

Abstract
Transposable elements (TEs) are known to influence the regulation of neighboring genes through a variety of mechanisms. Additionally, it was recently discovered that TEs can regulate non-neighboring genes through the trans-acting nature of small interfering RNAs (siRNAs). When the epigenetic repression of TEs is lost, TEs become transcriptionally active, and the host cell acts to repress mutagenic transposition by degrading TE mRNAs into siRNAs. In this study, we have performed a genome-wide analysis in the model plant Arabidopsis thaliana and found that TE siRNA-based regulation of genic mRNAs is more pervasive than the two formerly characterized proof-of-principle examples. We identified 27 candidate genic mRNAs that do not contain a TE fragment but are regulated through partial complementarity by the accumulation of TE siRNAs and are therefore influenced by TE epigenetic activation. We have experimentally confirmed several gene targets and demonstrated that they respond to the accumulation of specific 21 nucleotide TE siRNAs that are incorporated into the Arabidopsis Argonaute1 protein. Additionally, we found that one TE siRNA specifically targets and inhibits the formation of a host protein that acts to repress TE activity, suggesting that TEs harbor and potentially evolutionarily select short sequences to act as suppressors of host TE repression.
Keywords: transposable element, transposon, small interfering RNA, microRNA, epigenetics, post-transcriptional silencing, Arabidopsis
Introduction
Gene regulation occurs in plants and animals through the microRNA pathway, which is dependent upon the ability of an Argonaute family protein to bind small RNAs and guide the regulation of target mRNA transcripts (reviewed in ref. 1). In the model plant Arabidopsis thaliana, the protein Dicer-like1 (DCL1) processes stem-loop microRNA primary transcripts, and the resulting 21-22 nucleotide (nt) microRNAs are incorporated into the Argonaute1 (AGO1) protein.2,3 When a microRNA guides AGO1 to a fully or partially complementary target mRNA transcript, the result is either mRNA transcript cleavage or translational inhibition, both resulting in reduced protein production.4,5
Arabidopsis AGO1 is also involved in the regulation of gene expression through the plant-specific trans-acting small interfering RNA (tasiRNA) pathway.6 In this pathway, a microRNA-loaded AGO1 (or AGO7) protein cleaves a non-protein coding transcript, which is then targeted for double-stranded RNA (dsRNA) production by the RNA-dependent RNA Polymerase6 (RDR6) protein. This dsRNA is cleaved by DCL2 and DCL4 to produce multiple 21 nt tasiRNAs that are each loaded into AGO1 and can target complementary or partially complementary protein-coding mRNAs (reviewed in ref. 7). Gene regulation by the tasiRNA pathway is dependent on AGO1 activity and is necessary for proper cell type specification and development.8-10
In contrast to the roles of AGO1 in gene regulation, the other Argonaute proteins (Arabidopsis has ten Argonaute family proteins) AGO4, AGO6, and possibly AGO9 (but not AGO1), function to keep repetitive regions of the Arabidopsis genome in a transcriptionally silenced state. These other AGO proteins function in RNA-directed DNA methylation (RdDM) to establish and maintain transposable element (TE) silencing.11-13 Virtually all TEs in the plant body of Arabidopsis are epigenetically silenced, and the small RNAs produced from TEs by the RdDM pathway are 24 nt in length, which is larger than the 21-22 nt size of microRNAs and tasiRNAs that are incorporated into AGO1 and function to regulate genes14 (reviewed in ref. 15). Therefore, it has long been assumed that AGO1 does not function at TE loci or incorporate TE-derived small RNAs. However, it was recently discovered that upon TE epigenetic activation and RNA Polymerase II (Pol II) transcription, an endogenous RNAi pathway degrades TE mRNAs in an AGO1-dependent fashion.16 In this endogenous RNAi pathway, RDR6, DCL2, and DCL4 act to degrade Pol II-derived TE transcripts into 21-22 nt small interfering RNAs (siRNAs) that are incorporated into AGO1, similar to the tasiRNA gene-regulatory pathway mechanism. Thus, AGO1 does incorporate TE siRNAs; however, this occurs only when TEs are transcriptionally active and produce 21‒22 nt siRNAs.
