

Abstract
Removal of apoptotic cells and cellular debris by phagocytosis is essential for development, tissue homeostasis and resolution of inflammation. Eat-me signals control the initiation of phagocytosis, holding a key to our understanding of phagocyte biology. Due to lack of functional cloning strategy, eat-me signals are conventionally identified and characterized on a case-by-case basis. To investigate the feasibility of functionally cloning eat-me signals by phage display, we characterize the biological behavior of T7 phages displaying two well-known eat-me signals: growth arrest-specific gene 6 (Gas6) and milk fat globule-EGF8 (MFG-E8). Gas6-phage binds to all three known Gas6 receptors, Mer, Axl and Tyro3 receptor tyrosine kinases. Gas6-phage and MFG-E8-phage are capable of binding to phagocytes and non-phagocytes. However, both phages stimulate phage uptake only in phagocytes, including macrophages, microglia and retinal pigment epithelium (RPE) cells, but not in non-phagocytes. Furthermore, functional phage selection by phagocytosis in phagocytes enriches both Gas6-phage and MFG-E8-phage, suggesting that phage display can be used as a tool to functionally identify unknown eat-me signals from phage display cDNA library.
Keywords: Eat-me signals, phagocytosis, phage display, Gas6, MFG-E8
Introduction
Programmed cell death is essential for many aspects of the immune response, morphogenesis, cell homeostasis and tissue repair. As an estimated 1010 cells die every day in our body,1, 2 removal of apoptotic cells by phagocytosis is a critical biological process. Phagocytosis is a complex event with many signaling molecules involved.3, 4 The entire event is initiated with the binding of “eat-me” signals or recognition ligands on apoptotic cells to phagocytic receptors on phagocytes. In this regard, eat-me signals hold a key to our understanding of biological roles of phagocytosis.
Among a limited number of known eat-me signals, phosphatidylserine (PS) is the most well-characterized and is normally located on the inner leaflet of the plasma membrane in healthy cells. During apoptosis, PS flips across the plasma membrane, displays on the surface of apoptotic cells, and serves as an eat-me signal by eliciting phagocytosis either directly or indirectly. PS may directly bind to its receptors on phagocytes, such as BAI1 and TIM4,5, 6 or indirectly induce phagocytosis through bridging molecules like growth arrest-specific gene 6 (Gas6), protein S and milk fat globule-EGF8 (MFG-E8).3, 7 These bridging molecules have two binding domains, one for PS and the other for their cognate receptors on phagocytes. For example, Gas6 and protein S with similar molecular structure bind to PS through their N-terminal 11 γ-carboxyglutamic acid residues and interact with the members of Axl receptor tyrosine kinase (RTK) family through the C-terminal two globular laminin G-like (2 × LG) domains. Axl RTK family includes Mer, Axl and Tyro3.8 Mutations in Mer cause defect in retinal pigment epithelium (RPE) cell phagocytosis, leading to retinal degeneration in humans and the Royal College of Surgeons (RCS) rats.9, 10 Deletion of Mer, Axl and/or Tyro3 results in severe defect in phagocytosis and autoimmune diseases.11 MFG-E8 recognizes PS through its C-terminal discoidin-like domain and induces phagocytosis by binding to αvβ3 or αvβ5 integrin through its Arg-Gly-Asp (RGD) motif.7, 12 Several proteins, such as intercellular adhesion molecule 3 (ICAM3) and calreticulin, have been characterized as PS-independent eat-me signals by directly binding to their cognate receptors on phagocytes.4, 13 So far, limited number of known eat-me signals were all identified on a case-by-case basis. As a result, it is a daunting challenge to identify unknown eat-me signals. Developing a functional cloning strategy to systemically identify unknown eat-me signals, including the essential Mer-binding ligand(s) other than Gas6 to prevent retinal degeneration,14 is critical for our understanding of phagocyte biology and will facilitate drug discovery for phagocytosis-related disorders.
A few recent studies showed that short peptides or single-chain fragment variable (scFv) antibodies (Abs) with internalization capacity in cancer cell lines can be identified by phage display from random peptide libraries or scFv libraries.15–18 The main purpose of these studies was to develop guiding peptides or scFvs capable of delivering therapeutic reagents into cancer cells. We speculate that this approach may be applicable to identify eat-me signaling molecules in phagocytes from phage display cDNA libraries. In this study, we demonstrate that both Gas6- and MFG-E8-expressing phages are capable of stimulating phagocytosis in phagocytes, but not in non-phagocytes. Moreover, phagocytosis selection specifically enriched the phages encoding the eat-me molecules. These results suggest that phage display with cDNA library is a feasible approach to functionally clone unknown eat-me signals.
