

Abstract
Mutation in the tubby gene causes adult-onset obesity, progressive retinal and cochlear degeneration with unknown mechanism. In contrast, mutations in tubby-like protein 1 (Tulp1), whose C-terminus is highly homologous to tubby, only lead to retinal degeneration. We speculate that their diverse N-terminus may define their distinct disease profile. To elucidate the binding partners of tubby, we used tubby N-terminus (tubby-N) as bait to identify unknown binding proteins with open-reading-frame (ORF) phage display. T7 phage display was engineered with 3 improvements: high-quality ORF phage display cDNA library, specific phage elution by protease cleavage and dual phage display for sensitive high throughput screening. The new system is capable of identifying unknown bait-binding proteins in as fast as ~4–7 days. While phage display with conventional cDNA libraries identifies high percentage of out-of-frame unnatural short peptides, all 28 tubby-N-binding clones identified by ORF phage display were ORFs. They encode 16 proteins, including 8 nuclear proteins. Fourteen proteins were analyzed by yeast two-hybrid assay and protein pull-down assay with ten of them independently verified. Comparative binding analyses revealed several proteins binding to both tubby and Tulp1 as well as one tubby-specific binding protein. These data suggest that tubby-N is capable of interacting with multiple nuclear and cytoplasmic protein binding partners. These results demonstrated that the newly-engineered ORF phage display is a powerful technology to identify unknown protein-protein interactions.
Keywords: Tubby, ORF phage display, protein-protein interaction, tubby-binding proteins, T7 phage, cDNA library
INTRODUCTION
Obesity is a global pandemic and a major health concern. With more than 400 million adults obese worldwide, genetic factors play an important role in the epidemic (Lee, 2009; Low et al., 2009). For example, tubby gene mutation in Tubby mice causes adult-onset obesity, progressive retinal and cochlear degeneration with unknown mechanism (Carroll et al., 2004; Ikeda et al., 2002; Noben-Trauth et al., 1996). Tubby belongs to a well-characterized tubby protein family with four members (tubby, tubby-like protein 1, 2 and 3), which share a highly conserved C-terminal “tubby domain” of ~260 amino acids (Carroll et al., 2004; Ikeda et al., 2002). Tubby has been demonstrated as a putative membrane-bound, G protein-activated transcription factor (Santagata et al., 2001), although the target gene(s) regulated by tubby is yet to be described. Unlike tubby, mutations in tubby-like protein 1 (Tulp1) only lead to retinal degeneration with no other clinical manifestation (Abbasi et al., 2008; Hagstrom et al., 1998; Ikeda et al., 2000). It is speculated that the diverse N-terminal half of the proteins may impart their distinct functional specificities and disease profiles. Thus, identification of the proteins binding to the N-terminus of tubby (tubby-N) will help elucidate its pathological mechanism. In this study we identified a panel of new tubby-N-binding proteins by phage display.
Phage display has been widely used to identify bait-binding antibodies (Abs) or short peptides from Ab or random peptide libraries (Kehoe and Kay, 2005; Paschke, 2006). However, phage display with cDNA libraries is rare and inefficient. The critical issue is possible reading frame shifts in the cDNA repertoires fused to the N-terminus of filamentous phage gene III coat protein (pIII). Ab libraries with predictable reading frames can be conveniently fused to pIII without problem, whereas a cDNA library with unpredictable reading frames and stop codons may interfere with pIII expression. To circumvent the problem, various strategies of C-terminal display have been explored, including pJuFo phagemid (Crameri and Suter, 1993; Jestin, 2008). Moreover, T7 phage display vector was engineered with cDNA library proteins displayed at the C-terminus of capsid 10B (Danner and Belasco, 2001). However, C-terminal display cannot ensure that the cDNA library is expressed in the correct reading frames. Unlike yeast two-hybrid system (Y2H), out-of-frame phage clones encoding unnatural short peptides tend to outgrow clones with open reading frames (ORFs) through multiple rounds of selection and amplification. For example, only less than 10% (24/243) of clones identified from a conventional C-terminal cDNA library of T7 phage display were ORFs (Kalnina et al., 2008). Another study showed similar results with ~6% (8/130) of identified clones encoding real proteins (Lin et al., 2007). Among nearly 4,000 literature citations related to phage display, only a few deal with cDNA libraries, mostly reporting merely one or two identified proteins without elaborating the high frequencies of non-ORFs. Despite recent improvements on phage display cDNA libraries (Ansuini et al., 2002; Faix et al., 2004; Pavoni et al., 2004), it remains a daunting challenge for the technology to be applied to protein-protein interactions with an efficiency comparable to Y2H and mass spectrometry-based functional proteomics.
Here we describe an efficient, convenient and versatile technology of ORF phage display to identify unknown bait-binding proteins in as fast as ~4–7 days. The new system of ORF phage display consists of 3 improvements: high-quality ORF phage display cDNA libraries, specific elution of bound phages by protease cleavage and dual phage display for sensitive high throughput screening. We use the technology to identify a panel of unknown tubby-N-binding proteins. All 28 identified phage clones encode real proteins. More importantly, identified tubby-N-binding proteins are independently verified by Y2H and protein pull-down assay.
MATERIALS AND METHODS
Library construction
The linker sequence was generated by PCR with overlapping primers, digested with BamHI and SalI, and ligated into T7Select10-3b vector (Novagen) at BamHI and XhoI sites to yield T7Bio3C vector, as described previously (Caberoy et al., 2009b). Total RNA was extracted from the eyes of 40 mice (20 C57BL/6 and 20 CB6F1, 4–8 weeks old) using TRIzol reagent (Invitrogen). Orientation-directed cDNA library was generated as previously described (Caberoy et al., 2009a), and ligated into T7Bio3C vector that was pre-digested with NotI, XhoI and EcoRV. Triple digestion of the phage vector was to minimize vector self-ligation. After packaging with T7 phage package extract (Novagen), the library was amplified once on BLT5615 bacterial plates, enriched for biotinylated phages by binding to streptavidin-coated paramagnetic beads (Promega) and eluted with 3C protease (10 units in 400 μl) at 4°C for 16 h to generate ORF libraries. The library was amplified once on bacterial plates with a final titer of ~1 × 1010 pfu/ml. All procedures for T7 phage display, including phage DNA purification, digestion, ligation, phage packaging, plaque assay, phage liquid amplification and plating in BLT5615 bacterial plates, were carried out as previously described (Caberoy et al., 2009b).
Library characterization
Individual phage clones were randomly picked from phage plates of the cDNA libraries. cDNA inserts were amplified by PCR with T7SelectUp and T7SelectDown primers (Novagen), and analyzed on 2% agarose gel. The actual cDNA insert sizes were calculated by subtracting 239 bp of the sequences for the phage backbone and tagged random primers.