Since AGO1 function is associated with gene regulation, we wondered if AGO1 could differentiate between an incorporated gene-regulating microRNA/tasiRNA and a TE-derived siRNA of the same size produced from a TE mRNA. Because TE 21‒22 nt siRNA production is amplified by the activity of RDR6,16,17 the potential exists for high levels of TE 21‒22 nt siRNAs to be incorporated into AGO1. AGO1 could utilize TE-derived 21‒22 nt siRNAs in a manner similar to a tasiRNA for the regulation of genic mRNAs that have full or partial complementarity. However, this regulation of genes by TE siRNAs would only occur when TEs are epigenetically active, providing an additional layer of influence of TE epigenetic regulation on genes. TEs are known to have regulatory effects on genes; however, these effects are only understood for the genes that flank TEs (by read-through transcription, for example) or are at least linked to the gene and represent a cis effect of TE regulation on gene expression. Some of these TE-induced cis effects can occur at large distances, particularly in mammalian genomes, where TEs have been shown to influence promoter and enhancer regions over 100 kb away from a gene’s protein coding region.18,19 Alternatively, through our proposed tasiRNA-like mechanism, non-TE neighboring genes could be indirectly regulated in trans by an unlinked TE during conditions of stress or other diverse environmental influences that are known to affect the epigenetic regulation of TEs.20,21
We recently demonstrated that one particular TE siRNA, termed siRNA854, is derived in its 21‒22 nt form from the Arabidopsis Athila6A family of centromeric gypsy LTR retrotransposons only when TEs are globally epigenetically activated and transcribed by Pol II (in particular Arabidopsis mutants, stressed long-term cell cultures, or in the chromatin-decondensed pollen vegetative nucleus).16 In contrast to the 24 nt siRNAs TEs produce when silenced, the 21‒22 nt versions of siRNA854 are produced upon transcriptional activation and are incorporated into AGO1. We have shown that the TE-derived siRNA854 inhibits translation of the UBP1b genic mRNA, which contains partial complementarity to siRNA854 in its 3′ UTR. Thus UBP1b protein accumulation is regulated by the epigenetic control of a TE, and the TE Athila6A can regulate a gene in trans through the production of a gene-regulating siRNA. In addition to Arabidopsis, a Drosophila TE piwi-interacting RNA (piRNA) has also been recently shown to regulate a genic mRNA.22 Although these studies have demonstrated the proof of principle that TE small RNAs can regulate genes in trans, a genome-wide analysis of this regulation has not been performed, and many critical unanswered questions about this type of gene regulation remain.23
We performed a genome-wide small RNA and gene expression analysis in a mutant background with the maximum possible TE transcriptional activation in order to best observe the effect of active TEs on gene regulation. We aimed to answer three critical questions. First, which genes are regulated by TE siRNAs? Second, how many genes are regulated by this mechanism? Third, is this regulation evolutionarily random/stochastic, or do TEs carry sequences to specifically target particular mRNAs? We have informatically identified and experimentally confirmed genic targets of TE siRNA regulation, and are now able to estimate how many genes are affected by this mechanism. We have also concluded that for at least one TE, the siRNA it produces and gene it regulates provides a clear benefit to the TE, providing direct evidence that TEs specifically encode siRNAs to target particular mRNAs and act as suppressors of host repression.
Results
When TEs are transcriptionally activated, AGO1 incorporates high levels of Athila TE siRNAs
To determine which genic mRNAs are regulated by TE siRNAs on a genome-wide level, we began by immunoprecipitating the AGO1 protein (AGO1-IP) from wild-type Columbia (wt Col) and ddm1 mutant inflorescences. We used an AGO1-specific antibody that has previously been shown to interact with AGO1 and no other Argonaute family proteins.16,24 The DDM1 gene encodes a swi2/snf2 family chromatin remodeling protein that uses ATP hydrolysis to compact histones and is responsible for the formation of heterochromatin.25,26 In homozygous recessive ddm1 mutant plants, heterochromatic marks such as CG methylation and histone 3 lysine 9 di-methylation are lost from the TE portion of the genome, resulting in global TE transcriptional activation.26-28 As the recessive ddm1 mutation is homozygous for increasing generations, TE mRNA expression and siRNA levels increase,16 while these plants accumulate developmental phenotypes and become less fertile.29 We used ddm1 plants that were homozygous for six generations (ddm1 F6), which accumulate high levels of TE transcripts and siRNAs16,30 in order to best observe any TE siRNA regulation of genic mRNAs. We performed three biological replicates (bioreps) of the AGO1-IP from each genotype (wt Col and ddm1 F6), and isolated and deep sequenced the AGO1-bound small RNAs. We obtained a minimum of 1,795,708 genome-matched and filtered reads for each biorep (see Materials and Methods). To ensure that the AGO1 protein is not incorporating small RNAs after the cell is ruptured, we spiked lysed inflorescence cell extracts with purified small RNAs from seedlings and subsequently performed an AGO1-IP and northern blot (Fig. S1). This experiment demonstrated that incorporation of a seedling-specific microRNA into AGO1 does not occur in vitro and, therefore, our AGO1-IP deep sequencing data represents AGO1 binding in vivo and not after cell lysis.
We first pooled the AGO1-IP bioreps for each genotype and compared each small RNA library to a total small RNA (non-IP) library made from the same genotypes and tissue.31 We determined that, compared to the total small RNA libraries, 21 nt small RNAs are 2.44-fold enriched in the AGO1-IPs (Fig. 1A), confirming previous findings that AGO1 binds primarily 21 nt small RNAs.32 In the total small RNA libraries, ddm1 has an increase in the number of 21 nt small RNAs compared to wt Col (Fig. 1A). The increase in 21 nt small RNAs is derived from degraded TE mRNAs (Fig. 1B), produced from the combined activity of DCL2, DCL4, and RDR6.16 In the ddm1 F6 AGO1-IP small RNA dataset, an increase in the 21 nt size class is not apparent (Fig. 1A). However, when we characterized the AGO1-IP small RNA library composition, we identified a 4.5-fold increase [337,779 reads per million (RPM)] in the amount of TE siRNAs present in the ddm1 F6 AGO1-IP compared to wt Col AGO1-IP (Fig. 1B). Therefore, we conclude that TE siRNAs are specifically enriched in AGO1 protein complexes when TEs are transcriptionally activated and processed into 21‒22 nt siRNAs.

Figure 1A–D. Analysis of AGO1 immunoprecipitated small RNA deep sequencing. (A) The accumulation of 21, 22, and 24 nucleotide (nt) small RNAs. Both total small RNAs and AGO1-immunoprecipitated (AGO1-IP) small RNAs were sequenced from wild-type (wt) Col plants with epigenetically silenced transposable elements (TEs) and ddm1 F6 mutants that have transcriptionally activated TEs. Library size is normalized by calculating reads per million (RPM) of 18-28 nt perfectly genome-matched small RNAs. (B) Categorization of small RNAs by genome annotation feature. In the AGO1-IP libraries, ddm1 F6 has a decrease in microRNAs and an increase in TE-derived siRNAs compared to wt Col. (C) Analysis of reproducibility of AGO1-IP small RNA deep sequencing biological replicates by microRNA R2 values. Pair-wise comparisons were done both between biological replicates (green squares) of the same genotype, and between genotypes (yellow squares). (D) Analysis of sequence diversity in AGO1-IP small RNA libraries. The diversity of individual sequences that accumulate over 10 RPM (left) or 100 RPM (right) in the ddm1 F6 AGO1-IPs increases 3-fold.
