

Abstract
MicroRNAs and AU Rich element (ARE)-mediated degradation of transcripts are thought to be two independent means of gene regulation at the post-transcriptional level. However, since their site of action is the same (3‘UTR of mRNA), there exists a high probability that specific miRNAs may bind to AREs and, thus, interact with ARE-binding proteins (ARE-BPs) to regulate transcript levels. In this study, we have characterized AREs as potential targets of hsa-miR-3134. An analysis of the global gene expression profile of breast cancer cell line MCF7 overexpressing miR-3134 revealed the presence of at least one AUUUA element in the 3′-UTRs of 63% of miR-3134 regulated protein coding genes. Quantitative RT-PCR or 3′UTR luciferase assays show that miR-3134 mediates an up to 4–8-fold increase in the levels of ARE bearing transcripts-SOX9, VEGFA, and EGFR, while mutated miR-3134 shows a decreased effect. The miR-3134-mediated increase in transcript levels was unaffected by treatment with transcription inhibitor (actinomycin D), indicating that miR-3134 enhances transcript stability. To investigate a possible interplay between miR-3134 and a prototype ARE-BP, HuR, we compared their overexpression transcriptome profiles. Interestingly, up to 80% of miR-3134-regulated genes were also regulated by HuR. Overexpression studies of HuR alone or in combination with miR-3134 shows that wt miR-3134 but not a mutated miR-3134 promotes stabilization of HuR-regulated transcripts SOX9, VEGFA, and EGFR as confirmed by qRT-PCR or RNA-immunoprecipitation experiments. Overall, this report suggests that collaboration between ARE-binding microRNAs and ARE-binding proteins could be a general mechanism of 3′-UTR mediated regulation of gene expression in human cells.
Keywords: MicroRNA, miR-3134, AU rich elements, HuR, Breast cancer
Introduction
Gene expression in eukaryotes is subject to extensive post-transcriptional regulation. It is well documented that mRNA decay pathways play an important role in regulation of various transcripts involved in oncogenesis, inflammatory pathways, hypoxia regulation, and Alzheimers disease.1,2 This underlines the importance to understand the mechanisms of transcript stability control. This work was undertaken to identify novel regulators of mRNA decay. There are two well-known mechanisms for regulating the transcript turnover. The first one involves the A/U-rich elements (ARE) and their binding proteins (ARE-BP),1-4 whereas, the second one is mediated by microRNAs (miRNA).5-7 Since both pathways act through conserved elements present in the 3′UTR, they may interact with each other. So far, there has been little research on the cross-talk between these pathways.8,9 Two reports showed an interaction of HuR with miR-122 and miR-16 in regulation of CAT-1 and COX-2 transcript levels, respectively.10,11 Vasudevan et al., demonstrated miR-369-3p-mediated recruitment of the AGO2-FXR1 complex (associated with micro-ribonucleoproteins) in translational activation of TNF-α.12 An interesting tristetraprolin (TTP) and miR-16 crosstalk was shown to be essential for ARE mediated degradation of TNF α.8 However, this claim was challenged recently by Helfer et al., who show that AREs can function independently of miRNAs in mouse and Drosophila cells.13
We report an effort to identify the miRNAs that bind to AREs where they may enhance or inhibit the action of ARE-BPs to regulate ARE bearing transcripts. The knowledge of such regulatory interaction will be important in understanding miRNA:gene control and may shed light on various diseases, including cancer. Our results suggest that ARE binding miRNAs exercise a strong effect on a subset of human ARE bearing transcripts and, thus, may perhaps aid the design and development of specific miRNA-based therapeutics modulating the stability of ARE bearing transcripts involved in tumorigenesis.
