

Abstract
The compound class of 3-carboranyl thymidine analogues (3CTAs) are boron delivery agents for boron neutron capture therapy (BNCT), a binary treatment modality for cancer. Presumably, these compounds accumulate selectively in tumor cells via intracellular trapping, which is mediated by hTK1. Favorable in vivo biodistribution profiles of 3CTAs led to promising results in preclinical BNCT of rats with intracerebral brain tumors. This review presents an overview on the design, synthesis, and biological evaluation of first- and second-generation 3CTAs. Boronated nucleosides developed prior to 3CTAs for BNCT and non-boronated N3-substituted thymidine conjugates for other areas of cancer therapy and imaging are also described. In addition, basic features of carborane clusters, which are used as boron moieties in the design and synthesis of 3CTAs, and the biological and structural features of TK1-like enzymes, which are the molecular targets of 3CTAs, are discussed.
Boron neutron capture therapy (BNCT) is a binary cancer treatment modality that is based on the selective accumulation of 10B in tumor cells followed by irradiation with low-energy (thermal) neutrons. Neutron capture by 10B produces high linear energy transfer α-particles (4He2+) and 7Li3+ nuclei, both of which can selectively destroy tumor cells. Concurrently, nearby healthy cells are spared because of the limited path lengths of 5–9 μm of these particles. Approximately 109 10B atoms per tumor cells and tumor-to-normal tissue ratios of >3 to 1 are required for successful BNCT. Natural boron consists of stable 10B- (~20%) and 11B-isotopes (~80%). Only the former is suitable for BNCT [1,2].
Boronophenylalanine (1) and sodium borocaptate (2) are two boron delivery agents that are/ were used clinically in 10B-enriched form in, for example, Japan, the USA, Europe and Argentina (Figure 1) [1,2]. The development of novel improved tumor selective boron delivery vehicles has been one of the most pressing tasks of BNCT research because of suboptimal clinical results obtained with BPA and BSH [1]. These novel boron delivery agents include amino acids, polyamines [3–5], DNA-binding agents [6,7], porphyrins and related structures [8–10], carbohydrates [11–14], monoclonal antibodies [15–17] and liposomes [18–20].
Figure 1.
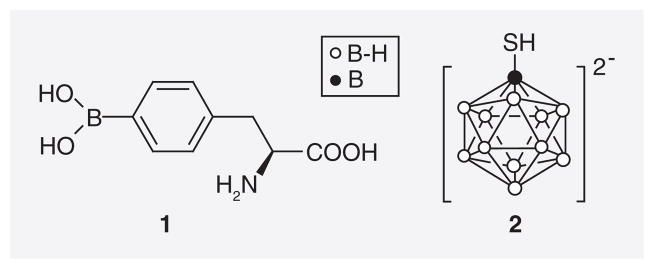
Boronophenylalanine (1) and sodium borocaptate (2).
Endogenous nucleosides have also attracted considerable attention as potential boron delivery vehicles for BNCT [21]. These structures are known to accumulate selectively in rapidly proliferating tumor cells, as they are building blocks for DNA and RNA synthesis [22]. The same is true for artificial nucleoside derivatives, such 3′-[18F] fluoro-3′-deoxythymidine ([18F]FLT), which is widely used in PET imaging of cancer [23].
In recent years, the development of boronated nucleosides for BNCT has mainly focused on 3-carboranyl thymidine analogues (3CTAs) [21,24], which possibly localize selectively in tumor cells by a process described as kinase-mediated trapping (KMT) [25]. In this review, we will present an overview on the design, synthesis and biological evaluation of first- and second-generation 3CTAs. We will also describe the biological and structural features of TK1-like enzymes, which are the molecular targets of 3CTAs, as well as the structural and chemical properties of carborane clusters, which are used as boron moieties in the design and synthesis of 3CTAs. Another topic of this review will be boronated nucleosides that have been the focus of agent development efforts in BNCT prior to 3CTAs.
The general concept of using thymidine modified at the N3-position with a therapeutic or imaging entity in the context of KMT was initially developed and explored using the example of 3CTAs. However, in recent years numerous non-boronated N3-modified thymidine bioconjugates were successfully developed for areas of cancer therapy other than BNCT as well as for tumor imaging, thereby validating this concept [26–30]. Thus, these conjugates will also be discussed in this review.
Carboranes
Three highly hydrophobic closo-carborane clusters with the chemical formula C2B10H12 have been used widely in the design and synthesis of 3CTAs. These are 1,2-closo-carborane (ortho), 1,7-closo-carborane (meta) and 1,12-closo-carborane (para) [31]. The former two cages can be transformed easily to anionic nido-carboranes by the action of fluoride anions or bases (Figure 2) [31]. Both nido-o-carborane and nido-m-carborane were also used in the synthesis of 3CTAs [21,24,32].
Figure 2. Carborane structures.

(A) closo-o-carborane, (B) closo-m-carborane, (C) closo-m-carborane, (D) nido-o-carborane, (E) nido-m-carborane.
Generated by Chem 3D, Perkin Elmer, Cambridge, USA.
Closo- and nido-carborane cages have found diverse applications not only in the development of boron delivery agents for BNCT [1,24]. The hydrophobic character of the closo-carboranes and the hydrophilic character of the nido-carboranes combined with their rigid geometry, high physiological stability, and synthetic versatility have made both species also attractive entities for other areas of medicinal chemistry, such as their use as pharmacophores in conventional drug design and as prosthetic groups for radiohalogens in molecular imaging applications [33–35].
TK1-like enzymes
TK1-like enzymes include hTK1, vaccinia virus thymidine kinase and several bacterial thymidine kinases [36–42]. Cytosolic hTK1 is an essential deoxynucleoside kinase of the salvage pathway of DNA synthesis [43,44]. It is the primary molecular target of 3CTAs. This enzyme phosphorylates thymidine to thymidine monophosphate, which is followed by the conversion of thymidine mono-phosphate to thymidine diphosphate by TMPK. Phosphorylation of thymidine diphosphate by NDPK leads to thymidine triphosphate, which is incorporated into DNA by DNA polymerase (Figure 3) [43,44]. There is a second mammalian thymidine kinase, designated TK2, which is localized in mitochondria. Based on amino acid sequences and substrate specificities, TK1 differs significantly from TK2 but also from other dNKs, such as dCK and dGK [44–48]. Human TK1 activity is cell-cycle dependent increasing in the G1/S phase. It declines rapidly in G2/M phase and is absent in resting cells. In contrast, TK2 activity is cell cycle independent [45–48].
Figure 3. Kinase-mediated trapping of N5.

NDPK: Nucleoside diphosphate kinase; TMPK: Thymidine monophosphate kinase.
Adapted with permission from The Royal Society of Chemistry [24].
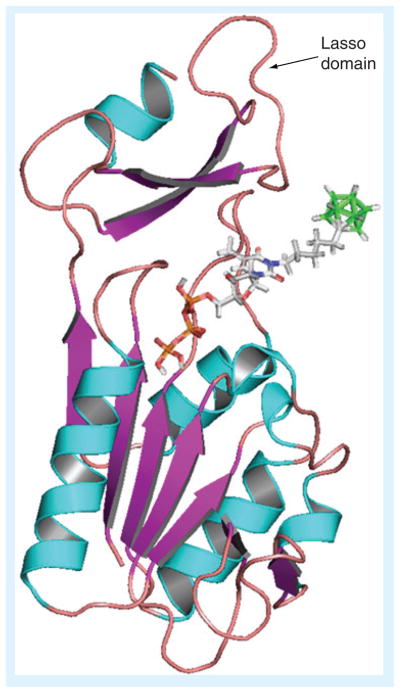
Cellular hTK1 is found in a dimeric form with low affinity for thymidine (Km = 15 μM) and a tetrameric form (Figure 4B) with high affinity for thymidine (Km = 0.7 μM) [49]. Substrate binding is demonstrated at the example of thymidine tri-phosphate to a monomeric subunit of hTK1 [36], which represents a holo form of the enzyme (Figure 4A). Hydrogen bonds between N3-H, C2=O and C4=O of the thymidine scaffold and carbonyl and amino groups of the peptide backbone primarily of the ‘lasso loop’ of hTK1 and other TK1-like enzymes are important for substrate binding [30–36]. As discussed by Welin et al. [36], the N3 nitrogen of thymidine appears to be positioned towards the surface of the enzyme, which could possibly allow bulky substituents at this position, albeit accompanied with loss of hydrogen bonding. A small hydrophobic pocket accommodates the 5-methyl group of thymidine [36] and also slightly larger ethyl and halogen substituents, as has been confirmed by various structure–activity relationship (SAR) studies [48,50]. The 3′-hydroxyl group of thymidine may also have some access to the surface of the enzyme [36], which explains some tolerance for small substituents at this position [44].