To perform an analysis of the total diversity of small RNA sequences within AGO1 protein complexes, we individually analyzed each biological replicate. First, using the microRNA distribution, we found that the similarity and reproducibility between individual bioreps and IPs within each genotype was very high (Fig. 1C). We performed pairwise biorep comparisons of the microRNA accumulation within each genotype and found that there was more variability within TE-activated ddm1 F6 mutants, where the lowest R2 value between two bioreps is 0.98936, while the lowest R2 value between two wt Col bioreps was 0.99757. In contrast, the similarity across genotypes was less (R2 = 0.9675-0.98261). We used the individual bioreps to determine that the increase in TE siRNAs in AGO1 protein complexes results in a statistically significant (P < 0.0001) 3-fold increase in the overall diversity of sequences within AGO1 protein complexes (Fig. 1D). Therefore, there is the potential to target more mRNAs for regulation in ddm1 F6 mutants due to the increased small RNA sequence diversity within AGO1 protein complexes.
We previously determined that the TE siRNAs that increase in abundance in ddm1 F6 mutant plants are not from all TEs, but rather from specific TE families.31 The three major contributor TE families to this increase are the Athila LTR retrotransposons, the AtGP1 LTR retrotransposons, and the AtENSPM5 family of DNA transposons. In addition, we characterized a control TE family, AtRep10D, a small non-autonomous Helitron element that produces large quantities of 24 nt siRNAs when silenced, but does not produce 21 nt siRNAs in ddm1 mutants (Fig. 1E).31 We annotated the TE siRNA portion of both the total and AGO1-IP small RNA libraries and characterized how many siRNAs these TE families contributed. We first annotated the distribution of these four TE families using 24 nt siRNAs, which are produced when TEs are associated with DNA methylation and are targets of RdDM. We find that of the 24 nt TE siRNAs present in either wt Col or ddm1 F6 total small RNA libraries, few are incorporated into AGO1 (Fig. 1E). The incorporation of a small amount of TE 24 nt siRNAs into AGO1 has been previously demonstrated in wt Col.32,33 We next focused on only the 21 nt siRNAs, as these are the size class that are specifically enriched in our AGO1-IP (Fig. 1A) and are known to regulate genes.14 We found that there are very few TE 21 nt siRNAs in wt Col total small RNA libraries or in wt Col AGO1-IPs. In ddm1 F6 mutants, the amount of 21 nt siRNAs associated with AGO1 increases 3-fold for AtGP1, 29-fold for AtENSPM5, and 133-fold for Athila compared to wt Col (Fig. 1E). We have previously shown that even though nearly all TE families are transcriptionally activated in ddm1 F6 mutants, only 15 TE families generate RDR6-dependent 21‒22 nt siRNAs when activated,31 and why only these TE families generate 21‒22 nt siRNAs remains enigmatic. Although siRNAs of all three of these TE families increase in AGO1 complexes when TEs are transcriptionally active, the 279,476 RPM increase in Athila 21 nt siRNAs (Fig. 1E) is the major contributor responsible for the increase in total TE siRNAs in AGO1-IPs from ddm1 F6 mutants (Fig. 1B). In contrast to the 24 nt siRNAs, most TE 21 nt siRNAs present in either wt Col or ddm1 F6 seem to be incorporated into AGO1, as Athila 21 nt siRNAs are enriched 1.8-fold in ddm1 F6 AGO1-IPs compared to the ddm1 F6 total small RNA library (Fig. 1E).

Figure 1E–G. (E) Analysis of TE family contribution to the small RNA libraries. The three TE families that accumulate the most 21‒22 nt siRNAs in ddm1 mutants are shown: Athila, AtGP1, and AtENSPM5. AtRep10D is shown as a control for a TE family that produces only 24 nt siRNAs. SiRNA accumulation is shown for both the total and AGO1-IP small RNA libraries, and split into 24 and 21 nt size fractions. (F) A decrease in AGO1 microRNA accumulation occurs in ddm1 F6 small RNA libraries compared to wt Col. Not all microRNAs experience a decrease in accumulation; those that statistically significantly decrease (P < 0.05) are listed. (G) Small RNA northern analysis of total small RNA (left) and AGO1-IP small RNA (right). The increase in Athila-derived small RNAs is evident in both the total small RNAs and AGO1-IP; whereas the decrease in microRNA166 (miR166) is also evident in both contexts.