To analyze the percentage of ORFs, the packaged phage cDNA libraries before and after the streptavidin enrichment were plated on BLT5615 bacterial plates at a similar density and lifted onto nitrocellulose membranes as described in Novagen’s T7Select System Manual (available at http://www.emdbiosciences.com/docs/docs/PROT/TB178.pdf). After blocking, the expression of the biotinylation tag in individual phage clones was analyzed by immunoblotting using streptavidin-conjugated horseradish peroxidase (HRP) and chemiluminescence detection. Positive signals indicated that the phage clones expressed the biotinylation tag with ORF cDNA inserts.
Phage selection
Phage selection was carried out as previously described (Caberoy et al., 2009b; Zhang et al., 2005) with the following modifications. ELISA plates (Corning Life Science; #2592) were coated with purified tubby-N [1 μg/100 μl/well in phosphate buffered saline (PBS)] at 4°C overnight or 37°C for 1 h, blocked for 1 h with 1% bovine serum albumin or 1% polyvinyl alcohol in PBST (PBS plus 0.1% Tween-20), and incubated with the phage library (~1 × 1010 pfu/ml, 0.1 ml/well) in the presence of 0.1% Tween-20 for 1 h at room temperature. The wells were washed with PBST for 6 min × 6. Bound phages were eluted with 3C protease (1–2 units/100 μl PBS/well) at 4°C for 1 h. An aliquot of eluted phages was diluted appropriately and quantified by plaque assay to determine total eluted phages. The remaining eluate was amplified in BLT5615 bacteria. The lysate was used as input for the next round of selection. After 3 rounds of selection, enriched phages were selected twice with immobilized streptavidin in a similar manner.
BLT7FLAG and BLT7Bio bacteria
A cDNA coding sequence for capsid 10A (22921–24001 bp in GenBank accession #V01146) was amplified from wild-type bacteriophage T7 by PCR, fused with a C-terminal FLAG tag by PCR with overlapping primers, digested with XbaI and EcoRI, and inserted into an engineered plasmid derived from pGEX-2T plasmid (GE Healthcare) to replace glutathione S-transferase (GST). The resulting plasmid was confirmed by DNA sequencing and transformed into BL21 bacteria to generate BLT7FLAG strain with carbenicillin selection. BLT7FLAG bacteria at an OD600 of 0.5 were induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) (1 mM) for 30 min to express capsid 10A-FLAG fusion protein and used as host bacteria for phage amplification. BLT7Bio bacterial strain was generated in a similar manner by fusing a biotinylation tag to the C-terminus of capsid 10A.
Colorimetric assay
Individual clones of enriched phages were randomly picked from phage plates and amplified in BLT7FLAG bacteria in 96-well plates by shaking at 37°C until lysis. After centrifugation at 1,400 g for 5 min, the supernatants were incubated with immobilized tubby-N or mock coated well in the ELISA plates in the presence of 0.1% Tween-20. After washing, bound phages were incubated with anti-FLAG M2 monoclonal antibody (mAb) (Sigma), followed with HRP-conjugated goat anti-mouse IgG. Bound HRP was visualized with o-phenylenediamine (OPD) (0.4 mg/ml) and H2O2 (0.01%) in citric acid buffer (20 mM citric acid, 40 mM NaH2PO4, pH 5.0), stopped with 1 M H2SO4 and quantified by a plate reader at 490 nm.
Recombinant proteins
The coding sequences for N-terminal (1M-242P) and full-length tubby were generated by reverse transcription PCR (RT-PCR) from mouse retina and cloned into pGEX-2T plasmid with a cleavage site for 3C protease immediately after GST. The fusion proteins were expressed in BL21(DE3) bacteria, purified with glutathione columns, eluted with reduced glutathione and dialyzed against PBS, as previously described (Li and Handschumacher, 2002). GST fusion protein of Tulp1-N (1M-298Q) was prepared in the same manner. GST-tagged 3C protease was expressed and purified in a similar manner, but without the 3C protease cleavage site between GST and 3C protease. Purified GST-tubby-N was cleaved with GST-tagged 3C protease at 4°C overnight. GST-free tubby-N was purified by depleting GST, GST-3C protease and other glutathione-binding impurities with a glutathione column. The purity of GST-free tubby-N was analyzed by SDS-PAGE.
FLAG-tagged tubby-F or green fluorescent protein (GFP) was generated by PCR and cloned into pAdTrack-CMV plasmid (He et al., 1998) at BglII and SalI sites. Recombinant adenovirus expressing FLAG-tubby-F or GFP-FLAG was generated as previously described (Chen et al., 2001) and used to infect HEK293 cells to express the recombinant protein. Both proteins were purified using anti-FLAG mAb affinity columns (Sigma), eluted with FLAG peptide (100 μg/ml), and dialyzed against PBS. Purified tubby-F and GFP were immobilized onto the ELISA plates (5 μg/ml, 100 μl/well) and used for phage binding assay.
Y2H assay
Tubby-N cDNA was cloned into pGBT9 plasmid (Clontech) at EcoRI and BamHI sites. The cDNA inserts of identified phages were amplified by PCR and cloned into pGAD424 plasmid (Clontech) at BamHI and SalI sites. Y2H assay was performed according to the manufacturer’s protocol. Briefly, S. cerevisiae CG-1945 strain was co-transformed with cDNA insert/pGAD424 and tubby-N/pGBT9 or pGBT9 control. The transformed yeast was selected on agar plates with double (-Leu/-Trp) or triple (-Leu/-Trp/-His) dropout of the essential amino acids. Dropout-resistant yeast clones were re-grown on YPD plates, lifted onto a filter paper and analyzed for galactosidase expression using X-gal as a substrate.
Protein pull-down assay
Full-length coding sequence of mouse Hmgb2 (Open Biosystems) was amplified by PCR and cloned into pcDNA3 with an N-terminal FLAG tag. HEK293 cells were transfected with the plasmid expressing FLAG-tagged Hmgb2 with Lipofectamine reagent (Invitrogen). The cells were harvested at 48 h post-transfection and washed. Cell lysate was prepared in PBS containing 0.5% Triton X-100, spun and incubated with purified GST-tubby-F or GST control, followed by glutathione resin. The resin was washed and analyzed by Western blot using anti-FLAG mAb.