Results and Discussion
Identification of microRNAs that can strongly hybridize with AU rich consensus sequence
The published literature was thoroughly surveyed to come up with an ARE consensus sequence for comparison with the miRNA database for complimentarity. Generally accepted core AU rich motifs present in 3′UTR of labile eukaryotic transcripts are the pentamer (AUUUA) and the nonamer (UUAUUUAUU).14-16 For our analysis, we chose the nonamer because of its bigger length, and the fact that AUUUA pentamer was its subset. Moreover, the nonamer has been shown to be minimal ARE consensus required for specific mRNA destabilization function. Finally, ARE consensus “UUAUUUAUU” was compared against the entire human micro RNA database using an RNA hybrid software.17 RNAhybrid is a tool for determining the minimum free binding energy of hybridization between a long and a short RNA sequence. The hybridization energy values for all human miRNAs were tabulated (Supplementary Data S1). A list of top 25 candidate miRNAs is shown in Figure 1A. miR-3609 showed the highest hybridization value with the ARE consensus, followed by miR-3134 (Fig. 1A). We focused on mir-3134 as the best possible candidate as putative ARE binding miRNA as it shows high energy binding to the ARE consensus and, in addition, the pairing of miR-3134 to the ARE is in the 5′region of the miRNA, i.e the seed region, which is believed to be critical for gene regulation (Fig. 1B).
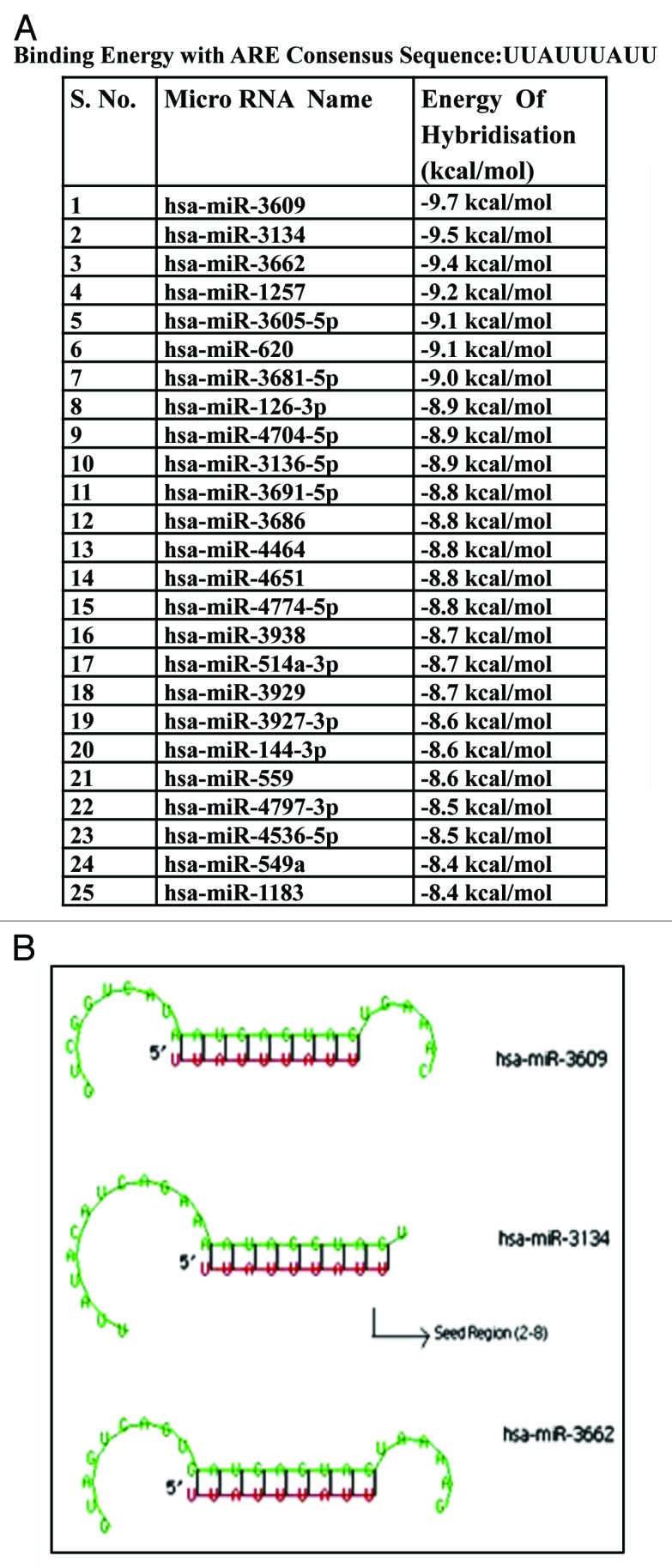
Figure 1. RNAhybrid results showing hybridization energy (A) and the binding (B) between human mature miRNAs and the ARE consensus sequence (UUAUUUAUU).