We next aimed to determine if AGO1 protein and/or transcript levels increased to accommodate the additional 434,171 RPM TE siRNAs incorporated into AGO1 in ddm1 F6 mutants compared to the wt Col AGO1-IP, or if particular small RNAs were flooded out of AGO1 by these TE siRNAs. We found that the levels of the AGO1 transcript, the levels of the AGO1-targeting microRNA168, the slicing of the AGO1 mRNA by microRNA168, and the AGO1 protein levels, all do not increase in ddm1 F6 mutant individuals (Fig. S2). Therefore, we conclude that the level of AGO1 does not change to accommodate the large volume of TE siRNAs incorporated in ddm1 F6 mutants. Instead, we found that TE siRNAs accumulate in AGO1 protein complexes at the expense of the exclusion of microRNAs (Fig. 1B). The levels of cDNA- and intron-matching siRNAs, as well as non-TE intergenic siRNAs, also decrease in ddm1 F6 (Fig. 1B); however, these are minor changes relative to the statistically significant (P < 0.0001) decrease of microRNA accumulation in the ddm1 AGO1-IPs (Fig. 1F). To determine which microRNAs are decreased in ddm1 AGO1-IPs, we calculated the accumulation for each microRNA in each biorep and performed individual statistical tests to determine which microRNAs are significantly (P < 0.05) decreased. Figure 1F lists the microRNAs significantly decreased in ddm1 AGO1-IPs. These microRNAs represent many of the most abundant microRNAs in wt Col AGO-IPs, and just these 11 microRNAs account for a 2.3-fold decrease (305,900 RPM) in ddm1 AGO1-IPs. We verified the increase in Athila TE siRNAs and decrease in microRNAs in AGO1-IPs using small RNA northern blots from total and AGO1-IP small RNAs (Fig. 1G). In Figure 1G, the decrease in microRNA166 (miR166) is apparent in both the AGO1-IP and total small RNA fractions in ddm1 F6 mutants. To determine if the reduction in microRNAs has an effect on the regulation of the microRNA-target mRNAs, we performed qRT-PCR and 5′ cleavage RACE RT-PCR to measure the abundance of un-cleaved and cleaved target mRNA. Using microRNAs with low, medium, and high levels of abundance in wt Col AGO1-IPs, we were unable to detect any change in the un-cleaved steady-state transcript levels or microRNA-directed cleavage patterns of the microRNA-target mRNAs (Fig. S3). Therefore, we conclude that there is still enough microRNA present in AGO1 or in other AGO complexes in ddm1 F6 mutants such that the decrease in microRNAs in AGO1 protein complexes does not disrupt the regulation of these target genes.
Identification of TE siRNAs incorporated into AGO1 and their predicted genic mRNA targets
To identify which TE siRNAs potentially regulate genes, we filtered the AGO1-IP datasets to identify TE siRNAs present in AGO1 protein complexes over 100 RPM. We used the 100 RPM as an arbitrary cut off level, as all known inflorescence-expressed and experimentally verified functional microRNAs and tasiRNAs accumulate over this level in our wt Col AGO1-IP dataset (the average microRNA accumulates to 12,521 RPM, and tasiRNA TAS1C-D4 to 2,604 RPM). SiRNAs that accumulate below this level likely would not alter the expression of their target mRNAs at a detectable level. In AGO1-IPs from wt Col, only five 21‒22 nt TE small RNAs accumulate to this level, and are from the Vandal4 (a combined 314 RPM), Athila4 (242 RPM), AtGP3 (228 RPM), RathE2_cons (166 RPM), and HelitronY1B (147 RPM) TE families (Table S1). These TE small RNAs have the potential to regulate a few target genic mRNAs (see below); however, in the ddm1 F6 mutant, 437 TE siRNAs accumulate to at least 100 RPM in AGO1 protein complexes (Fig. 2A; Table S1). Therefore, the gene regulatory potential increases due to TE siRNA accumulation when TEs are transcriptionally active. Of the 437 21‒22 nt TE siRNAs, 80.5% are 21 nt and 19.5% are 22 nt (Fig. 2B). In addition, 98.6% of the 437 siRNAs are derived from the Athila family of TEs, mostly from the Athila2 and Athila6A families, as the Athila siRNAs account for 184,005 RPM (18.4%) of all of the ddm1 F6 AGO1-IP siRNAs (Fig. 2C). These siRNAs are generated from the 3′ non-protein coding region of Athila, as well as the ancient and degraded envelope protein-coding domain (Fig. 2D). Interestingly, portions of this 3′ non-protein coding region of Athila elements display high nucleotide identity between Athila subfamily member elements (Fig. S4), suggesting this region is under positive selective pressure (see Discussion section). The average accumulation of an Athila 21‒22 nt siRNA from Table S1 is 427 RPM, but they can range over 10,000 RPM for an individual 21 nt siRNA. Compared to wt Col AGO1-IPs, the average fold-increase of the ddm1 F6 437 TE siRNAs is 989, with some 21 nt siRNAs having as high as a 5,000-fold increase, demonstrating that transcriptional activation of TEs results in particular TE siRNAs incorporating into AGO1 to high levels.

Figure 2. Analysis of the TE siRNA content in AGO1-IPs. (A) The total number of TE siRNAs that accumulate over 100 RPM in AGO1-IPs from wt Col and ddm1 F6. (B) The relative siRNA size distribution of the siRNAs from (A). (C) The TE family characterization of the TE siRNAs from (A). Nearly all of the TE siRNAs that accumulate in AGO1 over 100 RPM in ddm1 F6 mutants are from the Athila family of LTR retrotransposons. (D) Athila 21‒22 nt siRNA production comes primarily from the ancient envelope (env) protein coding region and the 3′ non-protein coding region of the Athila2 and Athila6A TE families. Twenty-one nt siRNAs are shown in red, while 22 nt siRNAs are depicted in green. (E) The number of predicted target genic mRNAs using the TE 21‒22 nt siRNAs sequenced in AGO1-IPs from (A). See manuscript for details on how the prediction of genic target mRNAs was performed.