Figure 4. hTK1.

(A) Monomer subunit of hTK1 with thymidine triphosphate. (B) Tetramer structure of hTK1.
Data taken from [39] (PDB: 1W4R).
5-carboranyl- & 3′-aminocyanoborane-substituted pyrimidine nucleoside analogues for BNCT
Early design strategies for boronated nucleosides for BNCT focused on the C5-position of 2′-deoxyuridine for attachment of boron moieties rather than on the N3-position of thymidine, as in the case of 3CTAs. These efforts were probably inspired by the knowledge that various 5-halo-2′-deoxyridines are rapidly incorporated into DNA, and, thus, selectively accumulate in proliferating tumor cells [51,52]. Schinazi and Prusoff prepared the first boronated nucleoside analogue, 5-(dihydroxyboryl)-2′-deoxyuridine (3; Figure 5) [53]. The compound did not appear to cause significant cytotoxicity in various mammalian cell lines. In vitro studies indicated that 3 was less effectively incorporated into DNA than 5-iodo- and 5-bromo-2′-deoxyuridine [54].
Figure 5. Boronated thymidine analogues synthesized and evaluated prior to the discovery of 3-carboranyl thymidine analogues.
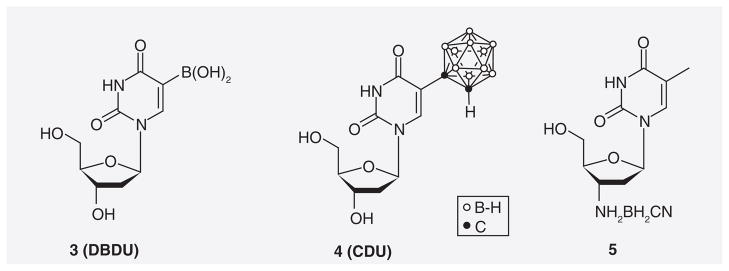
CDU: 5-(1-o-carboranyl)-2′-deoxyuridine; DBDU: 5-dihydroboryl-2′-deoxyuridine.
Yamamoto et al. synthesized the first 5-substituted carboranyl nucleoside, 5-(1-o-carboranyl)-2′-deoxyuridine (4) [55]. In vitro data indicated that 4 was overall more cytotoxic than its dihydroxyboryl counterpart 3. Studies by Lunato et al. demonstrated that 4 was a substrate of TK2 rather than TK1 [56], which is not surprising considering the limited tolerance of TK1 with respect to 5-substitution. Phosphorylation of radiolabeled 4 in vitro was observed in, for example, human T-cell leukemia and peripheral blood mononuclear cells [57].
Spielvogel et al. synthesized and evaluated 3′-aminocyanoborane-2′,3′-dideoxythymidine (5) [58]. Phosphorylation of thymidine analogues with minor modification at the 3′-position (e.g., F and N3) by TK1 is possible [44]. Compound 5 demonstrated significant cytotoxicity against various murine and human cancer cell lines, which was attributed to the inhibition of enzymes involved in both purine and pyrimidine nucleotide de novo synthesis [21,58].
3CTAs
First-generation 3CTAs & KMT
In 3CTAs, a carborane cage is linked through a spacer to the N3 position of thymidine, as represented by the examples of first-generation 3CTAs in Figure 6. In the case of 3CTAs 6–18, the attached cluster is o-carborane. These compounds are effectively 5′-monophosphorylated by TK1. This presumably causes the selective accumulation of 3CTAs in tumor cells [21,24,59–62] since the obtained negative charge should prevent passive diffusion out of the tumor cells and nucleoside transporters apparently do not accept nucleotides as substrates [63]. This process has been described as KMT (Figure 3) [25]. Theoretically, intracellular entrapment of 3CTAs could be further enhanced by di- and tri-phosphorylation followed by incorporation of the 3CTA–triphosphate into DNA (Figure 3). However, it is unknown if 3CTAs undergo these biochemical conversions.
Figure 6.
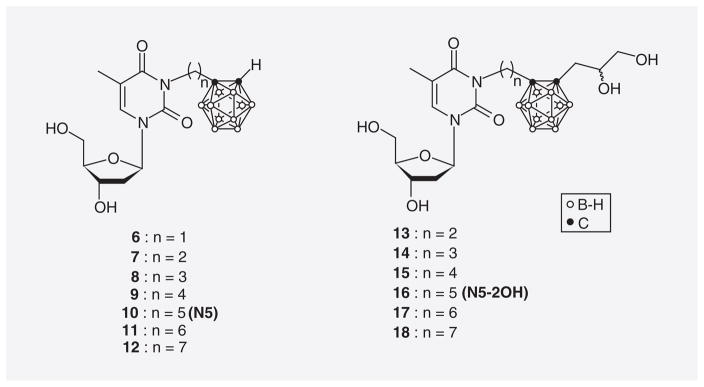
First-generation 3-carboranyl thymidine analogues.
The original design concept for 3CTAs was developed by Lunato et al. [56]. It is based on observations made during the purification of various kinases, including thymidine kinases, by affinity chromatography [64–66]. In these studies, kinase substrates were covalently linked to a solid matrix. In the case of thymidine, linkage occurred presumably through the N3-position. It was discovered that for a successful purification of the kinases, linkers of specific lengths between solid matrix and nucleoside/nucleotide scaffold were of crucial importance to overcome steric hindrance. This simple concept was adapted for the design of 3CTAs. Here, the solid support was replaced with the bulky carborane cluster hoping that the resulting construct would remain susceptible to phosphorylation by kinases. Fortuitously, 3CTAs eventually proved to be selective substrates of TK1 [21,24,59–61] confirming this design concept.
Among the first-generation 3CTAs represented in Figure 6, those with a pentylene linker between thymidine scaffold and carborane cluster were the best substrates of hTK1. These agents were designated as N5 (10) and N5-2OH (16). Their apparent relative kcat/Km values (rkcat/Kms) were 26.7 and 35.8%, respectively, which is comparable to clinically used hTK1 substrates, such as AZT [24]. kcat/Km values are a measure of the apparent catalytic efficiency of the enzyme for its substrates. In this case, the kcat/Km values for the 3CTAs are expressed relative to that of endogenous thymidine, which is arbitrarily set to 100% [67]. On the other hand, these 3CTAs did not appear to be substrates of TK2 and dCK [24]. Those 3CTAs with an ethylene spacer (7 & 13) were as good substrates of hTK1 as 10 and 16. However, they were apparently not as effective as the latter two agents in competing with endogenous thymidine at the substrate-binding site of hTK1 [59]. This was an important finding, as it is known that competition of endogenous nucleosides, such as thymidine, with nucleoside analogues used in imaging, such as [18F]FLT, negatively affects the uptake of the latter in tumor tissue [23]. In vitro uptake, retention and cytotoxicity of 10 (N5) and 16 (N5-2OH) were evaluated in F98 glioma, MRA 27 melanoma, and TK1(+)/TK1(−) L929 cells [59]. Both compounds were moderately cytotoxic in all four cell lines with IC50 values ranging from 18–70 μM. The cellular uptake of N5-2OH was highest in MRA 27 with 169 μg B/109 cells after 24 h incubation with 22% retention following additional 12 h of incubation in compound-free medium. More importantly, uptake/retention of N5-2OH in L929 TK(+) and L929 TK(−) differed significantly (92 μg B/109 cells/29% vs 9.3 μg B/109 cells/0%) indicating that KMT may be an important factor in the cellular accumulation of this compound. In L929 TK(+) and L929 TK(−) tumor-bearing nude mice, an uptake ratio of approximately three in favor of the TK1(+) tumor was found for N5-2OH (22.8 vs 8.4 μg B/g tumor tissue) [62]. Survival times of rats with intracranial RG2 glioma were increased by a factor of two by BNCT following prior intracranial (ic.) administration of N5-2OH compared with a control group of RG2 glioma bearing rats that were subjected to neutron radiation without prior injection of N5-2OH [62]. A group of rats without tumors that had received N5-2OH by ic. administration without neutron irradiation was monitored for signs of toxicity [62]. There was no evidence for neurological deficit. The animals lost initially <10% of their weight, which was, however, regained after a short period of time. A pathologic examination of the rat brains did not reveal any significant tissue alterations.