miR-3134 overexpression strongly affects ARE bearing transcriptome
As mir-3134 shows strong hybridization energy of binding with ARE consensus, we hypothesized that modulating miR-3134 levels may affect the ARE-bearing transcriptome subset. Thus, we overexpressed miR-3134 in MCF7 cells (Supplementary Data S2A), and analyzed the gene expression profile using Illumina Gene expression arrays. Genes showing more than 1.5-fold induction (p < 0.05) with respect to the vector control were considered to be differentially regulated. A total of 154 genes were found to be induced and expression of 66 genes was downregulated upon mir-3134 overexpression (Fig. 2A). A list of differentially expressed genes along with fold change is given in (Supplementary Data S3). Notably, based on the analysis done with the software AREscore, a total of 63% (114) of miR-3134 regulated protein coding genes (181) showed the presence of at least one AUUUA element in their 3′UTR (Fig. 2B; Supplementary Data S4).18 Two control sets of protein coding genes showed only 36.8% and 48.6% genes with AREs in their 3′UTR (Supplementary Data S4).

Figure 2. Microarray analysis of miR-3134-regulated genes. (A) Unsupervised hierarchial clustering of 220 statistically significant genes (> 1.5-fold, P < = 0.05) in response to miR-3134 overexpression (pc3134) as compared with control (pc) in MCF7 cell line. (A and B) refer to biological replicates. Genes are colored based on absolute expression levels of genes in pc- and pc3134-treated cells. (B) A total of 63% of mir-3134-regulated genes show the presence of at least one AUUUA (pentamer) element (AREscore database). A snapshot of top 20 genes with > eight AUUUA elements in 3′UTR is shown. (C) List of miR-3134-regulated genes showing differential expression in breast cancer based on The Genes-to-Systems Breast Cancer (G2SBC) Database. (D) Pie-chart distribution of significant gene ontology categories and pathways (P value < = 0.05) that were dysregulated by miR-3134-regulated genes.
Modulation of mRNA stability has been associated with various diseases including cancer. Since we used a breast cancer cell line, we compared the list of genes modulated by miR-3134 with those commonly mis-regulated in breast cancer using the G2SBC (The Genes-to-Systems Breast Cancer (G2SBC) Database) database and found that 50 out of 220 genes (22.7%) are shared between the two lists.19 These observations hint that miR-3134 regulated ARE transcriptome subset and, thus, levels of several cancer related genes may lead to breast cancer development/progression (Fig. 2C).
Further, an analyses of the differentially expressed genes was done with DAVID (http://david.abcc.ncifcrf.gov/) for Gene Ontology and Pathways, as described in Materials and Methods section (Fig. 2D). The gene ontology analysis of miR-3134-regulated genes showed genes involved in primary metabolic processes and response to extracellular stimuli, stress, and immune stress to be significantly regulated (p < 0.05). The pathway analysis found an enrichment of various signal transduction pathways like BMP receptor, C-MYC, and MAPK.20
miR-3134 promotes stability of VEGFA, SOX9, and EGFR transcripts
We confirmed the results obtained from gene expression profiling with real-time RT-PCR for a set of transcripts (VEGFA, EGFR, SOX9, DDIT3, and HLA-G) harboring AU-rich sequences in the 3′UTR, following transient and stable (polyclonal) overexpression of miR-3134 in MCF7 cells (Fig. 3A and B). We found that expression of VEGFA, SOX9, EGFR, and DDIT-3 was significantly induced by both transiently and stably overexpressed miR-3134 (Fig. 3A and B). While HLA-G levels remained unaffected in the transient overexpression assay, it showed up to 4-fold induction in stable cell line overexpressing miR-3134. Interestingly, introduction of a plasmid overexpressing miR-3134 carrying mutations in the seed region (pc3134mut) showed lower induction of these transcripts compared with the wild-type plasmid (pc3134) (Fig. 3A and B).