To determine which genes are potential targets of the five TE small RNAs that accumulate to over 100 RPM in wt Col, and the 437 TE siRNAs from ddm1 F6 AGO1-IPs, we used the psRNA microRNA target prediction software (see Materials and Methods).34 We identified 65 genic mRNAs as predicted targets of the five TE small RNAs that accumulate in wt Col AGO1-IPs, and 3,102 predicted genic mRNA targets of the 437 TE siRNAs in ddm1 F6 AGO1-IPs (Fig. 2E; Table S2). We did not continue our analysis of the 65 genes potentially regulated by the five TE small RNAs that accumulate in wt Col, as we do not currently understand the biogenesis of these small RNAs (whether they are TE-encoded microRNAs), and therefore experimental validation would be difficult. We focused instead on the 3,102 mRNA targets of siRNAs produced by transcriptionally active Athila TEs, as the regulation of these target mRNAs is under TE epigenetic control. Therefore, Athila is a suppressed repository of potential gene regulation by siRNAs, with a 47.7-fold increase in potential TE siRNA targets when epigenetically activated in ddm1 F6 mutants compared to wt Col. The predicted genic mRNA targets, the TE siRNA that is predicted to target the mRNA, and the E-value score of the siRNA/mRNA target match are shown in Table S2. Although we identified 3,102 predicted genic mRNA targets of TE siRNAs in ddm1 F6 mutants, 826 of these targets are predicted to be inhibited on the translational level and, therefore, their mRNA accumulation levels are not expected to change in ddm1 F6 mutants (Table S2). We focused on the other 2,276 genes, which are predicted to be cleaved by the TE siRNA, generating sliced transcripts without a 5′ cap that are subject to mRNA degradation and can be assayed by mRNA expression analysis.
Global gene expression analysis identifies putative genic mRNAs targeted by TE siRNAs
We next used global expression analysis to determine which genes potentially targeted by TE siRNAs (from Fig. 2E; Table S2) show decreased mRNA expression in ddm1 F6 mutants. We performed three bioreps of an Affymetrix ATH1 gene expression microarray of mRNA from both wt Col and ddm1 F6 inflorescences, the same genotypes and tissues used in the small RNA-seq experiments from Figure 1. We performed pair-wise comparisons of probe set expression values between normalized gene expression values for each biorep and found high degrees of similarity between bioreps of the same genotype (Fig. 3A). As a control for probe sets with increased expression in ddm1 F6 plants, we analyzed the 1,155 TE probe sets on the array (red points in Fig. 3B)35 and found 159 (13.8%) have a statistically significant (P <0 .05) >2-fold increase in expression. The TEs present on the array are not representative of the global TE distribution, as repetitive genome content was selected against when designing the array probes. We next identified 403 genes that decrease (> 2.0-fold) to a statistically significant level (P < 0.05) in ddm1 F6 mutants compared to wt Col (blue points in Fig. 3B; Table S3). Although these genes have decreased expression in ddm1 F6 mutants, the mechanism of their regulation may differ. Gene expression is influenced by the epigenetic regulation of neighboring TEs in both plant and animals (reviewed in refs. 15 and 36). Transcripts originating in TEs may generate alternative genic transcriptional start sites, express aberrant transcripts that result in the silencing of the gene, or the TE may act as a cis-regulatory element altering the regulation of the gene’s promoter (reviewed in ref. 37). To remove the genes that have decreased expression in ddm1 F6 mutants, but may be regulated by a neighboring TE (and, thus, not regulated by a TE siRNA in trans), we determined if each gene identified from our expression analysis contained a TE or TE fragment within its gene space. We defined the gene space as the exons, introns, untranslated regions, and 1 kb upstream and downstream of the transcriptional start and stop sites. We used the 1 kb arbitrary criteria for surrounding gene space because we calculated the median intergenic space of the gene-rich chromosome arms as 690 bp. Although individual examples of TEs acting on neighboring genes at large distances through interaction with distal enhancers are known,18,19 these few examples come from large mammalian genomes with greater intergenic distances, and expanding our definition of gene space in Arabidopsis would result in reduction of the number of testable genes. One hundred and five (26.1%) of the 403 genes that displayed decreased expression in ddm1 F6 plants contained a TE or TE fragment in their gene space (Table S3). The remaining 298 genes are not subject to regulation by a neighboring TE (at least within 1 kb), and are therefore candidates for decreased regulation in ddm1 F6 due to the activity of trans-acting TE siRNAs. These 298 genes represent 1.3% of the genes assayed on the ATH1 microarray.
