIP in both the nucleus and the cytoplasm constitutes an independent prognostic indicator of poor DFS in multivariate analysis. The role of p-RKIP in cancer is not well established. However, our data harmonize with a recent report from patients with lung cancer and melanoma [35,36]. Overall, these results indicate the importance of assessing both p-RKIP and RKIP expression levels to delineate RKIP role in different types of cancer.
We presented the only comprehensive analysis to date linking ER status to RKIP expression. At protein level, 82.5% of high nuclear and 80% of high cytoplasmic and nuclear RKIP expressing tumors were ER positive. This significant association was confirmed at mRNA level using two independent cohorts. 5α-Dihydrotestosterone has been shown to bind Androgen response elements on the RKIP promoter and to drive its expression in the prostate [37]. However, we found little evidence of ER binding consensus DNA sequences in the RKIP gene (data not shown). Moreover, Carroll et. al., have mapped all estrogen receptor and RNA polymerase II binding sites in MCF-7 cells using a genome-wide approach, and found no evidence for ER binding sites in the RKIP gene [38]. Therefore, we proposed that ER may indirectly influence RKIP expression. Indeed, the ER-MTA3-Snail axis has been previously described in relation to e-cadherin transcriptional silencing and EMT phenotype [29]. Our data includes RKIP as an additional key molecule in the aforementioned signaling dependent pathway in breast cancer. Our in vitro studies offer further support to this notion. ER-MTA3 appears to play only a part in RKIP activation since 48% of ER positive tumors had diminished RKIP expression indicating the existence of ER-independent RKIP silencing mechanisms in breast cancer. Equally, other factors may activate RKIP expression in ER negative tumors. This is not surprising given that RKIP expression is controlled at the transcriptional level by Snail [39,40]. Although Snail can be repressed transcriptionally by MTA3 [29], its transcription and stability can be modulated by NFκB [41] and GSK3β [42] respectively, while both can be modulated by RKIP [15,43]. Such important circuitry may explain the intricate molecular machinery controlling cancer growth, EMT, invasiveness and metastasis, especially in ER negative breast cancer.
The mitogenic effect of ER on breast tissue is well documented [44]. The wide use of selective ER modulators, particularly Tamoxifen, in the management of hormone receptor-positive breast cancer, has improved patients’ survival significantly. Moreover, 40% of Tamoxifen treated patients suffer from disease relapse. It would be of significant interest to examine if RKIP loss or diminution may be responsible for therapeutic resistance [45]. This proposition is not unrealistic given the existence of a large body of evidence linking the activation of Ras-Raf-MEK-ERK pathway with hormone therapy resistance [46,47] and the ability of signal transduction inhibitors in enhancing endocrine sensitivity [48]. The activation of the MAPK pathway in ERα positive breast cancer cells has been shown to induce molecular phenotypes reminiscent of ER negative breast cancer [49].
RKIP expression appears to vary across different breast cancer subtypes with the highest expression observed in Luminal A, and the lowest in Claudin-low subtype. Interestingly, the claudin-low subtype is characterized by low expression of tight junction proteins (claudin 3, 4, 7 and e-cadherin) with stem-cell like and intense EMT phenotypic features. This subtype expresses ZEB1, twist and Snail, which are markers for EMT and poor survival [1,24].
RKIP loss is highly permissive for β-catenin, Snail, SLUG, and vimentin expression, which are molecules constitutively expressed in EMT and ones that encourage cellular invasion and metastasis [15,16]. Therefore, our data offer additional leverage for the panoply of evidence pointing towards an intimate relationship between ER negativity, EMT [50] and their poor clinical outcome in breast cancer [51].
ERBB2-enriched breast tumors ranked second lowest in terms of RKIP expression after Claudin-low subtype. Independently, at protein level, we observed a similar reciprocal association between HER-2 overexpression/amplification and RKIP positivity at the protein level. These data are reminiscent of an earlier report confirming that RKIP was among 15 proteins downregulated in HER-2/neu positive compared to HER-2/neu negative breast cancer tissues [52]. We are currently actively investigating the prognostic efficacy of RKIP expression within each breast cancer subtype.