A hypothetical model for the binding of 3CTAs to the active site of hTK1 was developed by Tjarks et al. [24,61,68]. It is demonstrated at the example of a docked pose of N5-triphosphate in the active site of a homology model of hTK1 (Figure 7) [68]. The use of the triphosphate form of N5 proved to be useful in stabilizing the docked pose of the thymidine portion of the molecule within the substrate binding site [68]. This protein structure presumably represents an apo form of the enzyme because it was derived from an apo form of TmTK [40,68]. TK1-like enzymes respond with dramatic conformational changes upon ATP and/or thymidine binding, which was discussed in detail by Lavie et al. [40]. This is especially obvious in the case of the lasso loop, which takes a far more open conformation in apo forms [40] as compared to holo forms (depicted in Figure 4). As represented in Figure 7, docking of N5 to the substrate-binding site of this apo form can be accommodated because the bulky carborane remains outside of the protein. Similar results have been observed for the docking of N5-2OH [24] and its isomeric forms 20 and 21 [61] into apo forms of TK1-like enzymes. On the other hand, docking of 3CTAs into the active site of holo forms of TK1-like enzymes was not possible [24]. This rudimentary hypothetical binding mode of 3CTAs to TK1-like enzymes seems plausible. However, the exact binding mode of these structures to this type of enzymes remains unknown.
Figure 7. Docking pose of N5-triphosphate in a homology model of hTK1 (PDB: 1W4R), which was developed from TmTK (PDB: 2QPO).
[34].
Adapted with permission [63] © American Chemical Society (2012).
Extended synthetic & biological studies on N5-2OH (16)
Byun et al. reported the synthesis of both (R)-epimeric- (20) and (S)-epimeric (21) forms of N5-2OH (16) with natural abundance of boron isotopes as well as racemic N5-2OH (19) in 10B-enriched form (Figure 8) [61]. In addition, structural isomers of racemic N5-2OH (16) containing either m-carborane (22) or p-carborane (23) were prepared. The relative hTK1 phosphorylation rates (relative PRs) of 16 and 20–23 ranged from 32–45%. PRs are used to estimate the capacity of, for example, 3CTAs to function as substrates for hTK1 in a screening setting. As in the case of the kcat/Km values, PRs are expressed relative to that of endogenous thymidine, which is arbitrary set to 100% [67].
Figure 8.

10B-enriched racemic N5-2OH (19), (R)-N5-2OH (20), (S)-N5-2OH (21), m-carboranyl racemic N5-2OH (22), and p-carboranyl racemic N5-2OH (23).
Water-soluble amino acid ester prodrugs of 3CTAs
An approach focusing exclusively on improving the water solubilities of N5 and N5-2OH was recently reported by Hasabelnaby et al. [69]. A library of 3′-monosubstituted, 5′-mono-substituted, and 3′,5′-disubstituted L-valine-, L-glutamate-, and glycine-ester prodrugs of N5 and N5-2OH was synthesized and representative structures (24–27) are shown in Figure 9. These prodrugs were specifically designed for ic. administration to treat high-grade brain tumors, such as glioblastoma multiforme by BNCT. The interstitial fluid (ISF) of the brain appears to have significantly lower protein (enzyme) concentrations than normal serum [70,71]. This could impede cleavage of ester-type prodrugs that would rely on esterase activity following ic. injection into brain ISF. Therefore, amino acid esters were chosen as promoieties for N5 and N5-2OH, as it is known that amino acid esters of nucleoside derivatives are susceptible to both chemical and enzymatic hydrolysis [72–75]. Overall, 5′-L-glutamate– and glycine–ester prodrugs of N5 and N5-2OH (25–27) had the best prodrug properties among all synthesized compounds. Their half-lives in bovine cerebrospinal fluid, a medium that presumably mimics the protein (enzyme) concentration of brain ISF, ranged from 0.96 to 2.11 h. The authors indicated that these half-lives should be suitable for ic. adminstration. In addition, aqueous solubilities of the prodrugs 25, 26, and 27 improved by factors ranging from approximately 40 to approximately 2360 compared with parental N5.
Figure 9.
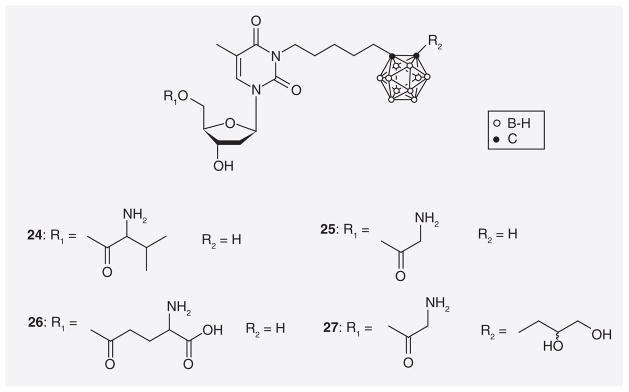
Amino acid ester prodrugs of N5 and N5-2OH.
Carborane cluster halogenated & radiohalogenated 3CTAs
The synthesis and biological evaluation of cage-halogenated and radiohalogenated derivatives of N5 were recently described by Tiwari et al. (28–34; Figure 10) [68]. Radiolabeled compounds, such as 32 and 33, have the potential to be used for tumor imaging and radiotherapy [35]. They may also serve in the real-time monitoring of compound concentrations in tumors during BNCT. Compounds 30/31 and 32/33 were synthesized by palladium-catalyzed halogen or isotope exchanges using either Na79/81Br or Na125I as the halogen source. The relative hTK1 PRs of compounds 28/29 and 30/31 were 29.6% and 36.9%, respectively, which was comparable with that of N5 in this specific study (38%). Thus, the addition of iodine or bromine atoms to the carborane cluster does not seem to have a significant effect on the phosphorylation of these 3CTAs by hTK1. In vitro uptake studies with N5, 28/29 and 30/31 in L929 TK1(+) and L929 TK1(−) cells resulted in preferential uptake of the compounds in the former cell line.
Figure 10.

Halogenated and radiohalogenated 3-carboranyl thymidine analogues.
Second-generation 3CTAs
3CTAs with ethyleneoxide moieties
Major shortcomings of N5-2OH (16) and other first-generation 3CTAs that were recognized during biological studies included suboptimal competition with endogenous thymidine at the substrate-binding site of hTK1 [24] and lack of water-solubility [69]. In addition, the capacity of first-generation 3CTAs to function as substrates for hTK1 seemed to be in need of improvement. Thus, Johnsamuel et al. initially attempted to enhance the pharmacodynamic and physiochemical properties of 3CTAs by using hydrophilic and highly flexible ethyleneoxide moieties in their synthesis (Figure 11) [76]. Compounds with ethyleneoxide spacer between carborane and thymidine scaffold (35–39) demonstrated slightly better relative hTK1 PRs (37–42%) than 3CTAs 40–45 (17–26%), which contained ethyleneoxide substituents at the second carbon of the carborane cluster. Overall, however, compounds 35–39 did not appear to be superior to N5-2OH as substrates of hTK1.
Figure 11.

3-carboranyl thymidine analogues containing ethyleneoxide moieties.
3CTAs containing polyhydroxylated functions
Tjarks et al. designed and synthesized a small library of 3CTAs containing cyclic and acyclic polyhydroxylated functions in the spacer between a p-carborane cluster and thymidine (46) or attached to the second carbon of this cage (47–49; Figure 12) [77,78]. Compounds 46–49 demonstrated relative hTK1 PRs ranging from 44–67%. In addition the rkcat /Km value of 46 was 57.4%, which was significantly higher than that previously reported for N5-2OH (35.8%) [59]. Unfortunately, compound 46 was a far less potent inhibitor of thymidine phosphorylation by hTK1 than N5-2OH (92 vs 17 μM).
Figure 12.

3-carboranyl thymidine analogues containing cyclic and acyclic polyhydroxylated functions.
3CTAs with triazole & tetrazole linkers
Lesnikowski et al. [79,80] synthesized a series of 3CTAs utilizing click chemistry [81] to incorporate a 1,2,3-triazole linker between carborane moiety and thymidine scaffold (50 & 51; Figure 13). The triazole group may participate in hydrogen bond formation between amino acids of the lasso loop and substrate thereby improving binding affinity. Compound 50 contains a metallacarborane moiety whereas 51 is substituted with nido-o-carborane. An evaluation of both compounds as potential substrates of hTK1 has not yet been published. Agarwal et al. recently synthesized a series of 3CTAs with tetrazolylmethyl- and tetrazolyl linker (52–57) based on the hypothesis that these hydrogen acceptor-rich structures may improve the binding affinity of 3CTAs to the active site of hTK1 [67]. Relative hTK1 phosphorylation rates of 52–57 ranged from 39.5–63.7%. A detailed comparative enzymatic evaluation indicated that compound 57 might be slightly superior to N5-2OH as a hTK1 substrate.