RESULTS
Engineering of T7 phage vector
We chose T7 bacteriophage to develop ORF phage display because of its C-terminal display and robust growth rate. Moreover, it is not necessary for the proteins displayed on the surface of T7 phage to be secreted through the cell membrane (Kruger and Schroeder, 1981), an essential step in filamentous phage assembly (Russel, 1991). To optimize T7 phage display, we engineered the multiple cloning site in T7Select10-3b phage vector to generate T7Bio3C vector (Fig. 1a,b). A cleavage motif for human rhinovirus (HRV) 3C protease (Cordingley et al., 1990) was fused to the C-terminus of capsid 10B, followed by a GS linker, cloning sites, a second GS linker and a biotinylation tag (Ansuini et al., 2002). A cDNA library was inserted at NotI and XhoI sites. The stop codon at the cloning sites is to prevent the expression of the biotinylation tag in the absence of cDNA insert. The C-terminal biotinylation tag is not expressed unless the cDNA insert is an ORF. Thus, ORF phages, but not non-ORF phages, are biotinylated by E. coli BirA ligase and can be enriched by binding to immobilized streptavidin. 3C protease cleavage at 4°C specifically eluted the phages bound to immobilized streptavidin or bait through the tagged library proteins, but not through other phage surface proteins (Fig. 2). Two flexible GS linkers were designed to improve the accessibility of 3C protease cleavage site, library proteins and biotin tag. 3C protease eluted ~80 times more of bound phages than the standard elution method of 1% SDS (Fig. 2c), suggesting that the protease cleavage site is readily accessible in the library.
Fig. 1.

T7 phage vector engineering. (a) Sequence of T7Bio3C phage display vector. (b) Illustration of T7 phage display. Each engineered phage has 415 copies of capsid 10A and/or 10B with ~5–15 copies of the ORF cDNA library protein fused to the C-terminus of capsid 10B. A 3C protease cleavage site is located between capsid 10B and the library protein. A biotinylation tag is expressed at the C-terminus of the library proteins.
Fig. 2.
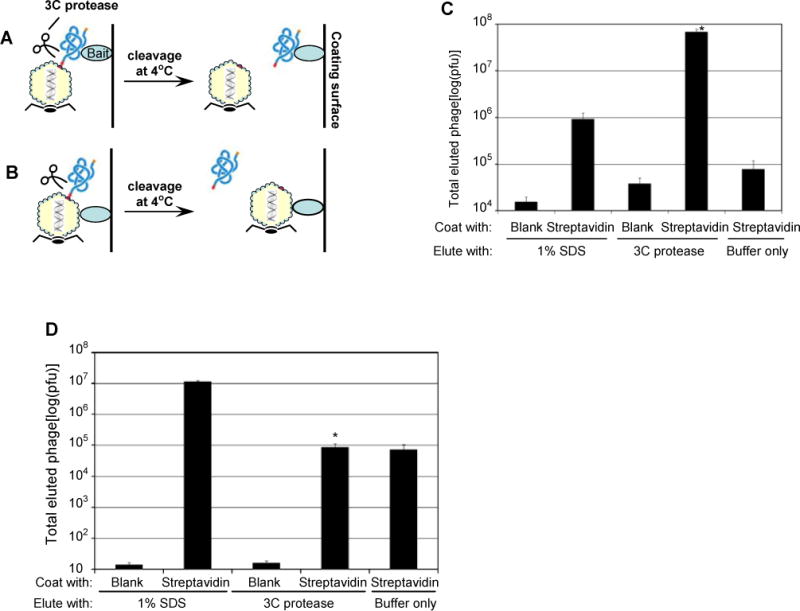
Specific phage elution by 3C protease. (a) Phages bound through the displayed protein can be released by 3C protease cleavage at 4°C. (b) Phages bound through other surface proteins cannot be eluted by 3C protease. (c) Specific elution by 3C protease. The ORF phage display library was bound to immobilized streptavidin through the biotin tag at the C-terminus of the library protein, washed and eluted with 1% SDS or 3C protease. (± SEM; n=3; *p<0.001; vs. streptavidin/1% SDS elution; t-test; throughout). (d) Elution of non-specifically bound phages by SDS, but not by 3C protease. As an illustration, the phage library was amplified in BLT7Bio bacteria to label capsid 10A with biotin (see Fig. 4 for detail), bound to immobilized streptavidin, washed and eluted with either 1% SDS or 3C protease.
ORF phage display cDNA libraries
ORF cDNA libraries were generated based on the fact that non-ORF cDNA has stop codon(s). Database analysis revealed that ~96% of 200-bp non-ORF cDNAs have at least one stop codon (Garufi et al., 2005). This number drastically increases to 99.6% for non-ORF cDNAs with 300 bp. We constructed an orientation-directed cDNA library between 300 bp – 1.5 kb from adult mouse eyes by the random priming method. Total library titer immediately after the phage packaging was ~2 × 107 pfu. The library was amplified once in BLT5615 bacteria, bound to immobilized streptavidin and eluted by 3C protease cleavage to generate ORF library. Size distribution analysis showed that >75% of the cDNA library had inserts longer than 300 bp (Fig. 3a). Immunoblotting assay revealed that >90% of the streptavidin-enriched ORF library had ORF cDNA inserts (Fig. 3b). Thus, the library is the best size-defined, C-terminal tagged ORF phage display cDNA library reported.
Fig. 3.
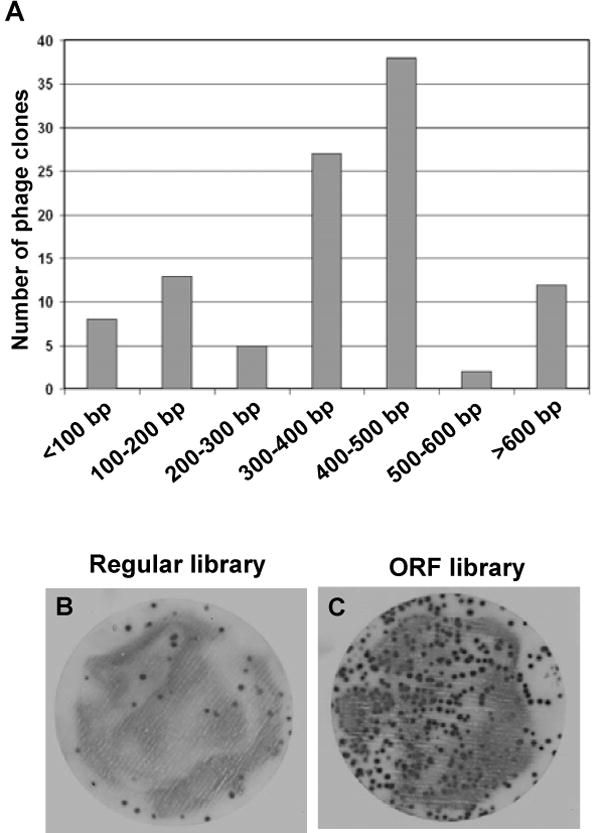
Characterization of ORF phage display cDNA library. (a) The size distribution of cDNA inserts in the library. A total of 105 phage clones were randomly picked from the library phage plates. Their cDNA inserts were amplified by PCR using T7SelectUp and T7SelectDown primers. The sizes of PCR products were analyzed on agarose gel. ~75% of the clones had cDNA insert longer than 300 bp. (b) and (c) Analysis of ORF library by immunoblotting. The packaged phage cDNA library was enriched with immobilized streptavidin to generate ORF cDNA library. The library before (b) and after (c) the enrichment was plated on BLT5615 bacterial plates at a similar density. The expression of the biotinylation tag in individual phage clones was analyzed by immunoblotting using streptavidin-conjugated HRP and chemiluminescence detection. Positive signals indicated that the phage clones expressed the biotinylation tag with ORF cDNA inserts. More than 90% of phage clones in the ORF library had ORF inserts, whereas only <10% of the unenriched clones had ORF inserts.