Future studies will be needed in order to decipher the particular functional significance of the many pathways modulated by RKIP or its phosphorylated form. Nevertheless, one may envision a therapeutic intervention aimed at inducing RKIP and its phosphorylated form in the treatment of Claudin-low or ERBB2-enriched breast cancer. Indeed, one study has shown that RKIP induction inhibited the in vitro growth, invasiveness and metastatic capability of human triple negative MDA-MB-231 cells [53].
Acknowledgements
The authors thank Dr. Brian Rowan of Tulane University and Paul Wade of NIH for expression vectors for siER and MTA3, respectively. This study was fully supported by the Kuwait Foundation for Advancement of Sciences; Contract grant number: 2011-1302-06 given to Prof. Fahd Al-Mulla and by NIH grant (RO1CA133479) to K. C. Yeung.
References
- 1.Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13:2329–2334. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- 2.Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2010;15(Suppl 5):39–48. doi: 10.1634/theoncologist.2010-S5-39. [DOI] [PubMed] [Google Scholar]
- 3.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 4.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polyak K, Metzger Filho O. SnapShot: breast cancer. Cancer Cell. 2012;22:562, e1. doi: 10.1016/j.ccr.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T, Zhou W, Berry L, Murray L, Amler L, Belvin M, Friedman LS, Lackner MR. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15:4649–4664. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- 8.Santarpia L, Qi Y, Stemke-Hale K, Wang B, Young EJ, Booser DJ, Holmes FA, O’Shaughnessy J, Hellerstedt B, Pippen J, Vidaurre T, Gomez H, Valero V, Hortobagyi GN, Symmans WF, Bottai G, Di Leo A, Gonzalez-Angulo AM, Pusztai L. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res Treat. 2012;134:333–343. doi: 10.1007/s10549-012-2035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 10.Trakul N, Menard RE, Schade GR, Qian Z, Rosner MR. Raf kinase inhibitory protein regulates Raf-1 but not B-Raf kinase activation. J Biol Chem. 2005;280:24931–24940. doi: 10.1074/jbc.M413929200. [DOI] [PubMed] [Google Scholar]
- 11.Trakul N, Rosner MR. Modulation of the MAP kinase signaling cascade by Raf kinase inhibitory protein. Cell Res. 2005;15:19–23. doi: 10.1038/sj.cr.7290258. [DOI] [PubMed] [Google Scholar]
- 12.Yeung K, Janosch P, McFerran B, Rose DW, Mischak H, Sedivy JM, Kolch W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–3085. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem. 2003;278:13061–13068. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- 14.Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426:574–579. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- 15.Al-Mulla F, Bitar MS, Al-Maghrebi M, Behbehani AI, Al-Ali W, Rath O, Doyle B, Tan KY, Pitt A, Kolch W. Raf kinase inhibitor protein RKIP enhances signaling by glycogen synthase kinase- 3beta. Cancer Res. 2011;71:1334–1343. doi: 10.1158/0008-5472.CAN-10-3102. [DOI] [PubMed] [Google Scholar]
- 16.Al-Mulla F, Bitar MS, Taqi Z, Rath O, Kolch W. RAF kinase inhibitory protein (RKIP) modulates cell cycle kinetics and motility. Mol Biosyst. 2011;7:928–941. doi: 10.1039/c0mb00208a. [DOI] [PubMed] [Google Scholar]
- 17.Al-Mulla F, Bitar MS, Taqi Z, Yeung KC. RKIP: much more than Raf kinase inhibitory protein. J Cell Physiol. 2013;228:1688–1702. doi: 10.1002/jcp.24335. [DOI] [PubMed] [Google Scholar]
- 18.Kirkegaard T, Edwards J, Tovey S, McGlynn LM, Krishna SN, Mukherjee R, Tam L, Munro AF, Dunne B, Bartlett JM. Observer variation in immunohistochemical analysis of protein expression, time for a change? Histopathology. 2006;48:787–794. doi: 10.1111/j.1365-2559.2006.02412.x. [DOI] [PubMed] [Google Scholar]