Figure 3. miR-3134 regulates stability of ARE transcripts-VEGFA, SOX9, and EGFR. (A) qRT-PCR data showing increase in transcript levels of VEGFA, SOX9, EGFR, and DDIT-3, in response to miR-3134 overexpression (pc3134) as compared with empty vector (pc) at 48 h post-transfection. GAPDH is used for normalization. The graphical data points represent mean ± SD of at least three independent experiments (*P < 0.05, **p > 0.1). (B) qRT-PCR data showing higher mRNA levels of VEGFA, SOX9, EGFR, DDIT3, and HLA-G in a polyclonal miR-3134 overexpressing stable cell line (PB3134) as compared with control (pBabe) cell line. GAPDH is used for normalization. The graphical data points represent mean ± SD of at least three independent experiments (*P < 0.05). (C) MCF7 cells were transfected with empty vector (pc) or plasmid overexpressing miR-3134 (pc3134) or mutated miR-3134 (pc3134mut) plasmid in presence of actinomycinD and transcript levels were measured at 1, 2, and 3 h post-treatment. Graphs showing actinomycin D time courses measuring VEGFA, SOX9, and EGFR mRNA decay kinetics in MCF7 cell line. mRNA half-lives resolved from the experiment are given in the text. The data points represent mean ± SD of at least three independent experiments (*P < 0.05). (D) Graph showing luciferase activity of 3′UTR luciferase constructs of VEGFA, SOX9, and EGFR co-transfected with plasmid overexpressing miR-3134 (pc3134) or mutated miR-3134 (pc3134mut), as compared with empty vector control. The graphical data points represent mean ± SD of at least three independent experiments (*P < 0.05). (E) Effect of wild-type (pc3134) or mutated (pc3134mut) miR-3134 overexpression as compared with empty vector control (pc) on EGFR 3′UTR-luciferase constructs bearing ARE (EGFR) or an ARE deletion (EGFRdel). The graphical data points represent mean ± SD of at least three independent experiments (*P < 0.05).
To determine if miR-3134 acts at the transcriptional/post-transcriptional level, we transfected MCF7 cells with control plasmid pcDNA3.1 (pc), miR-3134 overexpressing plasmid (pc3134) or mutated miR-3134 overexpressing plasmid (pc3134mut). The transfected cells were treated with transcription inhibitor, actinomycin-D for up to 3 h. The decay curves revealed that miR-3134 overexpression markedly stabilized VEGFA, SOX9, and EGFR transcripts in MCF7 cells as compared with the control or the mutated miR-3134 (Fig. 3C). The steady-state levels of the three transcripts declined to 20–35% (VEGFA t1/2 = ~1.3 h; SOX9 t1/2 = ~1.5 h; EGFR t1/2 = ~2.4 h) at 3 h post-actinomycin D addition in the control cells, whereas the transcript levels in miR-3134-overexpressing cells still remained at greater than 90%. These results indicate that miR-3134 exerts its effect largely at post-transcriptional level and promotes transcript stability.
To confirm that miR-3134 affects target transcript levels through the AREs in the 3′UTR, we performed 3′UTR luciferase assays. For this, we cloned 3′UTR fragments bearing the AREs of three miR-3134 inducible transcripts (SOX9, VEGFA, and EGFR) in luciferase reporter vector (pMIR-report), followed by transfection into MCF7 cells with or without pc3134 or pc3134mut. There was 2- to 3.5-fold increase in luciferase activity in response to miR-3134 overexpression and a decline in activity with pc3134mut. This suggests that the miR-3134-mediated increase of SOX9, VEGFA, and EGFR transcripts is through their 3′UTRs (Fig. 3D). We also performed ARE deletions. Since VEGFA and SOX9 carry a large number of AREs, we used our EGFR 3′UTR construct having a single ARE. Overexpression of miR-3134 did not increase the luciferase activity of the 3′UTR ARE deleted EGFR mutant (EGFRdel), confirming that miR-3134-mediated increase in EGFR levels is indeed ARE mediated (Fig. 3E).