Dual phage display
Unlike filamentous phages, few Abs are available for quantification of T7 phage. Consequently, T7 phage is traditionally quantified by tedious plaque assays. To improve the efficiency, we developed dual T7 phage display to replace the plaque assays. T7 phage consists of 415 copies of capsid 10A and/or 10B. Each engineered phage displayed ~5–15 copies of library protein fused to the C-terminus of capsid 10B with the remaining copies of capsid 10A provided by host bacteria. In addition to the displayed cDNA library proteins, we fused a FLAG or biotinylation tag to the C-terminus of capsid 10A, which was expressed by newly-engineered BLT7FLAG or BLT7Bio bacterial strain, respectively. Co-display of ~5–15 copies of capsid 10B-library fusion proteins and >400 copies of FLAG- or biotin-tagged capsid 10A on the same phage surface not only allowed the functional analysis of library proteins, but also enabled the sensitive quantification of bound phages with enzyme-conjugated streptavidin or anti-FLAG mAb (Fig. 4). The surface detection moiety can be conveniently switched by amplifying the same phage in bacterial strains expressing capsid 10A with different C-terminal tags (Fig. 4a).
Fig. 4.
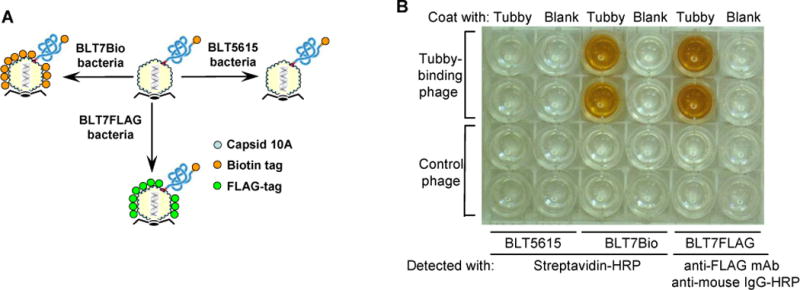
Dual phage display. (a) Schematic representation of dual phage display and surface detection moiety switching. T7 phages co-display capsid 10B-library fusion protein with a C-terminal biotinylation tag and capsid 10A with a C-terminal tag, which can be switched by amplifying the phages in different bacterial strains. (b) Colorimetric assay. A tubby-N-binding phage clone (see Fig. 5) was amplified in BLT5616, BLT7Bio or BLT7FLAG bacteria and bound to immobilized tubby-N. Bound phages were quantified by colorimetric assay using streptavidin-conjugated HRP or anti-FLAG mAb as indicated. Bound HRP was detected by colorimetric assay.
Identification of tubby-N-binding proteins
To identify proteins specifically binding to tubby-N, we expressed GST-tubby-N fusion protein in E. coli, purified GST-free tubby-N (Fig. 5a), immobilized the purified protein on ELISA plates as bait, performed 3 rounds of phage selection with the eye library (Fig. 5b), followed by 2 rounds of selection with immobilized streptavidin. A total of 108 individual phage clones were randomly isolated from phage plates, amplified in BLT7FLAG bacteria in 96-well plates and analyzed for their tubby-N-binding activity by colorimetric assay with dual phage display (Fig. 5c). Positive clones were further verified for their tubby-N-binding activities by plaque assay (Table 1). cDNA inserts of tubby-N-binding clones were amplified by PCR and identified by DNA sequencing. With the improved system of ORF phage display, each round of phage selection usually took less than 4 h, and the entire procedure can be completed manually in a regular laboratory setting within ~4–7 days.
Fig. 5.
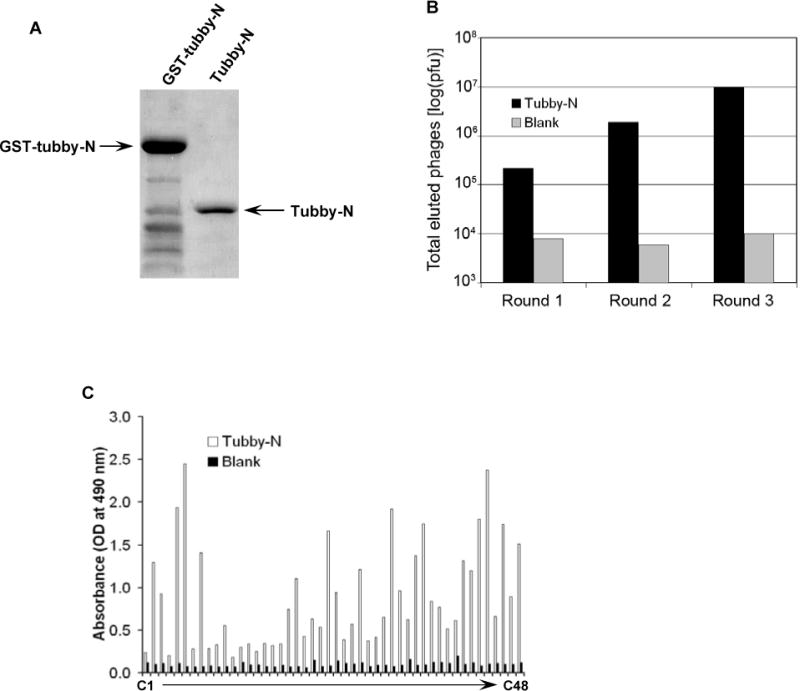
Identification of tubby-N-binding proteins. (a) Purification of GST-free tubby-N. GST-tubby-N was expressed in BL21(DE3) bacteria and purified using a glutathione column. Purified GST-tubby-N was digested with GST-tagged 3C protease. After dialysis, GST, GST-3C and minor glutathione-binding impurities were depleted with a glutathione column. Samples were analyzed for their purity by SDS-PAGE. Lane 1, purified GST-tubby-N; Lane 2, GST-free tubby-N. (b) Enrichment of tubby-N-binding phages by phage panning. The library was incubated with immobilized tubby-N in ELISA plates, washed, eluted with 3C protease, amplified and used as input for the next round of selection. Phage enrichment was analyzed by plaque assay. Blank control was included to monitor possible enrichment of plate-binding phages or phages binding to blocking reagents. (c) Screening for tubby-N-binding clones. Individual phage clones at round 3 were analyzed for their binding activities to immobilized tubby-N or mock coated well by colorimetric assay, as described in Fig. 4. C4 is a control phage without cDNA insert.