Figure 13.

3-carboranyl thymidine analogues with triazole and tetrazole spacers.
3CTAs containing amidinyl & guanidyl linkers
Agarwal et al. synthesized a series of 3CTAs with amidinyl and guanidyl groups in the spacer between the N3 position of thymidine and a m-carborane cluster (Figure 14) [67]. In this case, the hypothesis was that these groups might improve the binding affinity of the 3CTAs to the active site of hTK1 due to their hydrogen bond donating capacity. In addition, the highly basic properties of the amidinyl and guanidyl groups (58–62) should render these compounds water soluble under physiological aqueous conditions. Unfortunately, compounds 58–62 proved to be unstable under prolonged storage conditions and during enzyme assays, which prevented an accurate evaluation of their enzymatic and solubility properties. However, in the case of compound 61, phosphorylation by hTK1 could be confirmed by MS.
Figure 14.
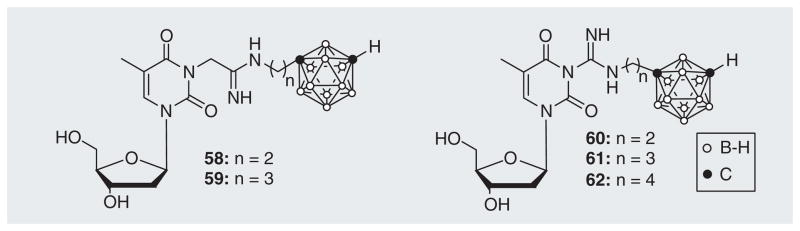
Amidinyl and guanidyl 3-carboranyl thymidine analogues.
3CTAs with amino & ammonium functions attached to the carborane cluster
In another attempt to improve the enzymatic properties and water solubility of 3CTAs, Byun et al. [32] synthesized a library of 12 analogues either with an amino group attached to closo-m-carborane (63–67,73; Figure 15) or an ammonium function attached to nido-m-carborane (68–72,74). Thus, the latter 3CTAs are zwitterionic structures. Both subseries appeared to be sufficiently lipophilic to enter into tumor cells via passive diffusion. In particular the compounds with ethylene linkers (63 & 68) had among the highest relative PRs observed for any 3CTAs (71.9% and 89.4%, respectively). In addition, compound 63 was approximately 103 times more soluble than N5-2OH in phosphate-buffered saline at pH 7.4 [HK Agarwal, Pers. Comm.]. Biodistribution studies of 69 in mice with subcutaneous L929 TK1(+) and L929 TK1(−) tumors resulted in intratumoral boron concentrations of 21.7 and 7.0 μg B/g tumor, respectively (3:1 ratio). These data were comparable with those obtained for N5-2OH in the same study. Interestingly, HPLC studies revealed two close peaks for 68 in chromatograms. A similar splitting phenomenon was observed for various signals in both 1H- and 13C-NMR spectra of 68. On the other hand, such splitting patterns were not observed for any of the other 3CTAs of this library indicating steric hindrance between the bulky nido-m-carborane cluster and the thymidine scaffold resulting in the formation of atropoisomers. Overall, the compounds of this library appeared to be the most promising second generation 3CTAs.
Figure 15.

3-carboranyl thymidine analogues with amino and ammonium groups at the carborane cage.
Non-boronated N3-substituted thymidine analogues
N3-alkyl & haloalkyl thymidine analogues
Several thymidine analogues with small alkyl groups at the N3-position were evaluated with respect to their CNS depressant (75 & 76), anti-nociceptive (75 & 76) and antiviral activities (77 & 78) by Yamamoto, Kitade, and Grierson and their collaborators (Figure 16) [82–85]. In most cases, however, interest in the exploration of non-boronated N3-substituted thymidine analogues appeared to be triggered by discoveries related to 3CTA research. These studies primarily focused on the design, synthesis and biological evaluation of thymidine analogues with radiolabels at the N3-postion for cancer therapy and imaging (Figures 16 & 17). Alauddin et al. reported the synthesis of a library of compounds with chloro-, bromo-, and iodo-substitutions at various positions of a N3-phenylalkyl side chain at thymidine (79–81) [86]. These agents may have been synthesized in preparation of a later synthesis of their radiohalogenated counterparts. Relative TK1 phosphorylation rates for some compounds of this series ranged from 2–6% [26]. Members of the research teams of Toyohara, Alauddin and Balzarini synthesized and evaluated several analogues with 18F or 19F attached to N3-alkyl side chains (82–87) [27,28,86–89,101]. Balzarini et al. observed that 83 was a relatively poor inhibitor of thymidine phosphorylation by TK1 [28]. Toyohara reported that 82–85 were effectively monophosphorylated by TK1 with rPRs ranging from 26–47% [27]. However, studies by the same group indicated that administration of 86 (the 18F counterpart of 83) into LL/2 Lewis lung carcinoma bearing mice did not result in tumor selective accumulation of the compound [87]. In contrast, Alauddin et al. observed that 86 and 87 were selectively retained in H441Gl tumors implanted into mice [88]. However, a review by the same group discusses the possibility that 83 was not a substrate of TK1 [27]. Alauddin et al. also synthesized compounds 88–91 [90]. No biological data have been published for these compounds as yet. Smith et al. [29,102] employed click chemistry to link 18F via a triazole moiety to the N3 position of thymidine (92 & 93). The corresponding 19F derivatives were also synthesized. Only the 19F derivative of 93 was moderately phosphorylated by TK1 [29].
Figure 16.

Various non-halogenated, halogenated and 18F-labeled N3-alkyl thymidine analogues.
Figure 17.

N3-substituted metal complex–thymidine conjugates.
Thymidine analogues conjugated at the N3-position with metal chelating functions
The synthesis and biological evaluation of several thymidine analogues conjugated at the N3 position to metal chelating complexes have been reported in recent years for potential applications in the radiotherapy and imaging of cancer [30,91–97]. Schmid et al. introduced 1,4,7,10-tetraazacyclododecane-1,4,7-tris(acetic acid) (DO3A) at the N3 position of thymidine to yield 3-(5-DO3A-pentyl)thymidine (D3T) (94; Figure 17) [91]. Compound 94 was radiolabeled with 68Ga and 111In to produce [68Ga]D3T and [111In]D3T. Cellular uptake of the 111In-labeled compound in DoHH2 and HL60 cells was low and phosphorylation by TK1 was not observed. Celen et al. synthesized N3-monoaminomonoamidodithiol-substituted thymidine derivative labeled with 99mTc (95) [98]. The metal-free compound was a poor inhibitor of thymidine phosphorylation by TK1. However, the 99mTc-labeled compound demonstrated some tumor selectivity in fibrosarcoma-bearing mice.
Zubieta et al. synthesized and evaluated a series of N3-Re(CO)3–thymidine conjugates (96; Figure 17) [93,94]. These compounds were generally poor inhibitors of thymidine phosphorylation by hTK1. Some derivatives of this library demonstrated significant cytotoxicity in A549 lung carcinoma cells in vitro. A substantial number of structurally different N3-Re(CO)3 – and N3-99mTc(CO)3–thymidine conjugates were prepared by Schibli et al. [30,92,95–97]. A representative structure is compound 97. Many of these compounds were good substrates of TK1. In the case of one of these agents, monophosphorylation by TK1 was confirmed by MS [92]. In vitro uptake in various cancer cell lines was generally low to moderate [30,97].
Thymidine conjugated at the N3-position with cancer chemotherapeutic drugs
Aspland et al. synthesized a series of thymidine derivatives N3-conjugated with paclitaxel or vinblastine as potential cancer therapeutics (98–107; Figure 18) [25]. Many of these conjugates were phosphorylated by TK1 with rPRs ranging from 13–68%. Several conjugates retained the microtubule-binding activities of unconjugated paclitaxel or vinblastine.
Figure 18.

Paclitaxel or vinblastine conjugated with thymidine.