Materials and Methods
Materials
T7Select10-3b vector and T7Select phage packaging kit were purchased from Novagen (Madison, WI). All cell culture media and supplements were from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was from Hyclone (Logan, UT). All the restriction enzymes were from New England Biolab (Ipswich, MA). Mer-Fc, Axl-Fc and Tyro3-Fc (Dtk-Fc) were from R&D Systems (Minneapolis, MN). All other reagents were from Sigma (St. Louis, MO). J774, ARPE19, Neuro-2a and HeLa cells were obtained from ATCC (Manassa, VA). BV-2 cell was a gift from Dr. Jonathan Godbout (Ohio State Univ., Columbus, Ohio).
Cell culture
J774, BV-2, Neuro-2a and HeLa cells were cultured in Dulbecco’s modified essential medium (DMEM) supplemented with 10% FBS and 2 mM L-glutamine. ARPE19 cells were cultured in DMEM/F-12 (1:1) medium supplemented with 10% FBS, 2 mM L-glutamine and 0.32% sodium biocarbonate.
T7 phage display vector
T7Select10-3b vector was engineered as the following by attaching a C-terminal biotinylation tag to displayed proteins. The linker sequence of 5′-GCGGATCCGCGGCCGCTA TAACTCGAGGGTAGTGGGAGCGGA TTA AAT GAT ATA TTT GAG GCA CAA AAA ATT GAA TGG CATTAA GAATTCAAGCTTGTCGACAA-3′ (underlined sequences for BamHI, NotI, XhoI and SalI cutting sites, italic sequence for the biotinylation tag, bolded codon for internal stop codons) was generated by PCR using overlapping primers, digested with BamHI and SalI, ligated into T7Select10-3b vector at BamHI and XhoI sites to generate T7Bio phage vector (Fig. 1A). Control Biotin-phage expressing biotinylation tag was constructed by replacing the stop codon between NotI and XhoI with a glycine. The engineered phage vector was verified by sequencing.
Fig. 1.

(A) T7Bio phage vector. The sequence fused to the C-terminus of capsid 10B was BamHI and NotI sites, followed by stop codon, XhoI site, a flexible linker and a biotinylation tag. This vector had an in-frame stop codon between NotI and XhoI sites to prevent the expression of the biotinylation tag in T7Bio phage without cDNA insert. (B) Schematic representation of phagocytosis selection. Phages were incubated with phagocytes at 4°C for 30 min without phagocytosis. After washing, the cells were incubated at 37°C for 30 min to allow phagocytosis of bound phages to occur. Unphagocytosed surface-bound phages were stripped off. Phagocytosed phages were released by cell lysis, amplified in BLT5615 bacteria and used as input for the next round of selection. A aliquot of released phages can also be quantified by plaque assay to determine total phagocytosed phages at each round.
All procedures for T7 phage display, including phage DNA purification, digestion, ligation, phage packaging, plaque assay, phage liquid amplification and plating with BLT5615 E. coli, were performed according to Novagen’s T7Select system manual (available at http://www.emdbiosciences.com/docs/docs/PROT/TB178.pdf).
Gas6-phage and MFG-E8-phage
Full-length mouse Gas6 cDNA sequence was obtained from Open Biosystems (Huntsville, AL). The 2 × LG receptor binding domains (114D-674P) were amplified by PCR with primers 5′-CAGCGGCCGCTGGACCAGTGCACCCCAAACCCT-3′ and 5′-AC CTCGAGGGGGGTGGCATGCTCCACAG-3′. The PCR product was digested with NotI and XhoI, ligated into T7Bio vector at the same sites, and packaged using T7 phage packaging kit. Clonal Gas6-phage was isolated from phage plates and verified by DNA sequencing.
Full length cDNA of MFG-E8 long form7 was generated by PCR from mouse eye with primers 5′-CAGCGGCCGCTGATGCAGGTCTCCCGTGTGCTG-3′ and 5′-GTCTCGAGACA GCCCAGCAGCTCCAGGCG-3′. The PCR product was digested with NotI and XhoI, ligated into T7Bio vector at the same sites, and packaged into T7 phage. Clonal MFG-E8-phage was isolated and verified by DNA sequencing.