Table 1.
Tubby-binding proteins.
| Protein | Accession number | Matched aa residues | Freqa | Binding to
|
Verifiable by Y2Hd | ||
|---|---|---|---|---|---|---|---|
| tubby-Nb | Tulp1-Nb | tubby-Fc | |||||
| ORF matched to protein coding sequence | |||||||
| Zinc finger E-box binding homeobox 1 (Zeb1) (N) | NM_011546 | 903E-1021E | 2 | ~284.0X | ~56.7X | ~28.7X | ++ |
| Zinc finger protein 609 (Zfp609) (N) | NM_172536 | 1M-183E | 2 | ~53.2X | ~24.3X | ~2.3X | +/− |
| CCCTC-binding factor (Ctcf) (N) | NM_181322 | 136G-400T | 1 | ~117.4X | ~7.7X | ~44.9X | + |
| PC4 and SFRS1 interacting protein 1 (Psip1) (N) | NM_133948 | 152K-219K | 4 | ~196.3X | ~24.1X | ~24.0X | + |
| High mobility group (Hmg) box 2 (Hmgb2) (N) | NM_008252 | 89K-188E | 5 | ~312.4X | ~55.2X | ~21.7X | + |
| Hmg. box 2-like 1 (Hmgb2l1) (N) | NM_178017 | 170K-305G | 1 | ~29.8X | ~10.5X | ~5.0X | + |
| Hmg. nucleosomal binding domain 1 (Hmgn1) (N) | NM_008251 | 1M-49K | 2 | ~61.4X | ~35.0X | ~9.5X | − |
| Hmg. nucleosomal binding domain 2 (Hmgn2) (N) | NM_016957 | 1M-56K | 1 | ~33.4X | ~37.8X | ~9.5X | + |
| IQ motif containing GTPase activating protein 2 (Iqgap2) | NM_027711 | 1232E-1368E | 1 | ~53.2X | ~1.3X | ~1.8X | NT |
| Paf1/RNA polymerase II complex component (Rtf1) | XM_917502 | 69N-186K | 1 | ~6.4X | ~15.6X | ~6.2X | +/− |
| Tubby like protein 1 (Tulp1) | NM_021478 | 125D-243E | 1 | ~49.0X | ~32.8X | ~29.1X | +++ |
| ATP-binding cassette F1 (Abcf1) | NM_013854 | 57K-196G | 2 | ~68.2X | ~33.4X | ~27.2X | ++ |
| Structure specific recognition protein 1 (Ssrp1) | NM_182990 | 486E-619G | 2 | ~156.9X | ~39.7X | ~4.5X | + |
| Microtubule-associated protein 7 (Mtap7) | NM_008635 | 23A-114R | 1 | ~3.5X | ~5.6X | ~3.8X | − |
| Regulated endocrine-specific protein 18 (Resp18) | NM_009049 | 68V-134K | 1 | ~10.3X | ~2.9X | ~4.8X | + |
| Armadillo repeat containing, X-linked 4 | BC128507 | 419S-587N | 1 | ~40.1X | ~15.6X | ~3.1X | NT |
| Non-ORF | 0 | ||||||
Clone frequency. All clones, including 8 nuclear proteins, were independent. Only the minimal overlapping amino acid sequences are shown for the proteins with multiple clones.
The binding specificity of clonal phages vs. control phage. GST-free tubby-N and GST-Tulp1 were used for the assays.
The binding specificity to FLAG-tubby-F vs. to GFP-FLAG. Phage binding activities were representatives of three independent experiments.
+++, strong signal for X-gal staining; ++, intermediate signal; +, weak signal; +/−, triple dropout-resistant yeast clones with barely detectable signal; −, double-dropout resistant clones with no signal; NT, not tested. N: nuclear protein.
A total of 28 tubby-N-binding clones were sequenced (Table 1). All the sequenced clones were ORFs, and matched to protein sequences in the GenBank database, including multiple independent clones matching to 7 proteins (Zeb1, Zfp609, Psip1, Hmgb2, Hmgn1, Abcf1 and Ssrp1) with overlapping, but not identical, amino acid sequences. Eight identified proteins were nuclear proteins. Interestingly, one of the identified proteins was Tulp1 in the same family. Sequence analyses revealed no obvious amino acid or domain similarity among identified tubby-binding proteins.
Independent validation
To confirm their binding activities to tubby rather than to possible bacterial contaminants in the purified tubby-N, we expressed FLAG-tagged full-length tubby (tubby-F) and GFP in HEK293 cell line, purified both proteins with anti-FLAG mAb affinity columns. The binding analyses with the bait proteins purified from a different source and with a different method verified that most of the identified phage clones preferentially bound to immobilized tubby-F, rather than GFP control (Table 1). These data suggest that identified proteins bind to tubby-N instead of possible contaminants.
Moreover, we took the advantage of the availability of cDNA sequences in the identified phage clones and independently analyzed tubby-binding proteins by Y2H. Among 14 proteins tested, 10 tubby-binding proteins were independently verified by Y2H, including tubby-Tulp1 interaction (Fig. 6a and Table 1). In addition, FLAG-tagged full-length Hmgb2 was independently validated by protein pull-down assay using GST-tubby-F (Fig. 6b). Thus, at least ~71% (10/14) of the identified tubby-binding proteins were independently verified.
Fig. 6.
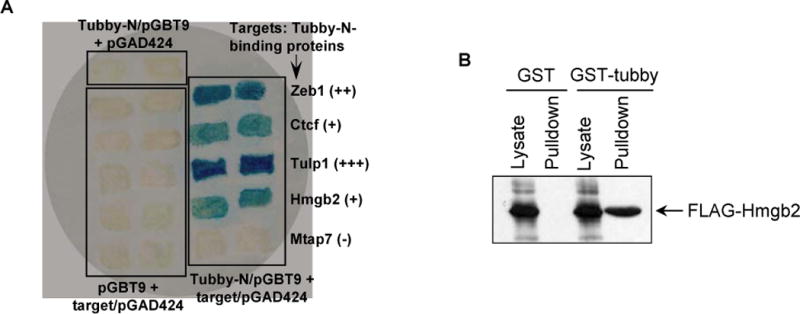
Independent verification of tubby-N-binding proteins. (a) Validation of tubby-N-binding proteins by Y2H assay. S. cerevisiae CG-1945 strain was co-transformed with pGBT9 plasmid expressing tubby-N and pGAD424 plasmid expressing indicated tubby-N-binding protein, and selected with -Leu/-Trp or -Leu/-Trp/-His dropout plates. Control plasmids were included. Dropout-resistant yeast colonies were re-grown on an YPD plate, lifted onto a filter and analyzed for β-galactosidase expression with X-gal. Blue color indicated that target proteins interact with tubby-N, leading to β-galactosidase expression. (b) Verification of tubby interaction with Hmgb2 interaction by protein pull-down assay. Cell lysate was prepared from FLAG-Hmgb2-expressing HEK293 cells, incubated with purified GST or GST-tubby, followed by glutathione resin. After washing, the resin was analyzed by Western blot using anti-FLAG mAb. Hmgb2 bound to GST-tubby, but not GST control.