- 19.Mohammed ZM, McMillan DC, Elsberger B, Going JJ, Orange C, Mallon E, Doughty JC, Edwards J. Comparison of visual and automated assessment of Ki-67 proliferative activity and their impact on outcome in primary operable invasive ductal breast cancer. Br J Cancer. 2012;106:383–388. doi: 10.1038/bjc.2011.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chatterjee D, Bai Y, Wang Z, Beach S, Mott S, Roy R, Braastad C, Sun Y, Mukhopadhyay A, Aggarwal BB, Darnowski J, Pantazis P, Wyche J, Fu Z, Kitagwa Y, Keller ET, Sedivy JM, Yeung KC. RKIP sensitizes prostate and breast cancer cells to drug-induced apoptosis. J Biol Chem. 2004;279:17515–17523. doi: 10.1074/jbc.M313816200. [DOI] [PubMed] [Google Scholar]
- 21.Hess KR, Anderson K, Symmans WF, Valero V, Ibrahim N, Mejia JA, Booser D, Theriault RL, Buzdar AU, Dempsey PJ, Rouzier R, Sneige N, Ross JS, Vidaurre T, Gomez HL, Hortobagyi GN, Pusztai L. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J. Clin. Oncol. 2006;24:4236–4244. doi: 10.1200/JCO.2006.05.6861. [DOI] [PubMed] [Google Scholar]
- 22.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 23.Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ, Fereday S, Lawrence M, Carter SL, Mermel CH, Kostic AD, Etemadmoghadam D, Saksena G, Cibulskis K, Duraisamy S, Levanon K, Sougnez C, Tsherniak A, Gomez S, Onofrio R, Gabriel S, Chin L, Zhang N, Spellman PT, Zhang Y, Akbani R, Hoadley KA, Kahn A, Kobel M, Huntsman D, Soslow RA, Defazio A, Birrer MJ, Gray JW, Weinstein JN, Bowtell DD, Drapkin R, Mesirov JP, Getz G, Levine DA, Meyerson M. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest. 2013;123:517–525. doi: 10.1172/JCI65833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, Datto MB, Kelley M, Mathey-Prevot B, Potti A, Nevins JR. A pathway-based classification of human breast cancer. Proc Natl Acad Sci U S A. 2010;107:6994–6999. doi: 10.1073/pnas.0912708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McShane LM, Altman DG, Sauerbrei W. Identification of clinically useful cancer prognostic factors: what are we missing? J Natl Cancer Inst. 2005;97:1023–1025. doi: 10.1093/jnci/dji193. [DOI] [PubMed] [Google Scholar]
- 28.van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 29.Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207–219. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 30.Eves EM, Shapiro P, Naik K, Klein UR, Trakul N, Rosner MR. Raf kinase inhibitory protein regulates aurora B kinase and the spindle checkpoint. Mol Cell. 2006;23:561–574. doi: 10.1016/j.molcel.2006.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li HZ, Wang Y, Gao Y, Shao J, Zhao XL, Deng WM, Liu YX, Yang J, Yao Z. Effects of raf kinase inhibitor protein expression on metastasis and progression of human epithelial ovarian cancer. Mol Cancer Res. 2008;6:917–928. doi: 10.1158/1541-7786.MCR-08-0093. [DOI] [PubMed] [Google Scholar]
- 32.Hagan S, Al-Mulla F, Mallon E, Oien K, Ferrier R, Gusterson B, Garcia JJ, Kolch W. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin Cancer Res. 2005;11:7392–7397. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- 33.Li HZ, Gao Y, Zhao XL, Liu YX, Sun BC, Yang J, Yao Z. Effects of raf kinase inhibitor protein expression on metastasis and progression of human breast cancer. Mol Cancer Res. 2009;7:832–840. doi: 10.1158/1541-7786.MCR-08-0403. [DOI] [PubMed] [Google Scholar]
- 34.Al-Mulla F, Hagan S, Behbehani AI, Bitar MS, George SS, Going JJ, Garcia JJ, Scott L, Fyfe N, Murray GI, Kolch W. Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. J. Clin. Oncol. 2006;24:5672–5679. doi: 10.1200/JCO.2006.07.5499. [DOI] [PubMed] [Google Scholar]