Phage binding assay
Streptavidin (Pierce, Rockford, IL) (10 μg/ml) or mock control was coated in phosphate buffered saline (PBS) on Enzyme-Linked ImmunoSorbent Assay (ELISA) plates with high binding capacity (Corning Life Sciences, Lowell, MA) at 4°C overnight, blocked for 1 h with 1% bovine serum albumin (BSA). Gas6-phage, MFG-E8 or T7Bio phage was amplified in BLT5615 cells until lysis. The debris was removed by centrifugation at 13,000 × g for 4 min. The phage lysates [100 μl/well, ~1 × 1011 plaque forming unit (pfu)/ml] were incubated with immobilized streptavidin in the presence of 0.2% of Tween-20 for 1 h, followed by washing with PBS containing 0.2% Tween-20 (PBST) for 5 min × 6 times. The bound phages were eluted with 1% sodium dodecyl sulfate (SDS) for 10 min, and quantified by plaque assay.
For receptor binding assay, Mer-Fc, Axl-Fc and Tyro3-Fc (the receptor extracellular domains fused to human IgG1 Fc fragment) were coated at 5 μg/ml on ELISA plates. The phage binding was carried out as described above.
Phage binding to cells
Phage lysates (0.5 ml, ~1 × 1011 pfu/ml) were mixed with 1/10 volume of 5 M NaCl, and centrifuged at 13,000 × g for 10 min. The supernatants were mixed with 1/6 volume of 50% polyethylene glycol (PEG)-8000, incubated on ice for 30 min and spun at 16,100 × g for 20 min at 4°C. The precipitated phages were resuspended in ice-cold complete media (0.5 ml/sample), added to the pre-chilled indicated cells at 60–80% confluency in 12-well plates, incubated at 4°C with gentle shaking for 30 min. The cells were washed with ice-cold PBS containing 1% FBS (PBS-FBS) for 10 min × 5 times. After washing, cells were solubilized with 0.5% Triton X-100 in PBS, and the bound phages were quantified by plaque assay. Total bound phages are expressed as binding index, which is normalized against control Biotin-phage; namely, binding index = (total bound clonal phage)/(total bound Biotin-phage).
Phage phagocytosis
All phages were precipitated from phage lysates (0.5 ml, ~1 × 1011 pfu/ml) as described above. Precipitated phages were incubated with indicated cells for 30 min at 4°C without phagocytosis. After removal of unbound phages by washing with ice-cold PBS, phage-cell complexes were incubated at 37°C in the complete medium for 30 min to allow phagocytosis to occur. Unphagocytosed surface-bound phages were stripped off by incubating the cells in the stripping buffer (100 mM glycine, pH 2.5, 150 mM NaCl, 200 mM urea, 2 mg/ml polyvinylpyrrolidone) for 2 min × 2 times at room temperature, followed by a quick wash with ice-cold PBS. Internalized phages were released by lysing the cells with the lysis buffer (1 mM triethylamine with 0.5% Triton X-100) for 1 min and immediately neutralized to pH 7.4 with 10 × PBS pre-mixed with diluted HCl. Since T7 bacteriophage is susceptible to acid and alkaline, it was critical to immediately neutralize the pH to 7.4. The released phages were quantified by plaque assay. Total phagocytosed phages are expressed as phagocytosis index, which is normalized against control Biotin-phage; namely, phagocytosis index = (total phagocytosed clonal phage)/(total phagocytosed Biotin-phage).
To determine the stripping efficiency, the mixture of Gas6-phage and MFG-E8-phage (1:1) was incubated with ARPE19 cells at 4°C without phagocytosis. After washing, the cells before or after stripping were solubilized with 0.5% Triton X-100 in PBS. Total bound phages were quantified by plaque assay and compared to total phagocytosed phages, which were quantified in experiments performed in parallel as described above.
Immunostaining of phagocytosed phages
ARPE19 cells with phagocytosed phages were fixed with 10% buffered formalin for 30 min, permeabilized with 0.2% Triton X-100 in PBS for 10 min, labeled with streptavidin-conjugated fluorescein isothiocyanate (FITC) to detect biotinylated Gas6-phage, MFG-E8-phage or control Biotin-phage. Nuclei were stained with 1% 4′,6-diamidino-2-phenylindole (DAPI). The intracellular fluorescence signals were analyzed by Leica (Bannockburn, IL) TCS SP5 confocal microscopy with diode laser for excitation at 405 nm and emission at 480 nm for DAPI, and argon laser excitation at 488 and emission at 510–570 nm for FITC. The percentage of FITC-positive cells was determined with at least 180 cells analyzed for each group.