To elucidate possible difference in molecular mechanisms between tubby and Tulp1, we analyzed the binding activity of identified phage clones to GST-Tulp1-N. The results showed that most of the identified proteins, including Zeb1, Psip1, Hmgb2, Hmgn2, Tulp1, Abcf1 and Ssrp1, had similar high binding activity to Tulp1-N. However, Ctcf was the only verified tubby-N-binding protein with minimal binding activity to Tulp1-N.
DISCUSSION
Tubby and Tulp1 are highly homologous in their C-terminal region, but diverse in the N-terminal half. Mutations in tubby and Tulp1 result in distinct disease profiles. Tubby mice with an autosomal recessive mutation in tubby gene develop adult-onset obesity, retinal and cochlear degeneration, whereas mutations in Tulp1 cause only retinal degeneration (Carroll et al., 2004; Ikeda et al., 2002). We speculated that their divergent N-termini may hold a key to elucidate the molecular mechanisms for different clinical manifestations. The identification and comparison of the proteins binding to their N-termini will shed light on their pathological mechanisms. For example, among 10 independently-verified tubby-N-binding proteins in this study, Ctcf is the only protein with relatively low binding activity to Tulp1-N and may be crucial to elucidate molecular mechanisms of tubby-related adult-onset obesity. On the other hand, the proteins binding to both tubby and Tulp1 may help delineate the common mechanisms for retinal degeneration. Moreover, among a total of 16 identified tubby-binding proteins, 8 tubby-binding proteins are nuclear proteins, suggesting that tubby likely interacts with multiple nuclear and cytoplasmic protein binding partners. These data are consistent with the previous report of tubby as a membrane-bound transcription factor (Santagata et al., 2001). Possible interaction between tubby and Tulp1 has been further verified by protein pull-down assay (unpublished data by N. Caberoy et al.). This further leads to the demonstration of their synergistic stimulation of retinal pigment epithelium (RPE) cell phagocytosis.
Given that proteins regulate almost every biological process, the exploration of protein interaction networks is expected to have major impacts on the understanding of biological systems, disease mechanisms and drug discovery (Colland and Daviet, 2004; Kelly and Stumpf, 2008). As a result, development of technologies to identify binding proteins is of major importance in functional proteomics (Timms, 2008). Y2H and mass spectrometry coupled with tandem affinity purification (MS-TAP) are two examples (Collins and Choudhary, 2008; Suter et al., 2008). However, these systems are limited by technical complexity, instrument requirements, labor and time commitments.
The improved ORF phage display is efficient to identify unknown bait-binding proteins. First, T7 bacteriophage has a robust growth rate. This substantially reduces the time needed to perform multiple rounds of phage selection and clone screening. Second, our dual phage display converts the cumbersome procedure of phage plaque assay into an efficient colorimetric assay for the binding analysis of individual clones. Although anti-T7 tag mAb is commercially available (Novagen) for Western blot and has been used for phage quantification by ELISA in literatures (Sheu et al., 2003), the mAb is not recommended by the company for ELISA due to limited sensitivity. Third, 3C protease cleavage further minimizes non-specific phage enrichment. Finally, high quality of the ORF phage display cDNA library is the key to ensure that the majority of identified phage clones encoded real proteins, rather than out-of-frame unnatural short peptides. ORF phage display integrated with these technical improvements is capable of rapidly identifying unknown bait-binding proteins.
Because of the availability of cDNA coding sequences for the bait-binding proteins in identified phage clones, protein-protein interactions can be conveniently and independently validated by different assay platforms, such as Y2H, co-immunoprecipitation or protein pull-down assay. On the contrary, cDNAs are not readily available for proteins identified by MS-TAP. Although co-immunoprecipitation coupled with Western blot or other detection methods is widely used to verify proteins identified by MS-TAP (Bauer and Kuster, 2003), co-immunoprecipitation itself is an integral part of MS-TAP (Collins and Choudhary, 2008). Thus, the validation in a sense is not truly independent.
Besides efficiency, sensitivity and accuracy are two other important parameters for the technologies of functional proteomics, a technology with low sensitivity will miss most of bona fide binding proteins. On the other hand, a highly sensitive technology with poor accuracy will identify a large number of false positives, which not only are misleading but also may consume enormous amount of time and effort to eliminate false positives. Although a previous study with tubby identified one tubby-binding protein from a C. elegans cDNA library by Y2H (Mukhopadhyay et al., 2005), it is unknown whether other identified tubby-binding proteins were not reported. Despite the identification of 16 tubby-binding proteins by analyzing 108 randomly picked phage clones in this study, it is possible to identify additional new tubby-binding proteins by screening more clones. While the error rate of the MS-TAP method is estimated at ~15%, the single epitope-tag method is ~50%, and Y2H is rated at 45–80% (Dziembowski and Seraphin, 2004). Our results showed that at least ~71% of tubby-binding proteins identified by ORF phage display are independently verified (i.e. <29% error rate). These data suggest that ORF phage display has competitive sensitivity and accuracy.
Compared with Y2H and MS-TAP, phage display is a more versatile technology, applicable not only to protein baits, but also to non-protein baits, including lipid, polysaccharide, DNA, RNA, and other molecules (Table 2) (Danner and Belasco, 2001; Sidhu, 2005; Young et al., 1997). Phage display with multimolecular baits, such as viruses, cells or even tissues or organs in in vitro or in vivo settings should be able to isolate biologically relevant endogenous proteins like viral receptors, and cell- or tissue-specific ligands (Valadon et al., 2006). Proteins with specific functions, including cell internalizing molecules and substrate degradomes of proteases could also be elucidated by phage display (Becerril et al., 1999; Sidhu, 2005). The caveat is that most of these applications have been previously described with Ab or random peptide libraries. Unfortunately, identified Abs and unnatural short peptides have minimal implications in protein biological networks. ORF phage display can efficiently identify real endogenous proteins with functional implications. For example, we have recently used phosphatidylserine as non-protein bait to identify 13 new phosphatidylserine-binding proteins from an embryonic ORF phage display library (Caberoy et al., 2009a). In another case, we used ORF phage display to identify several putative eat-me signals capable of stimulating RPE cell phagocytosis. Further characterization of one of the identified eat-me signals led to the elucidation of its novel activity as a ligand for a well-characterized phagocytic receptor, Mer receptor tyrosine kinase (unpublished data, Caberoy et al.). Unlike previous studies with random peptide libraries and Ab libraries (Becerril et al., 1999; Thapa et al., 2008), these studies illustrated that ORF phage display can efficiently identify real endogenous proteins with functional implications.