Significant overlap in ARE-BP (HuR) and ARE-BM (miR-3134) affected transcripts
It is well known that ARE-binding proteins (ARE-BPs), like HuR, AUF1, TTP, etc., bind to 3′UTRs, where they regulate transcript stability.3 Considering that both miR-3134 and ARE-BP may have common binding sites, we tested for overlapping regulation. HuR has been shown before to be involved in mediating the stability of VEGFA and SOX9 transcripts. We confirmed the same by evaluating the effect of HuR overexpression on our 3′UTR luciferase constructs (Supplementary Data S2D). To assess miR-3134 and HuR interaction, we overexpressed pcDNA3.1 or HuR in MCF7 cells and obtained gene expression profiles with Illumina Gene expression arrays (Sandor). A total of 1,011 genes were found to be induced and 859 downregulated upon HuR overexpression (Supplementray Data S2B and S5). Interestingly, a comparison of mir-3134 and HuR gene expression signature revealed that 139 out of 154 genes upregulated by mir-3134 were also found to be induced upon HuR overexpression. Similarly, among the downregulated genes 49 out of 66 genes were commonly downregulated by HuR and mir-3134. Thus, up to 80% of miR-3134 regulated genes were also regulated by HuR (Fig. 4A and B).

Figure 4. miR-3134 promotes stability of HuR regulated transcripts. (A) Venn diagram representation of distribution of genes up/downregulated by miR-3134 or HuR compared with pc based on microarray gene expression data. (B) Chi-square 2 × 2 contingency table showing genes individually or jointly regulated by pc3134 and HuR as compared with pc. Chi-square with Y-ates correction value equals 2,677.561 with 1 degree of freedom. The two-tailed P value is less than 0.0001 suggesting that association between genes regulated by HUR and 3134 is extremely statistically significant. (C) qRT-PCR data showing that increase in transcript levels of VEGFA, SOX9, and EGFR in response to HuR is further enhanced upon HuR and miR-3134 co-expression. miR-3134-mediated increase in transcript levels is completely inhibited by HuR siRNA coexpression. GAPDH is used for normalization. The graphical data points represent mean ± SD of at least three independent experiments (*P < 0.05, ** < 0.01). (D) RNA-IP analysis of VEGFA, EGFR, and SOX9 transcripts associated with HuR using GAPDH as loading control. MCF7 cell lysates were subjected to RNP IP followed by quantitative RT-PCR analysis to measure the enrichment of target transcripts in HuR alone as compared with HuR and pc3134/pc3134mut cotransfection IP. IgG is used as negative control antibody. The graphical data points represent average of at least three independent experiments.
We next checked whether any functional interaction exists between the ARE binding microRNA (ARE-BM, mir-3134) and the ARE binding protein (HuR). We thus, tested the effect of either HuR overexpression or silencing (using HuR siRNA) alone or in combination with miR-3134 on the levels of the three targeted transcripts SOX9, VEGFA, and EGFR. In all the cases, we found that levels of transcripts were higher upon HuR and miR-3134 co-expression, as compared with HuR overexpression alone (Fig. 4C; Supplementary Data S2B and C). Also, a miR-3134-mediated increase in transcript levels was completely inhibited by HuR siRNA expression (Fig. 4C). These results suggest that miR-3134 promotes the stabilization effect of HuR.
To investigate effect on RNA binding of HuR with or without miR-3134 overexpression, we performed RNA-immunoprecipitation (RNA-IP) using FLAG antibody to pull down the transcripts associated with FLAG-HuR. MCF7 cells were transfected with FLAG-HuR alone or in combination with miR-3134 or mut-miR-3134. RNA protein complexes were pulled down using the anti-FLAG antibody, followed by qRT-PCR for the SOX9, VEGFA, or EGFR transcripts. In accordance with the previous results, we found a much higher transcript level associated with the HuR protein, when transfection was done along with miR-3134, as compared with HuR alone or with mut-miR-3134 (pc3134mut), thus supporting our hypothesis that miR-3134 promotes stability of specific HuR-regulated transcripts (Fig. 4D).
In summary, we report the discovery that a human miRNA (miR-3134) strongly affects the AU-rich sub-transcriptome. Its interaction with an ARE-BP (HuR) in promoting stability of the target transcripts is engaging though needs further exploration. To our knowledge this is the first report of an ARE-binding miRNA shown to significantly affect a subgroup of ARE transcriptome. Previous reports were case studies of a single target ARE:ARE-BP dynamics affected by binding of miRNAs at/close to the ARE.8,10-12,21 Another ARE-binding miRNA reported is miR-16. However, its effect has been studied only in the context of TNF-α or COX-2 and not explored further.8,11
This study lays foundation for further research by raising several questions: Apart from miR-3134, which other miRNAs bind to AREs? Similarly, do other ARE-BPs in addition to HuR interact with miR-3134? What are the mechanistic details of miR-3134 interaction with HuR? Whether miR-3134 directly binds to the ARE and then promotes HuR binding, or HuR binds to miR-3134 and subsequent structural changes promotes its binding to ARE. Are miR-3134 levels differentially regulated in cancer cells? Is there any significance of miR-3134-HuR interaction in stress or cancer biology?