Phage enrichment by phagocytosis
The principle of phage phagocytosis selection is depicted in Fig. 1B. Gas6-phage or MFG-E8 phage was diluted with control Biotin-phage at a pfu ratio of 1:1,000,000. The mixed phages (2.5 ml, ~1 × 1011 pfu/ml) were precipitated, resuspended in the complete medium, incubated with ARPE19 or J774 in 60-mm culture dishes for 30 min at 4°C, followed by phagocytosis at 37°C as described above. After stripping off unphagocytosed surface-bound phages, phagocytosed phages were released by cell lysis, amplified in BLT5615 E. coli, and used as inputs for the next round of selection. A total of five rounds of selection were performed. Total phagocytosed phages at each round were quantified by plaque assay. Phage lysates at each round were monitored for the enrichment of Gas6-phage or MFG-E8-phage by PCR using following gene specific primers: T7SelectUp (5′-GGAGCTGTCGTATTCCAGTC-3′) and Gas6-R (5′-TCCCACTCCTGCCCGCCTGT-3′) primers for Gas6-phage; and T7SelectUp and MFG-E8-R (5′-GGGTGGGGACGGCAGTATTG-3′) primers for MFG-E8-phage. Phage lysates (1 μl per sample) were used in 20 μl of total PCR reaction mixture to detect the ligand-expressing phages with 30-cycle amplification. The PCR products were analyzed with 1% agarose gel.
Data analysis
All experiments were repeated independently for five times. Data are expressed as mean ± s.d. Student’s t-test is used to analyze statistical significance.
Results
Characterization of phage binding activity
Both Gas6-phage and MFG-E8-phage expressed an additional C-terminal biotinylation tag, which was spontaneously biotinylated by E. coli BirA ligase. To validate the expression of the proteins on phage surface, both phages were analyzed for their binding activity to immobilized streptavidin. The results showed that Gas6-phage and MFG-E8-phage had significant higher binding activity to streptavidin than T7Bio phage (Fig. 2A), which had an internal stop codon to prevent the tag expression. These results suggest that the biotinylated fusion proteins were expressed and displayed on phage surface.
Fig. 2.
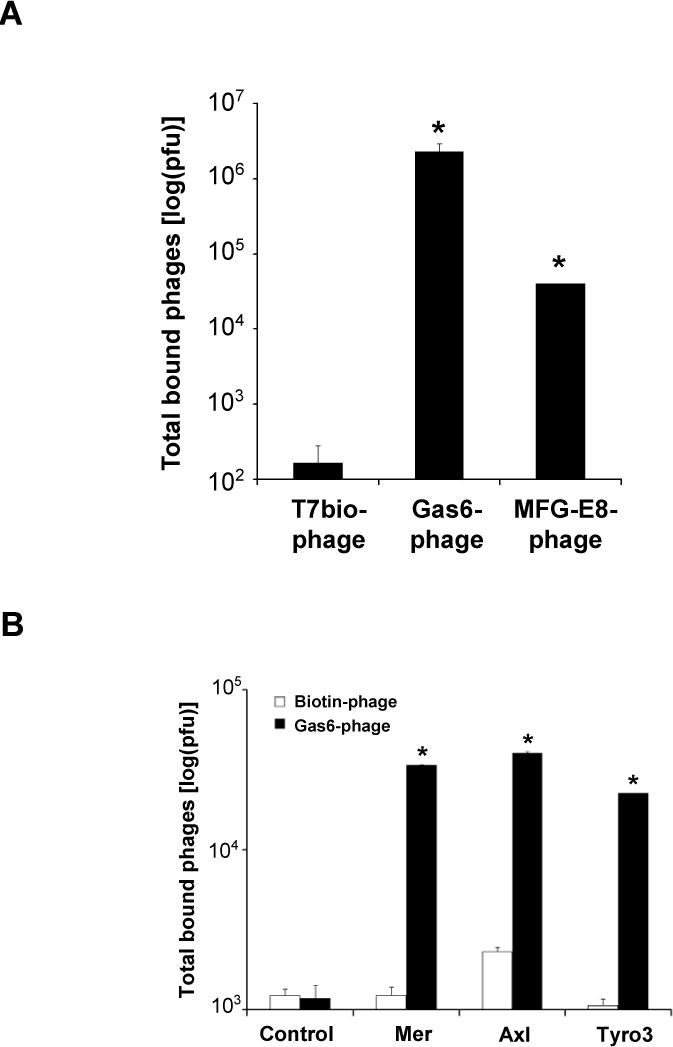
Phage binding to immobilized streptavidin and receptors. (A) The expression of Gas6 and MFG-E8 on phage surface is verified by the detection of C-terminal biotin displayed on both Gas6-phage and MFG-E8-phage. Control T7Bio phage had an internal stop codon to prevent the expression of C-terminal biotinylation tag. All phages were analyzed for their binding activity to immobilized streptavidin. Bound phages were quantified by plaque assay (± s.d., * p<0.001, vs. T7Bio phage, n=5). (B) Gas6-phage binding to immobilized receptors. Mer-Fc, Axl-Fc and Tyro3-Fc were directly immobilized on 96-well plates. Gas6-phage or control Biotin-phage (1 × 1010 pfu) was incubated with the immobilized receptors or mock control. After washing, bound phages were eluted, and quantified by plaque assay (± s.d., * p<0.001, vs. Biotin-phage, n=5).