Table 2.
Comparison of different technologies for functional proteomics
| ORF Phage Display | Y2H | MS-based approach | |
|---|---|---|---|
| Capacity of the technologies | |||
| Protein-protein interaction | Yes | Yes | Yes |
| Protein-polysaccharide interaction | Yes | No | Yes |
| Protein-lipid interaction | Yes | No | Yes |
| Protein-antibody interaction | Yes | No | Yes |
| Protein-RNA interaction | Yes | No | Yes |
| Protein-DNA interaction | Yes | Yesa | Yes |
| Protein-virus interaction | Yes | No | Nob |
| Protein-cell interaction | Yes | No | Nob |
| Protein-tissue interaction | Yes | No | Nob |
| In vivo selection | Yes | No | No |
| Protease substrate identification | Yes | No | Yes |
| Eat-me signal identification | Yes | No | No |
| Features of the technologies | |||
| Required time | ~2–7 daysc | ~1–4 monthsb | ~4–6 daysb, d |
| Screening scale | ≥1011 pfu | ~106−107 clones | Entire proteomeb |
| Sensitivity | Single clone | Single clone | Subfemtomole |
| Re-verification | |||
| Binding activity | Convenient | Convenient | Difficultb |
| Binding specificity | Convenient | Difficultb | Difficultb |
| cDNA for identified proteins | Available | Available | Not available |
| Instrument requirement | Minimal | Minimal | Demanding |
| Subcloning | Not required | Required | Requiredb |
| Skill required | Minimal | Demanding | Demandingb |
| Cost | Minimal | Costly | Costlyb |
Yeast one-hybrid system.
Open to interpretation.
A fully-automated system of ORF phage display could identify unknown bait-binding proteins in as fast as ~2 days.
Estimated time for MS-TAP with ectopic gene expression in mammalian cells.
The procedures for phage panning and colorimetric binding analysis of individual clones in ORF phage display are similar to ELISA with minimal requirement for skills and instrument, and can be conveniently adapted by individual laboratories. Compared with Y2H and MS-TAP, ORF phage display is a convenient technology for protein-protein interactions. On the other hand, it can be fully automated for high throughput screening to delineate global and pathway-specific protein interaction networks and therapeutic targets. A fully-automated system could identify bait-binding proteins in as fast as ~2 days.
In conclusion, we have made 3 technical improvements to phage display with cDNA library. Together, these improvements transform the conventional phage display into a powerful technology of functional proteomics with efficiency, versatility and convenience comparable to other well-established technologies. We have used the improved technology to efficiently identify a panel of new tubby-N-binding proteins. The technology is broadly applicable to bait proteins or non-protein molecules with the potential for far-reaching impact on our understanding of protein biological networks, disease mechanisms and drug discovery.
Acknowledgments
This project was supported by NIH R01EY016211, P30-EY014801 and Research to Prevent Blindness. NBC is a recipient of a Fight for Sight postdoctoral fellowship. YZ is supported by a pre-doctoral fellowship from China Scholarship Council. We thank Drs. W. Kong and X. Song for technical assistance, and Dr. A Hackam for the critical reading of the manuscript.
References
- Abbasi AH, Garzozi HJ, Ben-Yosef T. A novel splice-site mutation of TULP1 underlies severe early-onset retinitis pigmentosa in a consanguineous Israeli Muslim Arab family. Mol Vis. 2008;14:675–682. [PMC free article] [PubMed] [Google Scholar]
- Ansuini H, Cicchini C, Nicosia A, Tripodi M, Cortese R, Luzzago A. Biotin-tagged cDNA expression libraries displayed on lambda phage: a new tool for the selection of natural protein ligands. Nucleic Acids Res. 2002;30:e78. doi: 10.1093/nar/gnf077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A, Kuster B. Affinity purification-mass spectrometry. Powerful tools for the characterization of protein complexes. Eur J Biochem. 2003;270:570–578. doi: 10.1046/j.1432-1033.2003.03428.x. [DOI] [PubMed] [Google Scholar]
- Becerril B, Poul MA, Marks JD. Toward selection of internalizing antibodies from phage libraries. Biochem Biophys Res Commun. 1999;255:386–393. doi: 10.1006/bbrc.1999.0177. [DOI] [PubMed] [Google Scholar]
- Caberoy NB, Zhou Y, Alvarado G, Fan X, Li W. Efficient identification of phosphatidylserine-binding proteins by ORF phage display. Biochem Biophys Res Commun. 2009a;386:197–201. doi: 10.1016/j.bbrc.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberoy NB, Zhou Y, Li W. Can phage display be used as a tool to functionally identify endogenous eat-me signals in phagocytosis? J Biomol Screen. 2009b doi: 10.1177/1087057109335679. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll K, Gomez C, Shapiro L. Tubby proteins: the plot thickens. Nat Rev Mol Cell Biol. 2004;5:55–63. doi: 10.1038/nrm1278. [DOI] [PubMed] [Google Scholar]
- Chen J, Huber BT, Grand RJ, Li W. Recombinant adenovirus coexpressing covalent peptide/MHC class II complex and B7-1: in vitro and in vivo activation of myelin basic protein-specific T cells. J Immunol. 2001;167:1297–1305. doi: 10.4049/jimmunol.167.3.1297. [DOI] [PubMed] [Google Scholar]
- Colland F, Daviet L. Integrating a functional proteomic approach into the target discovery process. Biochimie. 2004;86:625–632. doi: 10.1016/j.biochi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Collins MO, Choudhary JS. Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr Opin Biotechnol. 2008;19:324–330. doi: 10.1016/j.copbio.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Cordingley MG, Callahan PL, Sardana VV, Garsky VM, Colonno RJ. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J Biol Chem. 1990;265:9062–9065. [PubMed] [Google Scholar]
- Crameri R, Suter M. Display of biologically active proteins on the surface of filamentous phages: a cDNA cloning system for selection of functional gene products linked to the genetic information responsible for their production. Gene. 1993;137:69–75. doi: 10.1016/0378-1119(93)90253-y. [DOI] [PubMed] [Google Scholar]
- Danner S, Belasco JG. T7 phage display: a novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc Natl Acad Sci U S A. 2001;98:12954–12959. doi: 10.1073/pnas.211439598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziembowski A, Seraphin B. Recent developments in the analysis of protein complexes. FEBS Lett. 2004;556:1–6. doi: 10.1016/s0014-5793(03)01357-7. [DOI] [PubMed] [Google Scholar]