Conclusion
During the last 15 years, 3CTAs have become one of the most widely studied pre-clinical BNCT agents. First-generation 3CTAs, such as N5 and N5-2OH, exhibited good relative hTK1 PRs, selective in vitro uptake in TK1(+) versus TK1(−) cells, and favorable in vivo biodistribution profiles leading to selective accumulation in tumors. N5-2OH proved to be effective in preclinical BNCT studies with rats bearing intracerebral brain tumors. Techniques were developed to use the carborane cluster of N5 as a platform for radiohalogens. Disadvantages of first-generation 3CTAs are that they demonstrate poor water solubility and limited capacity to compete with endogenous thymidine at the substrate-binding site of hTK1. These shortcomings could be overcome partially with the development of prodrug strategies and second-generation 3CTAs; the most promising, to date, appear to be those with amino substituents at the carborane cage. Research related to 3CTAs has sparked significant efforts in the development of thymidine conjugates with radiolabels in the N3 side chain. Promising biological data have also been reported in this new area of bioconjugate development for therapeutic and imaging applications. A remaining problem with second-generation 3CTAs and non-boronated radiolabeled N3-substituted thymidine analogues appears to be a very limited understanding of SARs, even within smaller sub-libraries of these compound classes. In addition, almost no information is available regarding the molecular interactions between N3-thymidine conjugates and their target, the hTK1 protein. In the case of some compounds conflicting data have been reported. These problems have to be addressed on the road to agents with improved capacities to act as substrates and inhibitors of hTK1.
Future perspective
3CTAs and other N3-substituted thymidine conjugates have significant potential in cancer therapy and imaging. However, an improved understanding of basic SARs has to be attained and structural biology and/or computational chemistry techniques have to be employed to gain insights into the molecular interactions between N3-thymidine conjugates and hTK1. The compound class might advance significantly towards clinical trials within the next 10 years provided these hurdles can be overcome and extended SAR information can be utilized to design novel derivatives with improved pharmacodynamic and pharmacokinetic properties.
Executive summary.
Background
The development of novel improved tumor-selective boron delivery vehicles has been one of the most pressing tasks of boron neutron capture therapy (BNCT) research.
3-carboranyl thymidine analogues (3CTAs), which, possibly, localize selectively in tumor cells by a process described as kinase-mediated trapping (KMT), are one of the most widely studied preclinical BNCT agents.
The general concept of using thymidine modified at the N3-position with a therapeutic or an imaging entity in the context of KMT has been validated successfully by recent studies on non-boronated N3-modified thymidine bioconjugates.
Carboranes
Hydrophobic closo-carborane and hydrophilic nido-carborane clusters have been used widely in the design and synthesis of 3CTAs.
Closo- and nido-carborane cages have found diverse applications not only in the development of 3CTAs.
TK1-like enzymes
hTK1 is the primary molecular target of 3CTAs.
hTK1 is only active in proliferating cells causing tumor-selective KMT of 3CTAs via 5′-monophosphorylation.
5-carboranyl- & 3′-aminocyanoborane-substituted pyrimidine nucleoside analogues for BNCT
These agents are the products of early design strategies for boronated nucleosides as boron delivery agents for BNCT.
First-generation 3CTAs
Among the first-generation 3CTAs, N5-2OH was the best substrate of hTK1 leading to successful preclinical BNCT studies with rats bearing intracerebral brain tumors.
Disadvantages of first-generation 3CTAs were poor water solubility, suboptimal competition with endogenous thymidine at the hTK1 binding site, as well as incomplete understanding of structure–activity relationships and molecular interactions between 3CTAs and hTK1.
Second-generation 3CTAs
3CTAs with various structural elements, including ethylene oxide, polyhydroxyl, triazolyl, tetrazolyl, amidinyl and guanidyl and amino moieties, were synthesized and evaluated for improved pharmacodynamic and pharmacokinetic properties.
3CTAs with amino and ammonium functions attached to the carborane cluster appeared to be the most promising second-generation 3CTAs.
Non-boronated N3-substituted thymidine analogues
The development of non-boronated N3-subtituted thymidine analogues for areas of cancer therapy other than BNCT as well as for tumor imaging is very promising but in an early stage.
Future perspective
3CTAs and non-boronated N3-substituted thymidine conjugates have great potential in cancer therapy and imaging provided currently encountered shortcomings can be overcome and missing information can be filled in.
Key Terms
- Kinase-mediated trapping
Selective uptake of cancer therapeutic and imaging agents in tumor cells through phosphorylation by intracellular kinases
- Lasso loop
Flexible region of human thymidine kinase 1 (hTK1) that covers the substrate binding site of the enzyme. It is comprised of the amino acids Leu166 to Lys 180
Footnotes
For reprint orders, please contact reprints@future-science.com
Disclaimer
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the other mentioned organizations and institutions.
Financial & competing interests disclosure
The preparation of the present paper was supported by funds from The Ohio State University College of Pharmacy, the Libyan Ministry of Higher Education and Scientific Research (per administration of the Canadian Bureau for International Education for T Ali), and NIH grant 5R01 CA127935. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Barth RF, Coderre JA, Vicente MG, Blue TE. Boron neutron capture therapy of cancer: current status and future prospects. Clin Cancer Res. 2005;11:3987–4002. doi: 10.1158/1078-0432.CCR-05-0035. [DOI] [PubMed] [Google Scholar]
- 2.Soloway AH, Tjarks W, Barnum BA, et al. The chemistry of neutron capture therapy. Chem Rev. 1998;98:1515–1562. doi: 10.1021/cr941195u. [DOI] [PubMed] [Google Scholar]
- 3.Das BC, Das S, Li G, Bao W, Kabalka GW. Synthesis of a water soluble carborane containing amino acid as a potential therapeutic agent. Synlett. 2001;9:1419–1420. [Google Scholar]
- 4.Cai J, Soloway AH, Barth RF, et al. Boron containing polyamines as DNA targeting agents for neutron capture therapy of brain tumors: synthesis and biological evaluation. J Med Chem. 1997;40:3887–3896. doi: 10.1021/jm960787x. [DOI] [PubMed] [Google Scholar]
- 5.Zhuo J-C, Cai J, Soloway AH, et al. Synthesis and biological evaluation of boron containing polyamines as potential agents for neutron capture therapy of brain tumors. J Med Chem. 1999;42:1282–1292. doi: 10.1021/jm980703f. [DOI] [PubMed] [Google Scholar]
- 6.Tietze LF, Griesbach U, Bothe U, Nakamura H, Yamamoto Y. Novel carboranes with a DNA binding unit for the treatment of cancer by boron neutron capture therapy. Chembiochem. 2002;3:219–225. doi: 10.1002/1439-7633(20020301)3:2/3<219::aid-cbic219>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 7.Crossley EL, Ziolkowski EJ, Coderre JA, Rendina LM. Boronated DNA-binding compounds as potential agents for boron neutron capture therapy. Mini Rev Med Chem. 2007;7:303–313. doi: 10.2174/138955707780059808. [DOI] [PubMed] [Google Scholar]
- 8.Isaac MF, Kahl SB. Synthesis of ether- and carbon-linked polycarboranyl porphyrin dimers for cancer therapies. J Organomet Chem. 2003;680:232–243. [Google Scholar]