Gas6 is one of the well-characterized ligands specifically binding to Mer, Axl and Tyro3 through its C-terminal 2 × LG domains. Mouse Gas6 has two potential glycosylation sites at position 417 and 488.19 The glycosylation was confirmed by the increase in molecular mass of the truncated mouse Gas6 (Asp115-Pro673) expressed in mouse myeloma cell line NSo (R&D Systems, Catalog #986-GS). It is unclear whether the glycosylation is necessary for its binding activity to the receptors. Since E. coli is deficient in glycosylation machinery, Gas6 displayed on phage surface should not be glycosylated. To investigate receptor binding activity of unglycosylated Gas6, we analyzed Gas6-phage binding to Mer-Fc, Axl-Fc and Tyro3-Fc immobilized on 96-well plates. The results indicated that Gas6-phage bound to immobilized RTKs, but not mock coated plates (Fig. 2B). Gas6-phage binding activities to the receptor were significantly higher than control Biotin-phage. These results suggest that unglycosylated Gas6-phage was fully capable of binding to the receptors.
Binding of Gas6-phage and MFG-E8-phage to phagocytes and non-phagocytes
Gas6-phage and MFG-E8-phage were characterized for their binding to phagocytes and non-phagocytes. J774 is murine macrophage cell line. BV-2 is microglial cell line with phagocytic activity.20 ARPE19 cell is derived from human RPE cells whose phagocytic activity is critical for the regeneration of photoreceptor outer segments (POS).21 All these three phagocyte cell lines have been used for in vitro phagocytosis studies. HeLa and Neuro-2a are non-phagocytes. To prevent non-specific internalization of bound phages, phage binding was performed at 4°C. Gas6-phage and MFG-E8-phage demonstrated higher binding activity to all cell lines than Biotin-phage (Fig. 3). J774, ARPE19 and Neuro-2a cells showed much higher binding activity to both phages. The highest binding activity of Gas6-phage was to Neuro-2a cells, but not to the phagocytes.
Fig. 3.

Cell binding activities of Gas6-phage and MFG-E8-phage. Both phages (5 × 1010 pfu) bound to J774, ARPE19, BV-2, Neuro-2a and HeLa cells at 4°C. After washing, bound phages were released by cell lysis with 0.5% Triton X-100 in PBS, and quantified by plaque assay. All the data were normalized to the control Biotin-phage and expressed as binding index (± s.d., * p<0.001, vs. Biotin-phage, n=5).
Gas6 and MFG-E8 stimulated phage uptake by phagocytes, but not by non-phagocytes
The capacity of Gas6 and MFG-E8 to stimulate phage phagocytosis was analyzed in both phagocytes and non-phagocytes. Gas6-phage and MFG-E8-phage facilitated phagocytosis in J774, BV-2 and ARPE19 cells, but not in HeLa and Neuro-2a cells (Fig. 4A). It is noteworthy that Gas6 minimally facilitated phage internalization in Neuro-2a cells, even though the ligand bound to the cells well (Fig. 3), suggesting that phage binding to surface receptor does not always elicit phagocytosis.
Fig. 4.

(A) Gas6-phage and MFG-E8-phage stimulate phagocytosis in phagocytes, but not in non-phagocytes. All phages (5 × 1010 pfu) were precipitated, incubated with the indicated cells, and phagocytosed as described in Fig. 1B. The phagocytosed phages were released by cell lysis and quantified by plaque assay. All the data were normalized to the control Biotin-phage and expressed as phagocytosis index (+ s.d., * p<0.001, vs. Biotin-phage, n=5). (B) The stripping efficiency. The mixture of Gas6-phage and MFG-E8-phage (1:1) was incubated with ARPE19 cells at 4°C without phagocytosis. After washing, the total surface-bound phages were quantified by plaque assay with or without stripping. Phagocytosed mixed phages in ARPE19 cells were quantified in a parallel experiment as described in (A).