- Faix PH, Burg MA, Gonzales M, Ravey EP, Baird A, Larocca D. Phage display of cDNA libraries: enrichment of cDNA expression using open reading frame selection. Biotechniques. 2004;36:1018–1022. 1024, 1026–1019. doi: 10.2144/04366RR03. [DOI] [PubMed] [Google Scholar]
- Garufi G, Minenkova O, Lo Passo C, Pernice I, Felici F. Display libraries on bacteriophage lambda capsid. Biotechnol Annu Rev. 2005;11:153–190. doi: 10.1016/S1387-2656(05)11005-9. [DOI] [PubMed] [Google Scholar]
- Hagstrom SA, North MA, Nishina PL, Berson EL, Dryja TP. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nat Genet. 1998;18:174–176. doi: 10.1038/ng0298-174. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda A, Nishina PM, Naggert JK. The tubby-like proteins, a family with roles in neuronal development and function. J Cell Sci. 2002;115:9–14. doi: 10.1242/jcs.115.1.9. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Shiva N, Ikeda A, Smith RS, Nusinowitz S, Yan G, Lin TR, Chu S, Heckenlively JR, North MA, et al. Retinal degeneration but not obesity is observed in null mutants of the tubby-like protein 1 gene. Hum Mol Genet. 2000;9:155–163. doi: 10.1093/hmg/9.2.155. [DOI] [PubMed] [Google Scholar]
- Jestin JL. Functional cloning by phage display. Biochimie. 2008;90:1273–1278. doi: 10.1016/j.biochi.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Kalnina Z, Silina K, Meistere I, Zayakin P, Rivosh A, Abols A, Leja M, Minenkova O, Schadendorf D, Line A. Evaluation of T7 and lambda phage display systems for survey of autoantibody profiles in cancer patients. J Immunol Methods. 2008;334:37–50. doi: 10.1016/j.jim.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Kehoe JW, Kay BK. Filamentous phage display in the new millennium. Chem Rev. 2005;105:4056–4072. doi: 10.1021/cr000261r. [DOI] [PubMed] [Google Scholar]
- Kelly W, Stumpf M. Protein-protein interactions: from global to local analyses. Curr Opin Biotechnol. 2008;19:396–403. doi: 10.1016/j.copbio.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Kruger DH, Schroeder C. Bacteriophage T3 and bacteriophage T7 virus-host cell interactions. Microbiol Rev. 1981;45:9–51. doi: 10.1128/mr.45.1.9-51.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS. The role of genes in the current obesity epidemic. Ann Acad Med Singapore. 2009;38:45–43. [PubMed] [Google Scholar]
- Li W, Handschumacher RE. Identification of two calcineurin B-binding proteins: tubulin and heat shock protein 60. Biochim Biophys Acta. 2002;1599:72–81. doi: 10.1016/s1570-9639(02)00402-8. [DOI] [PubMed] [Google Scholar]
- Lin HS, Talwar HS, Tarca AL, Ionan A, Chatterjee M, Ye B, Wojciechowski J, Mohapatra S, Basson MD, Yoo GH, et al. Autoantibody approach for serum-based detection of head and neck cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:2396–2405. doi: 10.1158/1055-9965.EPI-07-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low S, Chin MC, Deurenberg-Yap M. Review on epidemic of obesity. Ann Acad Med Singapore. 2009;38:57–59. [PubMed] [Google Scholar]
- Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. C. elegans tubby regulates life span and fat storage by two independent mechanisms. Cell Metab. 2005;2:35–42. doi: 10.1016/j.cmet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Naggert JK, North MA, Nishina PM. A candidate gene for the mouse mutation tubby. Nature. 1996;380:534–538. doi: 10.1038/380534a0. [DOI] [PubMed] [Google Scholar]
- Paschke M. Phage display systems and their applications. Appl Microbiol Biotechnol. 2006;70:2–11. doi: 10.1007/s00253-005-0270-9. [DOI] [PubMed] [Google Scholar]
- Pavoni E, Vaccaro P, Pucci A, Monteriu G, Beghetto E, Barca S, Dupuis ML, De Pasquale Ceratti A, Lugini A, Cianfriglia M, et al. Identification of a panel of tumor-associated antigens from breast carcinoma cell lines, solid tumors and testis cDNA libraries displayed on lambda phage. BMC Cancer. 2004;4:78. doi: 10.1186/1471-2407-4-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel M. Filamentous phage assembly. Mol Microbiol. 1991;5:1607–1613. doi: 10.1111/j.1365-2958.1991.tb01907.x. [DOI] [PubMed] [Google Scholar]
- Santagata S, Boggon TJ, Baird CL, Gomez CA, Zhao J, Shan WS, Myszka DG, Shapiro L. G-protein signaling through tubby proteins. Science. 2001;292:2041–2050. doi: 10.1126/science.1061233. [DOI] [PubMed] [Google Scholar]
- Sheu TJ, Schwarz EM, Martinez DA, O’Keefe RJ, Rosier RN, Zuscik MJ, Puzas JE. A phage display technique identifies a novel regulator of cell differentiation. J Biol Chem. 2003;278:438–443. doi: 10.1074/jbc.M208292200. [DOI] [PubMed] [Google Scholar]
- Sidhu SS. Phage Display in Biotechnology and Drug Discovery. CRC Press; Boca Raton, FL: 2005. [Google Scholar]
- Suter B, Kittanakom S, Stagljar I. Two-hybrid technologies in proteomics research. Curr Opin Biotechnol. 2008;19:316–323. doi: 10.1016/j.copbio.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Thapa N, Kim S, So IS, Lee BH, Kwon IC, Choi K, Kim IS. Discovery of a phosphatidylserine-recognizing peptide and its utility in molecular imaging of tumour apoptosis. J Cell Mol Med. 2008;12:1649–1660. doi: 10.1111/j.1582-4934.2008.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms JF. Protein technologies. Curr Opin Biotechnol. 2008;19:313–315. doi: 10.1016/j.copbio.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Valadon P, Garnett JD, Testa JE, Bauerle M, Oh P, Schnitzer JE. Screening phage display libraries for organ-specific vascular immunotargeting in vivo. Proc Natl Acad Sci U S A. 2006;103:407–412. doi: 10.1073/pnas.0506938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AC, Valadon P, Casadevall A, Scharff MD, Sacchettini JC. The three-dimensional structures of a polysaccharide binding antibody to Cryptococcus neoformans and its complex with a peptide from a phage display library: implications for the identification of peptide mimotopes. J Mol Biol. 1997;274:622–634. doi: 10.1006/jmbi.1997.1407. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Davis JL, Li W. Identification of tribbles homolog 2 as an autoantigen in autoimmune uveitis by phage display. Mol Immunol. 2005;42:1275–1281. doi: 10.1016/j.molimm.2004.11.020. [DOI] [PubMed] [Google Scholar]