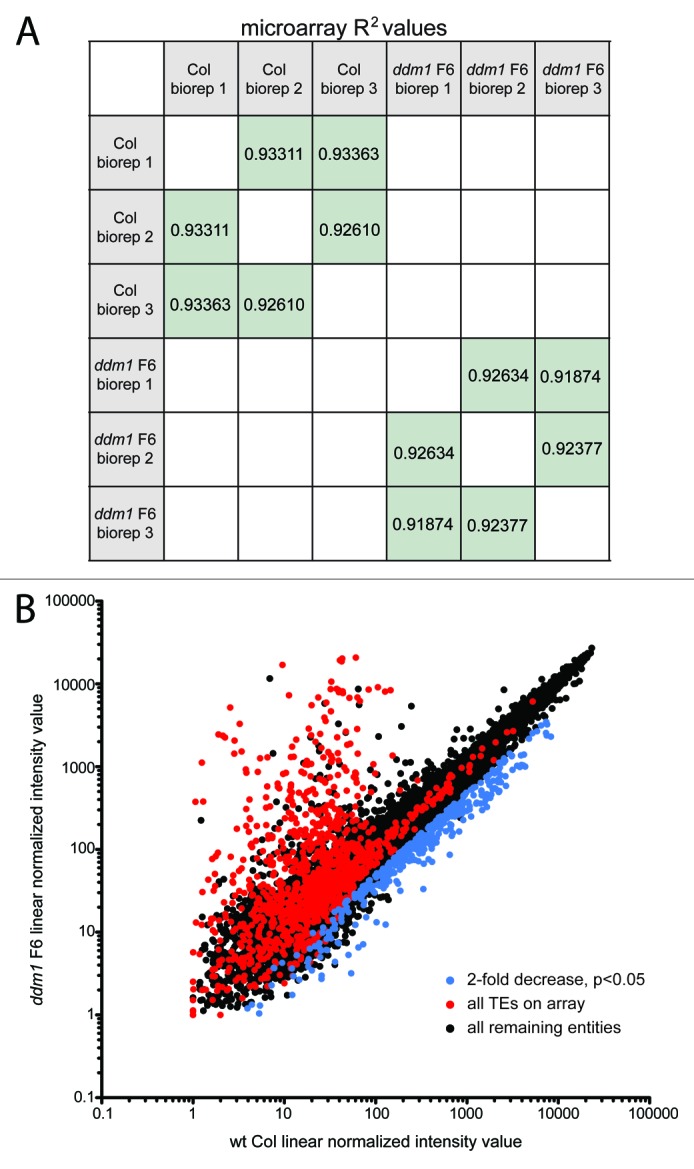
Figure 3. Gene expression analysis in ddm1 F6 mutants. (A) R2 values of ATH1 gene expression microarrays between biological replicates of the same genotype. Pair-wise comparisons were performed between biological replicates of the same genotype. (B) MAS5.0 linear normalized intensity values for each entity represented on the ATH1 array. The TEs present on the array are colored in red, and many of these show the predicted increase in expression in ddm1 F6 mutants. Genes with a decrease in expression (> 2-fold decrease, P < 0.05) are shown in blue.
To identify global targets of TE siRNAs, we compared the 2,276 mRNA targets predicted to be cleaved from TE siRNAs accumulating in the ddm1 F6 AGO1-IPs (Fig. 2E) to the 298 genes with decreased expression in ddm1 F6 (and no TE in the gene space). We found 27 genes that overlap in these datasets, which is not statistically different from the expected overlap between two sets of genes of these sizes. Table S4 lists the gene annotation, the targeting TE siRNA, and gene expression change in ddm1 F6 mutants for the 27 gene targets we informatically identified. All 27 of our identified genes are potentially targeted by 21 nt TE siRNAs, and the average accumulation of these siRNAs in ddm1 F6 AGO1-IPs is 279 RPM, while the accumulation of some TE siRNAs ranges as high as > 800 RPM. We utilized this gene list to draw candidate mRNA targets for experimental validation.
Validation of genic mRNA targets of TE siRNAs
We experimentally validated TE siRNA regulation of a subset of the target mRNAs from Table S4. We chose a subset of targets to test with a broad range of TE 21 nt siRNA accumulation in AGO1 protein complexes (123‒816 RPM) and a range of predicted strength of the complementarity between the TE siRNA and target mRNA (psRNA microRNA target prediction E value = 2‒4.5). We tested these targets in two assays. First, we performed qRT-PCR on the target genes in wt Col, ddm1 F6, and ddm1/rdr6 plants. This assay will confirm the expression decrease in ddm1 F6 plants compared to wt Col detected in the gene expression microarray from Figure 3. In addition, all 21‒22 nt siRNAs generated from the 3′ region of the Athila TE are dependent on a functional RDR6 protein for their production.16 Therefore, we predict that any mRNA targeted for cleavage by a TE siRNA in ddm1 F6 plants will no longer be targeted in a ddm1/rdr6 double mutant, as TE 21‒22 nt siRNA biogenesis is abolished. The mRNA levels of the target should decrease in ddm1 F6 single mutants, and return to near wt Col levels in ddm1/rdr6 double mutants. Although the comparison between wt Col, ddm1 F6, and ddm1/rdr6 gene expression levels is a strong indicator of TE siRNA regulation, using this assay we could not definitively conclude that our individual predicted TE siRNA was specifically regulating the genic mRNA. Expression analysis in ddm1/rdr6 double mutants cannot inform us about sequence specificity, as this double mutant approach is not specific to one siRNA/mRNA direct target, and gene regulation in a ddm1/rdr6 double mutant could be due to an indirect effect and not our predicted TE 21 nt siRNA. As a gold standard for the regulation of a genic mRNA by a TE siRNA, we tested the regulation of some targets by directly interfering with the accumulation of specific TE siRNAs using sequence-specific short tandem target mimic (STTM) transgenes. STTM transgenes express transcripts that are partially complementary to a microRNA or siRNA, sponging and degrading the small RNA, and specifically decreasing the interaction between the small RNA and the target mRNA in a sequence-dependent manner.38 We generated STTM transgenes specific to particular TE siRNA sequences and transformed them into ddm1 F5 mutant plants to produce ddm1 F6 plants with individual STTM transgenes. We have confirmed the targeting of the genic mRNAs At2g16910/AMS, At4g30850/HHP2, and At1g57790 by the TE siRNAs we predicted using STTM transgenes (Fig. 4A, Table 1). In each of these cases, the decreased expression detected on the ddm1 F6 microarray was independently confirmed by qRT-PCR, and the average decrease in expression for these three genes was 1.91, less than the 2.53 average fold decrease detected for these three genes detected on the microarray (Fig. 4A, Table 1). Importantly, the decrease in expression detected in ddm1 F6 plants is dependent specifically on the predicted TE siRNA. Therefore, the regulation of these genic mRNAs is not regulated by TE activity and expression alone, but specifically by TE siRNAs produced from a separate locus. In addition, for the genic mRNA At4g30850 that encodes the HHP2 transmembrane receptor protein, we were able to detect the accumulation of a distinct 5′ cleavage site in ddm1 F6 single mutants (with TE expression and siRNA production), but not in ddm1/rdr6 double mutants (with TE expression and no siRNA production) (Fig. 4B), demonstrating that this site is cleaved by the predicted TE siRNA. Sequencing of this 5′ cleavage product identified that all cleavage occurs at one specific location within the pairing between the siRNA and mRNA (Fig. 4B). The experimentally confirmed genic mRNAs targeted by epigenetically regulated TE siRNAs are shown in Table 1. The average TE siRNA accumulation level of these experimentally confirmed regulators of gene expression is 358 RPM, while the average qRT-PCR expression decrease of the target genes in ddm1 F6 is 2.77-fold.