It is finally intriguing that miR-3134, by promoting transcript stability, defies the general notion that miRNAs are involved in post-transcriptional gene silencing. With hardly any literature on miR-3134 to follow, it is too early to comment. However, discovery of this ARE-BM and its interplay with an ARE-BP has added new dimension to the ever-evolving field of mRNA decay. Further exploration of the mechanistic details of miR-3134-HuR collaboration may help in designing therapeutic strategies for regulating levels of ARE-bearing transcripts in various cancers.
Materials and Methods
Development of ARE consensus sequence to match with human miRNAs
A list of human mature miRNA sequences was obtained from miRBase, release 19.0. RNAhybrid was used to find matches of mature miRNA sequences with the ARE consensus (UUAUUUAUU).
Cell culture and plasmids
MCF7 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum with 1% Penstrep. The sequences (pre-miR-3134 with flanking sequences [~200 nt]), (mut-miR-3134 with flanking sequences [~200 nt]) and (the coding sequence of HuR tagged with N-terminal FLAG sequence) as well as (3′UTR sequences of specific genes bearing ARE elements) were PCR amplified from human genomic DNA using specific primers (Supplementary Data S6) and further cloned into pcDNA3.1/pBabe-puro/pmiR-Report vectors as described in the text. Plasmid EGFRdel was constructed by digesting the amplified fragment of the EGFR 3′UTR with HindIII, SpeI and DraI enzymes followed by religation of the HindIII-DraI and DraI-SpeI fragments (ARE containing DraI fragment was deleted). The religated fragment was cloned between the HindIII and SpeI sites of the pMIR-report vector. Control and HuR siRNAs were purchased from Sigma-Aldrich.
Luciferase assay
The 3′UTR luciferase assays were performed in MCF7, following the manual of the Dual Luciferase Reporter Assay kit (Promega).
Quantitative RT-PCR
Total RNA isolation was done using the Trizol reagent (Invitrogen). The levels of miR-3134 and RNU6B (used for normalization) were measured using the Taqman Assay (Applied Biosystem), while transcript levels of various genes were determined using Power SyBr Green master mix kit (Ambion) using specific primers (Supplementary Data S6). GAPDH was used as control for normalization of the data.
mRNA turnover assay
MCF7 cells were transfected with control (pc) or miR-3134-overexpressing vector (pc3134). Twenty-four hours post-transfection actinomycinD (to final conc-1 µm) was added to the culture media and cells were harvested at different time intervals (0–3 h) for RNA extraction with Trizol. cDNA was synthesized from purified RNA, and aRT-PCR was performed using specific primers as described above.
Ribonucleoprotein-immunoprecipitation
RNA-IP was performed for FLAG-tagged HuR-associated transcripts using antibodies against Flag Tag (Cell signaling, Cat. No-2368, 1:100 dilution) and IgG (Santa Cruz, Cat. No.-sc-2027, 1: 100 dilution). For detailed protocol, refer to Supplementary Data S6.
Microarray data processing and analysis
miR-3134 was cloned into overexpression vector pcDNA3.1, and MCF7 cells were transfected with overexpression plasmid (pc3134) or empty plasmid vector (pc) (Supplementary Data S2A). Total RNA was isolated 48 h post-transfection and sent for gene expression profiling using Illumina Gene expression arrays (Illumina.SingleColor.HumanHT-12_V4_0_R2_15002873_B, Sandor). The details of microarray data processing and analysis are given in Supplementary Data S6.
Gene ontology and pathway analysis
Biological analysis of differentially expressed genes was done for Gene Ontology and Pathways using DAVID (http://david.abcc.ncifcrf.gov/). Statistically significant ontologies and pathways were filtered based on P values < 0.05 (Obtained using Fischer Exact Test) with Benjamini Hochberg (FDR) correction.