- 9.Jori G, Soncin M, Friso E, et al. A novel boronated-porphyrin as a radio-sensitizing agent for boron neutron capture therapy of tumors: in vitro and in vivo studies. Appl Radiat Isot. 2009;67:S321–S324. doi: 10.1016/j.apradiso.2009.03.071. [DOI] [PubMed] [Google Scholar]
- 10.Renner MW, Miura M, Easson MW, Vicente MGH. Recent progress in the syntheses and biological evaluation of boronated porphyrins for boron neutron-capture therapy. Anti-Cancer Agents Med Chem. 2006;6:145–157. doi: 10.2174/187152006776119135. [DOI] [PubMed] [Google Scholar]
- 11.Tietze LF, Griesbach U, Schuberth I, Bothe U, Marra A, Dondoni A. Novel carboranyl C-glycosides for the treatment of cancer by boron neutron capture therapy. Chemistry. 2003;9:1296–1302. doi: 10.1002/chem.200390148. [DOI] [PubMed] [Google Scholar]
- 12.Bregadze V, Semioshkin A, Sivaev I. Synthesis of conjugates of polyhedral boron compounds with tumor-seeking molecules for neutron capture therapy. Appl Radiat Isot. 2011;69:1774–1777. doi: 10.1016/j.apradiso.2011.01.043. [DOI] [PubMed] [Google Scholar]
- 13.Bregadze VI, Sivaev IB. Polyhedral boron compounds for BNCT. In: Hosmane NS, editor. Boron Science. CRC Press; FL, USA: 2012. pp. 181–207. [Google Scholar]
- 14.Orlova AV, Kononov LO. Synthesis of conjugates of polyhedral boron compounds with carbohydrates. Russ Chem Rev. 2009;78:629–642. [Google Scholar]
- 15.Olsson P, Gedda L, Goike H, et al. Uptake of a boronated epidermal growth factor-dextran conjugate in CHO xenografts with and without human EGF-receptor expression. Anticancer Drug Des. 1998;13:279–289. [PubMed] [Google Scholar]
- 16.Yang W, Barth RF, Wu G, Tjarks W, Binns P, Riley K. Boron neutron capture therapy of EGFR or EGFRvIII positive gliomas using either boronated monoclonal antibodies or epidermal growth factor as molecular targeting agents. Appl Radiat Isot. 2009;67:S328–S331. doi: 10.1016/j.apradiso.2009.03.030. [DOI] [PubMed] [Google Scholar]
- 17.Yang W, Wu G, Barth RF, et al. Molecular targeting and treatment of composite EGFR and EGFRvIII-Positive gliomas using boronated monoclonal antibodies. Clin Cancer Res. 2008;14:883–891. doi: 10.1158/1078-0432.CCR-07-1968. [DOI] [PubMed] [Google Scholar]
- 18.Carlsson J, Kullberg EB, Capala J, Sjoberg S, Edwards K, Gedda L. Ligand liposomes and boron neutron capture therapy. J Neurooncol. 2003;62:47–59. doi: 10.1007/BF02699933. [DOI] [PubMed] [Google Scholar]
- 19.Justus E, Awad D, Hohnholt M, et al. Synthesis, liposomal preparation, and in vitro toxicity of two novel dodecaborate cluster lipids for boron neutron capture therapy. Bioconjug Chem. 2007;18:1287–1293. doi: 10.1021/bc070040t. [DOI] [PubMed] [Google Scholar]
- 20.Altieri S, Balzi M, Bortolussi S, et al. Carborane derivatives loaded into liposomes as efficient delivery systems for boron neutron capture therapy. J Med Chem. 2009;52:7829–7835. doi: 10.1021/jm900763b. [DOI] [PubMed] [Google Scholar]
- 21.Byun Y, Narayanasamy S, Johnsamuel J, et al. 3-Carboranyl thymidine analogues (3CTAs) and other boronated nucleosides for boron neutron capture therapy. Anticancer Agents Med Chem. 2006;6:127–144. doi: 10.2174/187152006776119171. [DOI] [PubMed] [Google Scholar]
- 22.Nerini-Molteni S, Seregni E, Crippa F, Maffioli L, Botti C, Bombardieri E. Uptake of tritiated thymidine, deoxyglucose and methionine in three lung cancer cell lines: deoxyglucose uptake mirrors tritiated thymidine uptake. Tumour Biol. 2001;22:92–96. doi: 10.1159/000050602. [DOI] [PubMed] [Google Scholar]
- 23.Zhang CC, Yan Z, Li W, et al. [18F]FLT-PET imaging does not always “light up” proliferating tumor cells. Clin Cancer Res. 2012;18:1303–1312. doi: 10.1158/1078-0432.CCR-11-1433. [DOI] [PubMed] [Google Scholar]
- 24.Tjarks W, Tiwari R, Byun Y, Narayanasamy S, Barth RF. Carboranyl thymidine analogues for neutron capture therapy. Chem Commun. 2007;47:4978–4991. doi: 10.1039/b707257k. [DOI] [PubMed] [Google Scholar]
- 25.Aspland SE, Ballatore C, Castillo R, et al. Kinase-mediated trapping of bi-functional conjugates of paclitaxel or vinblastine with thymidine in cancer cells. Bioorg Med Chem Lett. 2006;16:5194–5198. doi: 10.1016/j.bmcl.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh P, Pal A, Shavrin A, Bornmann W, Gelovani JG, Alauddi MM. Synthesis of N3-substituted thymidine analogues for measurement of cellular kinase activity. Med Chem. 2008;4:503–512. doi: 10.2174/157340608785700252. [DOI] [PubMed] [Google Scholar]
- 27.Toyohara J, Hayashi A, Gogami A, et al. Alkyl-fluorinated thymidine derivatives for imaging cell proliferation I. The in vitro evaluation of some alkyl-fluorinated thymidine derivatives. Nucl Med Biol. 2006;33:751–764. doi: 10.1016/j.nucmedbio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Balzarini J, Celen S, Karlsson A, De Groot T, Verbruggen A, Bormans G. The effect of a methyl or 2-fluoroethyl substituent at the N-3 position of thymidine, 3′-fluoro-3′-deoxythymidine and 1-beta-D-arabinosylthymine on their antiviral and cytostatic activity in cell culture. Antivir Chem Chemother. 2006;17:17–23. doi: 10.1177/095632020601700103. [DOI] [PubMed] [Google Scholar]
- 29.Smith G, Sala R, Carroll L, et al. Synthesis and evaluation of nucleoside radiotracers for imaging proliferation. Nucl Med Biol. 2012;39:652–665. doi: 10.1016/j.nucmedbio.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Desbouis D, Struthers H, Spiwok V, Kuster T, Schibli R. Synthesis, in vitro, and in silico evaluation of organometallic technetium and rhenium thymidine complexes with retained substrate activity toward human thymidine kinase type 1. J Med Chem. 2008;51:6689–6698. doi: 10.1021/jm800530p. [DOI] [PubMed] [Google Scholar]
- 31.Hosmane NS. Boron Science: New Technologies and Applications. CRC Press; FL, USA: 2012. [Google Scholar]
- 32▪▪.Byun Y, Yan J, Al-Madhoun AS, et al. Synthesis and biological evaluation of neutral and zwitterionic 3-carboranyl thymidine analogues for boron neutron capture therapy. J Med Chem. 2005;48:1188–1198. doi: 10.1021/jm0491896. Describes very promising second-generation 3-carboranyl thymidine analogues (3CTAs) with amino and ammonium functions attached to the carborane cluster. [DOI] [PubMed] [Google Scholar]
- 33.Issa F, Kassiou M, Rendina LM. Boron in drug discovery: carboranes as unique pharmacophores in biologically active compounds. Chem Rev. 2011;111:5701–5722. doi: 10.1021/cr2000866. [DOI] [PubMed] [Google Scholar]
- 34.Scholz M, Hey-Hawkins E. Carbaboranes as pharmacophores: properties, synthesis, and application strategies. Chem Rev. 2011;111:7035–7062. doi: 10.1021/cr200038x. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal HK, Hasabelnaby S, Tiwari R, Tjarks W. Boron cluster (Radio) halogenation in biomedical research. In: Hosmane NS, editor. Boron Science. CRC Press; FL, USA: 2012. pp. 107–144. [Google Scholar]
- 36.Welin M, Kosinska U, Mikkelsen NE, et al. Structures of thymidine kinase 1 of human and mycoplasmic origin. Proc Natl Acad Sci USA. 2004;101:17970–17975. doi: 10.1073/pnas.0406332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosinska U, Carnrot C, Eriksson S, Wang L, Eklund H. Structure of the substrate complex of thymidine kinase from Ureaplasma urealyticum and investigations of possible drug targets for the enzyme. FEBS J. 2005;272:6365–6372. doi: 10.1111/j.1742-4658.2005.05030.x. [DOI] [PubMed] [Google Scholar]