Figure 4. Validation of a subset of genes targeted by TE siRNAs. (A) qRT-PCR of three predicted target genes from Table S4. Their relative expression has been normalized to 1 in wt Col. In each, their expression decreases in ddm1 F6 mutants, and increases back to near wt Col levels in ddm1/rdr6 mutants that have TE activation but do not produce TE 21‒22 nt siRNAs. STTM transgenes in ddm1 F6 plants block the interaction between a TE siRNA and its mRNA target in a sequence-specific manner, and expression that was decreased in ddm1 F6 plants is increased back to wt Col levels. (B) 5′ cleavage RACE PCR analysis of the AT4g30850 (HHP2) gene. This gene is specifically cleaved by the TE siRNA in ddm1 F6 mutants, but not when the TE is epigenetically silenced (in wt Col) or when TEs are activated but do not produce 21‒22 nt siRNAs (in ddm1/rdr6 double mutants). ARF16 is shown as a control for consistent levels of sliced mRNA. The experimentally determined cleavage site of the HHP2 mRNA (arrow) is shown along with the number of times this cleavage site was sequenced and the total number of sequences analyzed. (C) Phenotypic analysis of pollen abortion in wt Col, ddm1 F6, ddm1/rdr6, and ddm1/STTM plants. The ddm1/STTM plants are the same as in (A) and specifically block the interaction between the At2g16910 (AMS) mRNA and the TE siRNA. The box plot “+” is the mean, the box is the 25th and 75th percentile, while the whiskers represent the 10th and 90th percentile. n, number of individual anthers analyzed. (D) qRT-PCR of three target genes that did not make the predicted list of targets (Table S4). See (A) for description of genotypes and normalization.
Table 1. Experimentally validated target genes of epigenetically-regulated TE siRNAs.

a The TE siRNA is on top and is drawn in the 5'→3' orientation. The target mRNA sequences is drawn on the bottom in the 3'→5' orientation. bPerfect base pair matches are shown as a line, imperfect non-canonical purine-pyrimidine base pairing is shown as dots.
Of the 27 predicted genic mRNAs targeted by TE siRNAs in AGO1-IPs, one of these has a well-characterized morphological phenotype. The gene Aborted Microspores (AMS) (At2G16910) is a basic helix-loop-helix transcription factor expressed in the immature floral inflorescence and in the developing anther that plays a critical role in pollen development. In ams loss-of-function mutants, pollen fails to develop and the plants are male sterile.39 We investigated the pollen abortion phenotype in wt Col and ddm1 F6 plants and showed that ddm1 F6 has statistically increased levels of pollen abortion (Fig. 4C). In ddm1/rdr6 double mutants, in which TEs are still transcriptionally activated but do not produce 21‒22 nt Athila siRNAs, pollen abortion decreases to near wt Col levels. Additionally, in ddm1 F6 plants that generate TE-derived 21‒22 nt siRNAs, and the AMS-targeting siRNA sequence is sequestered and/or degraded by a sequence-specific STTM transgene transcript, the interaction between the siRNA and AMS mRNA is blocked. This results in a reduction of the pollen abortion phenotype to near background levels (Fig. 4C). These data demonstrate that the regulation of the 50% reduction in expression of the AMS gene (Fig. 4A) manifests as a weak but apparent phenocopy of the ams loss-of-function mutation (Fig. 4C).
In addition to the predicted genic mRNA targets of TE siRNAs that we filtered using strict criteria (Tables S1‒4), there were additional mRNA targets that we felt were likely regulated by TE siRNAs, even though they were filtered out of our analysis. We have confirmed that three genic targets, which did not make our final list of 27 targets, are also directly targeted by TE siRNAs. These genic mRNAs include At4g07960/AtCLSC12 and At5g27600 (both filtered off the final list in Table S4 due to having a TE in its gene space) and At4g17610 (less than a 2-fold change by microarray analysis) (Fig. 4D, Table 1). Therefore, the current number of experimentally validated Arabidopsis genic mRNAs regulated by epigenetic TE activity and TE 21-22 nt siRNAs is seven, six of which are identified in Figure 4 plus UPB1b from McCue et al.16 UBP1b did not make our Table S4 list of predicted targets because the repression of UBP1b in ddm1 inflorescence is at the translational level,16 and the score of the strength of pairing between the Athila-derived siRNA854 and UBP1b relies on non-canonical base pairing40 that scores below the threshold we used. The lack of identification of UBP1b and the identification of three targets not on our final list (Fig. 4D) demonstrate that our identified 27 targets in Table S4 is an underestimate of the number of genic mRNAs regulated by TE siRNAs. In addition, for all seven confirmed gene targets of TE 21 nt siRNAs, we failed to detect any similarity between the genic mRNA and TE that extended beyond the short site of TE siRNA complementarity (data not shown), demonstrating that these target genes do not represent a class of missed TE annotation, but only share a small amount of identity to a TE, shown in Table 1.