Supplementary Material
Acknowledgments
RK acknowledges support from the Department of Biotechnology, GOI for a RGYI grant. We acknowledge Prof Alok Bhattacharya, School of Life Sciences, Jawaharlal Nehru University for extending the lab facilities funded and supported by the Department of Biotechnology for experimental work. SS1 thanks the Department of Biotechnology for a Junior Research Fellowship. SS2 acknowledges fellowship from the DBT-RA program. JKT acknowledges funding from NIPGR core grant. We acknowledge Dr Stefan Oehler for carefully editing this manuscript.
Glossary
Abbreviations:
- 3′UTR
3′ untranslated region
- AGO2
Argonaute 2
- ARE-BM-
AU rich element binding miRNA
- ARE-BP-
AU rich element binding protein
- AUF1
AU-rich element RNA-binding protein 1
- BMP
bone morphogenetic protein
- CAT-1
cationic amino acid transporter 1
- COX-2
Cyclooxygenase-2
- DAVID
database for annotation, visualization and integrated discovery
- DDIT3
DNA-damage-inducible transcript 3
- EGFR
epidermal growth factor receptor
- FXR1
fragile X mental retardation syndrome-related protein 1
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HLA-G
histocompatibility antigen, class I, G
- HuR
human antigen R
- MAPK
mitogen-activated protein kinase
- RNU6B
RNA U6B small nuclear
- SOX9
SRY (sex determining region Y)-box 9
- TNF-α
tumor necrosis factor-alpha
- TTP
Tristetraprolin
- VEGFA
vascular endothelial growth factor A
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/25482
References
- 1.Knapinska AM, Irizarry-Barreto P, Adusumalli S, Androulakis I, Brewer G. Molecular Mechanisms Regulating mRNA Stability: Physiological and Pathological Significance. Curr Genomics. 2005;6:471–86. doi: 10.2174/138920205774482954. [DOI] [Google Scholar]
- 2.Hollams EM, Giles KM, Thomson AM, Leedman PJ. MRNA stability and the control of gene expression: implications for human disease. Neurochem Res. 2002;27:957–80. doi: 10.1023/A:1020992418511. [DOI] [PubMed] [Google Scholar]
- 3.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–50. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–26. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 5.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 6.Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149:515–24. doi: 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee D, Shin C. MicroRNA-target interactions: new insights from genome-wide approaches. Ann N Y Acad Sci. 2012;1271:118–28. doi: 10.1111/j.1749-6632.2012.06745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–34. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 9.von Roretz C, Gallouzi IE. Decoding ARE-mediated decay: is microRNA part of the equation? J Cell Biol. 2008;181:189–94. doi: 10.1083/jcb.200712054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–24. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 11.Young LE, Moore AE, Sokol L, Meisner-Kober N, Dixon DA. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res. 2012;10:167–80. doi: 10.1158/1541-7786.MCR-11-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–4. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 13.Helfer S, Schott J, Stoecklin G, Förstemann K. AU-rich element-mediated mRNA decay can occur independently of the miRNA machinery in mouse embryonic fibroblasts and Drosophila S2-cells. PLoS One. 2012;7:e28907. doi: 10.1371/journal.pone.0028907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zubiaga AM, Belasco JG, Greenberg ME. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol. 1995;15:2219–30. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006;34(Database issue):D111–4. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lagnado CA, Brown CY, Goodall GJ. AUUUA is not sufficient to promote poly(A) shortening and degradation of an mRNA: the functional sequence within AU-rich elements may be UUAUUUA(U/A)(U/A) Mol Cell Biol. 1994;14:7984–95. doi: 10.1128/mcb.14.12.7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–17. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spasic M, Friedel CC, Schott J, Kreth J, Leppek K, Hofmann S, et al. Genome-wide assessment of AU-rich elements by the AREScore algorithm. PLoS Genet. 2012;8:e1002433. doi: 10.1371/journal.pgen.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mosca E, Alfieri R, Merelli I, Viti F, Calabria A, Milanesi L. A multilevel data integration resource for breast cancer study. BMC Syst Biol. 2010;4:76. doi: 10.1186/1752-0509-4-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobsen A, Wen J, Marks DS, Krogh A. Signatures of RNA binding proteins globally coupled to effective microRNA target sites. Genome Res. 2010;20:1010–9. doi: 10.1101/gr.103259.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.