- 38.Segura-Pena D, Lutz S, Monnerjahn C, Konrad M, Lavie A. Binding of ATP to TK1-like enzymes is associated with a conformational change in the quaternary structure. J Mol Biol. 2007;369:129–141. doi: 10.1016/j.jmb.2007.02.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birringer MS, Claus MT, Folkers G, Kloer DP, Schulz GE, Scapozza L. Structure of a type II thymidine kinase with bound dTTP. FEBS Lett. 2005;579:1376–1382. doi: 10.1016/j.febslet.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 40.Segura-Pena D, Lichter J, Trani M, Konrad M, Lavie A, Lutz S. Quaternary structure change as a mechanism for the regulation of thymidine kinase 1-like enzymes. Structure. 2007;15:1555–1566. doi: 10.1016/j.str.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kosinska U, Carnrot C, Sandrini MP, et al. Structural studies of thymidine kinases from Bacillus anthracis and Bacillus cereus provide insights into quaternary structure and conformational changes upon substrate binding. FEBS J. 2007;274:727–737. doi: 10.1111/j.1742-4658.2006.05617.x. [DOI] [PubMed] [Google Scholar]
- 42.El Omari K, Solaroli N, Karlsson A, Balzarini J, Stammers DK. Structure of vaccinia virus thymidine kinase in complex with dTTP: insights for drug design. BMC Struct Biol. 2006;6:22. doi: 10.1186/1472-6807-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Madhoun AS, Tjarks W, Eriksson S. The role of thymidine kinases in the activation of pyrimidine nucleoside analogues. Mini Rev Med Chem. 2004;4:341–350. doi: 10.2174/1389557043403963. [DOI] [PubMed] [Google Scholar]
- 44.Arner ES, Eriksson S. Mammalian deoxyribonucleoside kinases. Pharmacol Ther. 1995;67:155–186. doi: 10.1016/0163-7258(95)00015-9. [DOI] [PubMed] [Google Scholar]
- 45.Eriksson S, Munch-Petersen B, Johansson K, Eklund H. Structure and function of cellular deoxyribonucleoside kinases. Cell Mol Life Sci. 2002;59:1327–1346. doi: 10.1007/s00018-002-8511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Priego EM, Karlsson A, Gago F, Camarasa MJ, Balzarini J, Perez-Perez MJ. Recent advances in thymidine kinase 2 (TK2) inhibitors and new perspectives for potential applications. Curr Pharm Des. 2012;18:2981–2994. doi: 10.2174/138161212800672787. [DOI] [PubMed] [Google Scholar]
- 47.Kauffman MG, Kelly TJ. Cell cycle regulation of thymidine kinase: residues near the carboxyl terminus are essential for the specific degradation of the enzyme at mitosis. Mol Cell Biol. 1991;11:2538–2546. doi: 10.1128/mcb.11.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eriksson S, Kierdaszuk B, Munch-Petersen B, Oberg B, Johansson NG. Comparison of the substrate specificities of human thymidine kinase 1 and 2 and deoxycytidine kinase toward antiviral and cytostatic nucleoside analogs. Biochem Biophys Res Commun. 1991;176:586–592. doi: 10.1016/s0006-291x(05)80224-4. [DOI] [PubMed] [Google Scholar]
- 49.Hanan S, Jagarlamudi KK, Liya W, Ellen H, Staffan E. Quaternary structures of recombinant, cellular, and serum forms of thymidine kinase 1 from dogs and humans. BMC Biochem. 2012;13:12. doi: 10.1186/1471-2091-13-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johansson NG, Eriksson S. Structure-activity relationships for phosphorylation of nucleoside analogs to monophosphates by nucleoside kinases. Acta Biochim Pol. 1996;43:143–160. [PubMed] [Google Scholar]
- 51.Bodei L, Kassis AI, Adelstein SJ, Mariani G. Radionuclide therapy with iodine-125 and other auger-electron-emitting radionuclides: experimental models and clinical applications. Cancer Biother Radiopharm. 2003;18:861–877. doi: 10.1089/108497803322702833. [DOI] [PubMed] [Google Scholar]
- 52.Russo A, Gianni L, Kinsella TJ, et al. Pharmacological evaluation of intravenous delivery of 5-bromodeoxyuridine to patients with brain tumors. Cancer Res. 1984;44:1702–1705. [PubMed] [Google Scholar]
- 53.Schinazi RF, Prusoff WH. Synthesis of 5-(dihydroxyboryl)-2′-deoxyuridine and related boron containing pyrimidines. J Org Chem. 1985;50:841–847. [Google Scholar]
- 54.Goudgaon NM, Fulcrand E-KG, Schinazi RF. Boron containing pyrimidines, nucleosides, and oligonucleotides for neutron capture therapy. Nucleosides Nucleotides. 1994;13:849–880. [Google Scholar]
- 55.Yamamoto Y, Seko T, Nakamura H, et al. Synthesis of carboranes containing nucleoside bases. Unexpectedly high cytostatic and cytocidal toxicity towards cancer cells. Chem Comm. 1992;2:157–158. [Google Scholar]
- 56▪.Lunato AJ, Wang J, Woollard JE, et al. Synthesis of 5-(carboranylalkylmercapto)-2′-deoxyuridines and 3-(carboranylalkyl) thymidines and their evaluation as substrates for human thymidine kinases 1 and 2. J Med Chem. 1999;42:3378–3389. doi: 10.1021/jm990125i. Describes the synthesis and biological evaluation of the first 3CTAs. [DOI] [PubMed] [Google Scholar]
- 57.Lesnikowski ZJ, Shi J, Schinazi RF. Nucleic acids and nucleosides containing carboranes. J Organomet Chem. 1999;581:156–169. [Google Scholar]
- 58.Burnham BS, Chen SY, Sood A, Spielvogel BF, Miller MC, 3rd, Hall IH. The cytotoxicity of 3′-aminocyanoborane-2′, 3′-dideoxypyrimidines in murine and human tissue cultured cell lines. Anticancer Res. 1995;15:951–958. [PubMed] [Google Scholar]
- 59▪▪.Al-Madhoun AS, Johnsamuel J, Barth RF, Tjarks W, Eriksson S. Evaluation of human thymidine kinase 1 substrates as new candidates for boron neutron capture therapy. Cancer Res. 2004;64:6280–6286. doi: 10.1158/0008-5472.CAN-04-0197. Describes important enzymatic, in vitro and in vivo studies of first-generation 3CTAs. [DOI] [PubMed] [Google Scholar]
- 60▪▪.Barth RF, Yang W, Al-Madhoun AS, et al. Boron containing nucleosides as potential delivery agents for neutron capture therapy of brain tumors. Cancer Res. 2004;64:6287–6295. doi: 10.1158/0008-5472.CAN-04-0437. Describes important in vitro and in vivo studies of first-generation 3CTAs. [DOI] [PubMed] [Google Scholar]
- 61.Byun Y, Thirumamagal BT, Yang W, Eriksson S, Barth RF, Tjarks W. Preparation and biological evaluation of 10B-enriched 3-[5-{2-(2,3-dihydroxyprop-1-yl)-o-carboran-1-yl}pentan-1-yl]thymidine (N5–2OH), a new boron delivery agent for boron neutron capture therapy of brain tumors. J Med Chem. 2006;49:5513–5523. doi: 10.1021/jm060413w. [DOI] [PubMed] [Google Scholar]
- 62▪▪.Barth RF, Yang W, Wu G, et al. Thymidine kinase 1 as a molecular target for boron neutron capture therapy of brain tumors. Proc Natl Acad Sci USA. 2008;105:17493–17497. doi: 10.1073/pnas.0809569105. Describes important 3CTA-based preclinical boron neutron capture therapy studies of rats with intracerebral brain tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia. 2001;15:875–890. doi: 10.1038/sj.leu.2402114. [DOI] [PubMed] [Google Scholar]
- 64.Lowe CR, Incropera FP, DeWitt DP, Dean PDG. In: Affinity Chromatography. Lowe CR, Dean PDG, editors. Wiley-Interscience; NY, USA: 1974. [Google Scholar]
- 65.Lowe CR, Harvey MJ, Craven DB, Dean PD. Some parameters relevant to affinity chromatography on immobilized nucleotides. Biochem J. 1973;133:499–506. doi: 10.1042/bj1330499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grobner P, Loidl P. Thymidine kinase. A novel affinity chromatography of the enzyme and its regulation by phosphorylation in Physarum polycephalum. J Biol Chem. 1984;259:8012–8014. [PubMed] [Google Scholar]
- 67.Agarwal HK, McElroy CA, Sjuvarsson E, Eriksson S, Darby MV, Tjarks W. Synthesis of N3-substituted carboranyl thymidine bioconjugates and their evaluation as substrates of recombinant human thymidine kinase 1. Eur J Med Chem. 2013;60:456–468. doi: 10.1016/j.ejmech.2012.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tiwari R, Toppino A, Agarwal HK, et al. Synthesis, biological evaluation, and radioiodination of halogenated closo-carboranylthymidine analogues. Inorg Chem. 2011;51:629–639. doi: 10.1021/ic202150b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hasabelnaby S, Goudah A, Agarwal HK, Abd Alla MS, Tjarks W. Synthesis, chemical and enzymatic hydrolysis, and aqueous solubility of amino acid ester prodrugs of 3-carboranyl thymidine analogs for boron neutron capture therapy of brain tumors. Eur J Med Chem. 2012;55:325–334. doi: 10.1016/j.ejmech.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davson H, Segal Mb. Physiology of the CSF and blood–brain barriers. CRC Press; FL, USA: 1996. [Google Scholar]