The stripping efficiency was analyzed by comparing the total surface-bound phages before and after stripping without phagocytosis. The results revealed that more than 99.98% of surface-bound phages were stripped off (Fig. 4B), suggesting an excellent stripping efficiency. A parallel experiment revealed that ~11.5% of the total bound phages was phagocytosed.
Since all the phages used in phagocytosis had C-terminal biotinylation tag, internalized phages in ARPE19 cells were conveniently visualized with streptavidin-FITC. Confocal microscopy analysis revealed that Gas6-phage and MFG-E8-phage, but not Biotin-phage, were internalized (Fig. 5A). Compared with 8% of ARPE19 cells with internalized Biotin-phage, 66% and 57% of the cells had intracellular Gas6-phage and MFG-E8-phage (Fig. 5B), respectively. These data suggest that Gas6 and MFG-E8 displayed on phage surface specifically stimulated phage phagocytosis in the phagocytes.
Fig. 5.

Immunostaining for phagocytosed phages. Phage phagocytosis in ARPE19 was performed as described in Fig. 4A. (A) After phagocytosis, ARPE19 cells were fixed, permeabilized with Triton X-100, labeled with streptavidin-conjugated FITC for the phagocytosed phages, and analyzed by confocal microscopy. Nuclei were stained with DAPI. Panels in the left column (a, d and g) are in lower magnification, while all others are in higher magnification. The indicated size bars are 25 μm. (B) Percentage of cells with phagocytosed phage. At least 180 cells were analyzed for each group (± s.d., * p<0.001, vs. Biotin-phage, n>180).
Enrichment of Gas6-phage and MFG-E8-phage by phagocytosis selection
The higher internalization capacity of Gas6-phage and MFG-E8-phage suggests that they should be enrichable by multiple rounds of phagocytosis selection. As an illustration, we diluted Gas6-phage or MFG-E8 phage with Biotin-phage at a pfu ratio of 1:1,000,000. Diluted phages were added to J774 cells or ARPE19 cells to analyze the possible enrichment of Gas6-phage and MFG-E8 phage. The total phagocytosed phages were drastically increased at Round 3–5 (Fig. 6A and B). Enriched phages were analyzed by PCR using gene-specific primers. After 3–5 rounds of phagocytosis selection, Gas6-phage and MFG-E8 phage were detected by PCR, whereas both phages were not detectable in the diluted phage mixture before the selection (Fig. 6C). Additional selections further enriched both phages. These results showed that Gas6-phage and MFG-E8-phage were substantially enriched with multiple rounds of phagocytosis selection in J774 and ARPE19 cells.
Fig. 6.


Enrichment of Gas6-phage and MFG-E8-phage by phagocytosis. Gas6-phage or MFG-E8 phage was diluted with Biotin-phage at a pfu ratio of 1:1,000,000. The diluted phages were precipitated, incubated with ARPE19 or J774, phagocytosed, recovered by cell lysis, amplified in BLT5615, and used as inputs for the next round of selection. A total of five rounds of selection were performed. Total internalized phages for diluted Gas6-phage (A) and MFG-E8-phage (B) were quantified by plaque assay at each round. Biotin-phage was included as a negative control. The data are the representative of five independent experiments. (C) Phage lysates at each round were analyzed with gene-specific primers to detect Gas6-phage and MFG-E8 phage.
Discussion
Binding of eat-me signals on apoptotic cells to phagocytic receptors on phagocytes triggers signaling cascades in phagocytes and facilitates engulfment and ingestion of apoptotic cells. This process not only leads to the removal of apoptotic cells, but also results in suppression of inflammation and immune tolerance to self-proteins.2, 11 Despite its importance, eat-me signals on apoptotic cells have received relatively little attention. One of the reasons is that it is a daunting challenge to identify unknown eat-me signals. Limited number of eat-me signals, including PS, Gas6, MFG-E8, calreticulin, ICAM3, were identified on a case-by-case basis. However, it is unknown how many other eat-me signals are yet to be identified in various phagocytes and whether defect in eat-me signals may associate with diseases.