- 35.Cardile V, Malaponte G, Loreto C, Libra M, Caggia S, Trovato FM, Musumeci G. Raf kinase inhibitor protein (RKIP) and phospho-RKIP expression in melanomas. Acta Histochem. 2013 doi: 10.1016/j.acthis.2013.03.003. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 36.Huerta-Yepez S, Yoon NK, Hernandez-Cueto A, Mah V, Rivera-Pazos CM, Chatterjee D, Vega MI, Maresh EL, Horvath S, Chia D, Bonavida B, Goodglick L. Expression of phosphorylated raf kinase inhibitor protein (pRKIP) is a predictor of lung cancer survival. BMC Cancer. 2011;11:259. doi: 10.1186/1471-2407-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, Wu J, Keller JM, Yeung K, Keller ET, Fu Z. Transcriptional Regulation of RKIP Expression by Androgen in Prostate Cells. Cell Physiol Biochem. 2012;30:1340–1350. doi: 10.1159/000343323. [DOI] [PubMed] [Google Scholar]
- 38.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 39.Beach S, Tang H, Park S, Dhillon AS, Keller ET, Kolch W, Yeung KC. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene. 2008;27:2243–2248. doi: 10.1038/sj.onc.1210860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren G, Baritaki S, Marathe H, Feng J, Park S, Beach S, Bazeley PS, Beshir AB, Fenteany G, Mehra R, Daignault S, Al-Mulla F, Keller E, Bonavida B, de la Serna I, Yeung KC. Polycomb Protein EZH2 Regulates Tumor Invasion via the Transcriptional Repression of the Metastasis Suppressor RKIP in Breast and Prostate Cancer. Cancer Res. 2012;72:3091–3104. doi: 10.1158/0008-5472.CAN-11-3546. [DOI] [PubMed] [Google Scholar]
- 41.Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A, Larue L. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26:7445–7456. doi: 10.1038/sj.onc.1210546. [DOI] [PubMed] [Google Scholar]
- 42.Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005;280:11740–11748. doi: 10.1074/jbc.M413878200. [DOI] [PubMed] [Google Scholar]
- 43.Bonavida B, Baritaki S. Dual role of NO donors in the reversal of tumor cell resistance and EMT: Downregulation of the NF-kappaB/Snail/YY1/RKIP circuitry. Nitric Oxide. 2011;24:1–7. doi: 10.1016/j.niox.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Renoir JM, Marsaud V, Lazennec G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol. 2013;85:449–465. doi: 10.1016/j.bcp.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 45.Al-Mulla F, Bitar MS, Feng J, Park S, Yeung KC. A new model for raf kinase inhibitory protein induced chemotherapeutic resistance. PLoS One. 2012;7:e29532. doi: 10.1371/journal.pone.0029532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 47.Swanton C, Downward J. Unraveling the complexity of endocrine resistance in breast cancer by functional genomics. Cancer Cell. 2008;13:83–85. doi: 10.1016/j.ccr.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 48.Johnston SR, Martin LA, Leary A, Head J, Dowsett M. Clinical strategies for rationale combinations of aromatase inhibitors with novel therapies for breast cancer. J Steroid Biochem Mol Biol. 2007;106:180–186. doi: 10.1016/j.jsbmb.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 49.Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006;66:3903–3911. doi: 10.1158/0008-5472.CAN-05-4363. [DOI] [PubMed] [Google Scholar]
- 50.Al Saleh S, Al Mulla F, Luqmani YA. Estrogen receptor silencing induces epithelial to mesenchymal transition in human breast cancer cells. PLoS One. 2011;6:e20610. doi: 10.1371/journal.pone.0020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lapidus RG, Nass SJ, Davidson NE. The loss of estrogen and progesterone receptor gene expression in human breast cancer. J Mammary Gland Biol Neoplasia. 1998;3:85–94. doi: 10.1023/a:1018778403001. [DOI] [PubMed] [Google Scholar]
- 52.Zhang D, Tai LK, Wong LL, Putti TC, Sethi SK, Teh M, Koay ES. Proteomic characterization of differentially expressed proteins in breast cancer: Expression of hnRNP H1, RKIP and GRP78 is strongly associated with HER-2/neu status. Proteomics Clin Appl. 2008;2:99–107. doi: 10.1002/prca.200780099. [DOI] [PubMed] [Google Scholar]
- 53.Hao C, Wei S, Tong Z, Li S, Shi Y, Wang X, Zhu ZH. The effects of RKIP gene expression on the biological characteristics of human triple-negative breast cancer cells in vitro. Tumour Biol. 2012;33:1159–67. doi: 10.1007/s13277-012-0358-7. [DOI] [PubMed] [Google Scholar]