- 71.Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45:545–552. doi: 10.1016/j.neuint.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 72.Song X, Lorenzi PL, Landowski CP, Vig BS, Hilfinger JM, Amidon GL. Amino acid ester prodrugs of the anticancer agent gemcitabine: synthesis, bioconversion, metabolic bioevasion, and hPEPT1-mediated transport. Mol Pharm. 2005;2:157–167. doi: 10.1021/mp049888e. [DOI] [PubMed] [Google Scholar]
- 73.Tsume Y, Vig BS, Sun J, et al. Enhanced absorption and growth inhibition with amino acid monoester prodrugs of floxuridine by targeting hPEPT1 transporters. Molecules. 2008;13:1441–1454. doi: 10.3390/molecules13071441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vig BS, Lorenzi PJ, Mittal S, et al. Amino acid ester prodrugs of floxuridine: synthesis and effects of structure, stereochemistry, and site of esterification on the rate of hydrolysis. Pharm Res. 2003;20:1381–1388. doi: 10.1023/a:1025745824632. [DOI] [PubMed] [Google Scholar]
- 75.Sun Y, Sun J, Shi S, et al. Synthesis, transport and pharmacokinetics of 5′-amino acid ester prodrugs of 1-beta-D-arabinofuranosylcytosine. Mol Pharm. 2009;6:315–325. doi: 10.1021/mp800200a. [DOI] [PubMed] [Google Scholar]
- 76.Johnsamuel J, Lakhi N, Al-Madhoun AS, et al. Synthesis of ethyleneoxide modified 3-carboranyl thymidine analogues and evaluation of their biochemical, physicochemical, and structural properties. Bioorg Med Chem. 2004;12:4769–4781. doi: 10.1016/j.bmc.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 77.Thirumamagal BT, Johnsamuel J, Cosquer GY, et al. Boronated thymidine analogues for boron neutron capture therapy. Nucleosides Nucleotides Nucleic Acids. 2006;25:861–866. doi: 10.1080/15257770600793844. [DOI] [PubMed] [Google Scholar]
- 78.Narayanasamy S, Thirumamagal BT, Johnsamuel J, et al. Hydrophilically enhanced 3-carboranyl thymidine analogues (3CTAs) for boron neutron capture therapy (BNCT) of cancer. Bioorg Med Chem. 2006;14:6886–6899. doi: 10.1016/j.bmc.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 79.Semioshkin A, Laskova J, Wojtczak B, et al. Synthesis of closo-dodecaborate based nucleoside conjugates. J Organomet Chem. 2009;694:1375–1379. [Google Scholar]
- 80.Wojtczak BA, Andrysiak A, Gruener B, Lesnikowski ZJ. ‘Chemical ligation’: a versatile method for nucleoside modification with boron clusters. Chem Eur J. 2008;14:10675–10682. doi: 10.1002/chem.200801053. [DOI] [PubMed] [Google Scholar]
- 81.Padwa A. 1,3-Dipolar Cycloaddition Chemistry. Vol. 1. John Wiley and Sons; NY, USA: 1984. [Google Scholar]
- 82.Kimura T, Watanabe K, Tateoka Y, Kondo S, Ho IK, Yamamoto I. Preparation and pharmacological evaluation of N3-substituted thymidine derivatives as central depressants. Chem Pharm Bull. 1993;41:1180–1182. doi: 10.1248/cpb.41.1180. [DOI] [PubMed] [Google Scholar]
- 83.Shimizu T, Kimura T, Funahashi T, Watanabe K, Ho IK, Yamamoto I. Synthesis of N3-substituted uridine and related pyrimidine nucleosides and their antinociceptive effects in mice. Chem Pharm Bull. 2005;53:313–318. doi: 10.1248/cpb.53.313. [DOI] [PubMed] [Google Scholar]
- 84.Kitade Y, Suzuki A, Hirota K, et al. Synthesis and anti-human immunodeficiency virus type 1 (HIV-1) activity of 3-substituted derivatives of 3′-azido-3′-deoxythymidine (AZT), and inhibition of HIV-1 reverse transcriptase by their 5′-triphosphates. Chem Pharm Bull. 1992;40:920–924. doi: 10.1248/cpb.40.920. [DOI] [PubMed] [Google Scholar]
- 85.Adams DR, Perez C, Maillard M, et al. Preparation and anti-HIV activity of N-3-substituted thymidine nucleoside analogs. J Med Chem. 1997;40:1550–1558. doi: 10.1021/jm9600095. [DOI] [PubMed] [Google Scholar]
- 86.Alauddin MM, Ghosh P, Gelovani JG. Synthesis of [18F]-labeled N-3(substituted) thymidine analogs: N-3([18F]fluorobutyl) thymidine ([18F]-FBT) and N-3([18F] fluoropentyl) thymidine ([18F]-FPT) for PET. J Labelled Compd Radiopharm. 2006;49:1079–1088. [Google Scholar]
- 87.Toyohara J, Hayashi A, Gogami A, Fujibayashi Y. Alkyl-fluorinated thymidine derivatives for imaging cell proliferation II. Synthesis and evaluation of N3-(2-[18F] fluoroethyl)-thymidine. Nucl Med Biol. 2006;33:765–772. doi: 10.1016/j.nucmedbio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 88.Mukhopadhyay U, Soghomonyan S, Yeh HH, et al. N(3)-Substituted thymidine analogues V: synthesis and preliminary PET imaging of N(3)-[18F]fluoroethyl thymidine and N(3)-[18F]fluoropropyl thymidine. Nucl Med Biol. 2008;35:697–705. doi: 10.1016/j.nucmedbio.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 89.Ghosh P, Gelovani JG, Alauddin MM. N3-Substituted thymidine analogues. Part III: radiosynthesis of N3-[(4-[18F] fluoromethyl-phenyl)butyl]thymidine ([18F]-FMPBT) and N3-[(4-[18F] fluoromethyl-phenyl)pentyl] thymidine ([18F]-FMPPT) for PET. J Labelled Compd Radiopharm. 2007;50:1185–1191. [Google Scholar]
- 90.Ghosh P, Gelovani JG, Alauddin MM. N3-substituted thymidine analogues: radiosyntheses of N3-[4-(4-(2-[18F] fluoroethyl)phenyl)butyl]thymidine and N3-[5-(4-(2-[18F]fluoroethyl)phenyl)pentyl] thymidine for PET. Curr Radiopharm. 2009;2:2–8. [Google Scholar]
- 91.Schmid M, Neumaier B, Vogg AT, et al. Synthesis and evaluation of a radiometal-labeled macrocyclic chelator-derivatised thymidine analog. Nucl Med Biol. 2006;33:359–366. doi: 10.1016/j.nucmedbio.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 92.Mindt T, Struthers H, Garcia-Garayoa E, Desbouis D, Schibli R. Strategies for the development of novel tumor targeting technetium and rhenium radiopharmaceuticals. Chimia. 2007;61:725–731. [Google Scholar]
- 93.Bartholoma MD, Vortherms AR, Hillier S, et al. Synthesis, cytotoxicity, and insight into the mode of action of Re(CO)3 thymidine complexes. ChemMedChem. 2010;5:1513–1529. doi: 10.1002/cmdc.201000196. [DOI] [PubMed] [Google Scholar]
- 94.Bartholomae MD, Vortherms AR, Hillier S, et al. Synthesis, cytotoxicity and cellular uptake studies of N3 functionalized Re(CO)3 thymidine complexes. Dalton Trans. 2011;40:6216–6225. doi: 10.1039/c0dt01452d. [DOI] [PubMed] [Google Scholar]
- 95.Struthers H, Hagenbach A, Abram U, Schibli R. Organometallic [Re(CO)3]+ and [Re(CO)2(NO)]2+ labeled substrates for human thymidine kinase 1. Inorg Chem. 2009;48:5154–5163. doi: 10.1021/ic9000126. [DOI] [PubMed] [Google Scholar]
- 96.Struthers H, Spingler B, Mindt TL, Schibli R. “Click-to-chelate”: design and incorporation of triazole-containing metal-chelating systems into biomolecules of diagnostic and therapeutic interest. Chem Eur J. 2008;14:6173–6183. doi: 10.1002/chem.200702024. [DOI] [PubMed] [Google Scholar]
- 97.Struthers H, Viertl D, Kosinski M, Spingler B, Buchegger F, Schibli R. Charge dependent substrate activity of C3′ and N3 functionalized, organometallic technetium and rhenium-labeled thymidine derivatives toward human thymidine kinase 1. Bioconjug Chem. 2010;21:622–634. doi: 10.1021/bc900380n. [DOI] [PubMed] [Google Scholar]
- 98.Celen S, De Groot T, Balzarini J, et al. Synthesis and evaluation of a 99mTc-MAMA-propyl-thymidine complex as a potential probe for in vivo visualization of tumor cell proliferation with SPECT. Nucl Med Biol. 2007;34:283–291. doi: 10.1016/j.nucmedbio.2007.01.003. [DOI] [PubMed] [Google Scholar]
Patents
- 101.Bornmann W, Alauddin M, Gelovani J, et al. WO2008024826A2. 2008
- 102.Aboagye EO, Smith G. WO2010023457A1. 2010