The only strategy to functionally clone signaling molecules in phagocytosis is chemically-induced random genetic mutations in C. elegans, coupled with morphological screening for accumulation of unengulfed cell corpse by optics microscopy and cloning of the mutated genes.1, 22 This approach has yielded a number of genes involved in two independent signaling pathways of the engulfment in C. elegans.1 However, this strategy is technically complicated and time-consuming. More importantly, this approach is not applicable to other animals, which are not transparent for live screening of accumulated cell corpse by optics microscopy. Unlike mammals, C. elegans lacks professional phagocytes. Furthermore, this approach identifies not only eat-me signals, but also other intracellular signaling molecules. Therefore, random genetic mutation is not a feasible approach for functional cloning of eat-me signals for mammalian phagocytes.
Several previous studies identified short peptides and scFvs from phage display random peptide libraries and scFv libraries in different cancer cell lines, implicating that phage display may be feasible approach for functional cloning of unknown eat-me signals.15–18 However, cell internalization or endocytosis is a complicated process and includes both phagocytosis (cell eating) and pinocytosis (cell drinking).23 In general, phagocytosis arbitrarily refers to internalization of large particles (>200 nm), whereas pinocytosis refers to internalization of smaller particles or cell surface-bound ligands or molecules through different mechanisms.23 For example, the molecular mechanism of pinocytosis induced by epidermal growth factor (EGF) binding to EGF receptor (EGF-R) on non-phagocytes is very different than those for PS-, Gas6-, or MFG-E8-mediated phagocytosis of apoptotic cells in professional phagocytes. Internalization of EGF-expressing phage by binding to EGF-R on non-phagocytes does not necessarily validate the feasibility of phage display for phagocytosis study in professional phagocytes.16, 24 In this regard, we characterized the phagocytosis of Gas6- and MFG-E8-expressing phages in professional phagocytes and non-phagocytes.
Our results indicated that phages expressing eat-me signals can bind to both phagocytes and non-phagocytes. However, only phagocytes can phagocytose phages expressing eat-me signals. These results suggest that binding of eat-me signals to their cognate phagocytic receptors does not always trigger phagocytosis. Downstream signaling pathways in phagocytes, including receptor signaling, cytoskeleton protein rearrangement and engulfment, also play critical roles in the uptake. Our data also demonstrated that multiple rounds of phagocytosis selection result in enrichment of phages expressing eat-me signals, opening the opportunity for possible functional cloning of eat-me signals by phage display.
Mammals have developed professional phagocytes to facilitate the removal of apoptotic cells or cellular debris. Besides the well-known macrophages, microglia in the central nerve system (CNS), RPE cells in the retina and Sertoli cells in the testis are the well-characterized specialized phagocytes.21, 25, 26 Most known eat-me signals were identified and characterized in macrophages. Little is known about eat-me signals in other phagocytes. For example, RPE phagocytosis is critical for the maintenance of visual function.21 Photoreceptor outer segments (POS), which convert light to electrical impulses, are susceptible to photo-oxidation damage. As a part of repair process, photoreceptors shed the damaged POS at the tip of the outer segments in a diurnal rhythm. RPE cells underneath photoreceptors within close contact to the POS play a pivotal role in the POS regeneration by ingesting and recycling shed POS vesicles through phagocytosis. The importance of RPE phagocytosis has been implicated in the retinal degeneration model of RCS rats,9 whose defect in the removal of shed POS is due to a genetic mutation in Mer receptor tyrosine kinase on RPE cells. To identify unknown eat-me signals in RPE cells, we have recently generated an open-reading-frame (ORF) phage display cDNA library from mouse eye. Phagocytosis selection of the library by phage display resulted in the identification of nine putative eat-me signals. Among them was Gas6(43Q-148R) (unpublished data), which substantiates the functional cloning strategy for unbiased discovery of endogenous eat-me signals. Mutations in two novel eat-me signals associated with photoreceptor degeneration with unknown mechanism (unpublished data). However, one of the critical concerns is the legitimacy of phage display technology to study eat-me signals. The characterization of phage display as a valuable tool to study eat-me signals in this study lay a groundwork for functional cloning of eat-me signals in phagocytosis in various professional phagocytes.
A major concern is whether identified phage clones are false positives. Putative eat-me signals identified by phage display should be extensively validated and characterized. They should be vigorously analyzed for their stimulation on phagocytosis by means other than phage display, including phagocytosis of fluorescent microbeads covalently conjugated with purified eat-me signals or phagocytosis of membrane vesicles or apoptotic cells expressing the putative eat-me signals. Identification of their phagocytic receptors with expression on phagocytes will help verify their functional role in phagocytosis. Eat-me signals should have physiological or pathological access to the receptors. Cells or membrane vesicles from mice deficient of eat-me signals may have reduced capacity to induce phagocytosis.27 All these experiments will help validate a putative eat-me signals identified by phage display.
In conclusion, we have demonstrated in this study that two well-characterized eat-me signals displayed on phage surface can stimulate phagocytosis of the cognate phage by professional phagocytes, leading to the specific enrichment of the phage. These results suggest that phage display is a feasible approach for functional cloning of unknown eat-me signals.
Acknowledgments
This project was supported in part by NIH R01EY016211, EY014801 and an unrestricted grant from RPB. NBC is a recipient of Postdoctoral Fellowship from Fight for Sight.
References
- 1.Reddien PW, Horvitz HR. The engulfment process of programmed cell death in caenorhabditis elegans. Annu Rev Cell Dev Biol. 2004;20:193–221. doi: 10.1146/annurev.cellbio.20.022003.114619. [DOI] [PubMed] [Google Scholar]
- 2.Erwig LP, Henson PM. Immunological consequences of apoptotic cell phagocytosis. Am J Pathol. 2007;171:2–8. doi: 10.2353/ajpath.2007.070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964–74. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 4.Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell. 2004;14:277–87. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
- 5.Park D, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007 doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 6.Miyanishi M, et al. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007 doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 7.Hanayama R, et al. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–7. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 8.Hafizi S, Dahlback B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. Febs J. 2006;273:5231–44. doi: 10.1111/j.1742-4658.2006.05529.x. [DOI] [PubMed] [Google Scholar]
- 9.D’Cruz PM, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–51. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 10.Gal A, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–1. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 11.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–36. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandrot EF, et al. Essential role for MFG-E8 as ligand for alphavbeta5 integrin in diurnal retinal phagocytosis. Proc Natl Acad Sci U S A. 2007;104:12005–10. doi: 10.1073/pnas.0704756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008;15:243–250. doi: 10.1038/sj.cdd.4402184. [DOI] [PubMed] [Google Scholar]
- 14.Hall MO, Obin MS, Heeb MJ, Burgess BL, Abrams TA. Both protein S and Gas6 stimulate outer segment phagocytosis by cultured rat retinal pigment epithelial cells. Exp Eye Res. 2005;81:581–91. doi: 10.1016/j.exer.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Spring H, Schwab M. Neuroblastoma tumor cell-binding peptides identified through random peptide phage display. Cancer Lett. 2001;171:153–64. doi: 10.1016/s0304-3835(01)00575-4. [DOI] [PubMed] [Google Scholar]
- 16.Burg M, et al. Selection of internalizing ligand-display phage using rolling circle amplification for phage recovery. DNA Cell Biol. 2004;23:457–62. doi: 10.1089/1044549041474760. [DOI] [PubMed] [Google Scholar]
- 17.Goenaga AL, et al. Identification and characterization of tumor antigens by using antibody phage display and intrabody strategies. Mol Immunol. 2007;44:3777–88. doi: 10.1016/j.molimm.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao C, et al. De novo identification of tumor-specific internalizing human antibody-receptor pairs by phage-display methods. J Immunol Methods. 2003;274:185–97. doi: 10.1016/s0022-1759(02)00522-7. [DOI] [PubMed] [Google Scholar]
- 19.Manfioletti G, Brancolini C, Avanzi G, Schneider C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol Cell Biol. 1993;13:4976–85. doi: 10.1128/mcb.13.8.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller CD, et al. Functional beta-glucan receptor expression by a microglial cell line. Res Immunol. 1994;145:267–75. doi: 10.1016/s0923-2494(94)80015-4. [DOI] [PubMed] [Google Scholar]
- 21.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–81. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 22.Hedgecock EM, Sulston JE, Thomson JN. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science. 1983;220:1277–9. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Shapiro JI. Endocytosis and signal transduction: basic science update. Biol Res Nurs. 2003;5:117–28. doi: 10.1177/1099800403256860. [DOI] [PubMed] [Google Scholar]
- 24.Larocca D, Jensen-Pergakes K, Burg MA, Baird A. Receptor-targeted gene delivery using multivalent phagemid particles. Mol Ther. 2001;3:476–84. doi: 10.1006/mthe.2001.0284. [DOI] [PubMed] [Google Scholar]
- 25.D’Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging. 2004;25:675–83. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 26.Nakanishi Y, Shiratsuchi A. Phagocytic removal of apoptotic spermatogenic cells by Sertoli cells: mechanisms and consequences. Biol Pharm Bull. 2004;27:13–6. doi: 10.1248/bpb.27.13. [DOI] [PubMed] [Google Scholar]
- 27.Gardai SJ, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]