To determine if the genes regulated by TE 21‒22 nt siRNAs are part of a common pathway or if particular cellular functions are specifically targeted, we performed a gene ontology classification on the genes downregulated in ddm1 F6 mutants, as well as our predicted and confirmed lists of target genes. We found primary metabolic processes (p = 8.54E-36), particularly carbohydrate and lipid metabolism, and catalytic activity (p = 1.16E-26) over-enriched in the list of total genes downregulated in ddm1 F6 mutants (Table S3). The enrichment of these gene classifications persisted in our list of genes downregulated in ddm1 F6 without TEs in their gene space (Table S3), and the list of 27 predicted genes regulated by TE 21‒22 nt siRNAs (Table S4). In fact, of our final list of seven confirmed targets (six from Table 1 plus UPB1b), five are involved in primary metabolic processes, suggesting this broad biological process is a specific target of regulation by TE siRNAs when TEs become epigenetically activated.
Gene regulation via TE siRNAs can act as a suppressor of TE silencing
To determine why TE 21 nt siRNAs regulate some genes, we focused on the critical unresolved question of whether the regulation of genes by TE siRNAs is simply due to random partial complementarity between siRNAs and genes, or if this regulation imparts some advantage to the host cell or TE that may be evolutionarily selected for over time.23 To address this question, we used the Athila6-derived siRNA854 and its target UPB1b genic mRNA. UBP1b is a protein predicted to play a role in the formation of stress granules, sites of translational repression of mRNAs.41 The animal UBP1b homolog, TIA-1, acts to repress viral translation, and is targeted by viral microRNAs that act as suppressors of host viral silencing.42,43 In plants, UBP1b protein is located in the nuclei of unstressed cells, and translocates to punctate cytoplasmic bodies in stress conditions (such as dark grown or osmotic stress conditions) or during the loss of heterochromatic control and TE activation in ddm1 mutants.16 Therefore, we hypothesized that UBP1b, and potentially stress granules, may act to translationally inhibit TE activity, and that Athila6 has retained the siRNA854 sequence as a mechanism to suppress the host UBP1b-based translational silencing of TEs.23
To investigate the role UBP1b plays in silencing Athila translation, we first confirmed that during stress conditions, UBP1b is indeed localized to stress granules and not P-bodies, which are sites of mRNA decay (reviewed in ref. 44). We created plants with the UBP1b protein fused to GFP and either the DCP2 protein (a P-body marker) or the G3BP protein (a stress granule marker) fused to RFP. We transformed these markers into wt Col and observed their localization in stressed root meristem cells grown without light. We determined that UBP1b-GFP fluorescence overlaps with the G3BP protein, which specifically marks the sites of stress granules (Fig. 5A). In contrast, UBP1b fluorescence did not overlap with the DCP2-RFP marked P-bodies (Fig. 5A). This data demonstrates this for the first time in whole Arabidopsis organs that the UBP1b protein is indeed a component of stress granules. We next aimed to determine if TE protein levels increase in ubp1b mutants, which would indicate that the wt UBP1b protein acts to repress TE protein levels. We raised a polyclonal antibody to the GAG capsid protein of the Athila6 retrotransposon. We used this antibody for Westerns using protein extracted from inflorescence tissue in a panel of single, double and triple mutants with various combinations of the ddm1, rdr6, and upb1b mutations. A representative Western of three independent experimental and biological replicates is shown in Figure 5B, with the levels and variation of GAG accumulation normalized to Actin across these biological replicates shown in Figure 5C. In wt Col or rdr6 single mutant plants, Athila6 is transcriptionally silenced16 and the level of GAG protein is virtually undetectable. In ddm1 F2 or F6 single mutants, Athila6 is transcriptionally activated, and the levels of GAG protein increase 4-fold. We next investigated ubp1b mutants, which are not in the reference Col background as are the ddm1 and rdr6 mutations, but rather in a WS background.16 We tested this upb1b mutant in WS, as well after introgressing the ubp1b mutation into Col over six generations. Some individual bioreps of any genotype with a WS background show low levels of GAG protein accumulation (Fig. 5B), but over the course of the three biological replicates the level of GAG protein does not show significant changes in wt WS or upb1b single mutants (in Col or WS backgrounds) compared to wt Col (Fig. 5C). In rdr6/ubp1b double mutants (which have a mixed Col/WS background), Athila6 TEs are still transcriptionally silenced, and GAG protein does not accumulate. In ddm1/ubp1b mutants, Athila6 elements are transcriptionally active (due to the ddm1 mutation), but GAG protein levels decrease compared to ddm1 single mutants, and we are unable to conclude if this decrease is due to the upb1b mutation or the mixed Col/WS background. We generated two types of ddm1/rdr6 double mutants in which TEs are transcriptionally active but these transcripts are not degraded by RNAi via RDR6. One is in a pure Col background, and one is in a Col/WS mixed background. We found that the background does play a role in the accumulation of GAG protein, as the mixed background plant accumulates higher GAG protein levels compared to the pure Col background. Most importantly, in each of the three biological replicates, the ddm1/rdr6/upb1b triple mutant displayed the highest level of GAG protein accumulation (Fig, 5B and C). We observed a 2-fold increase in GAG protein accumulation compared to the mixed-background ddm1/rdr6 double mutant (P < 0.05). These data demonstrate that the UBP1b stress granule protein plays a role in repressing Athila6 GAG protein production when the TE is transcriptionally activated (in a ddm1 mutant) and RNAi is abolished (in the rdr6 mutant). Therefore, UBP1b serves as a third line of defense in the cell against the activity of TEs, and is only required once transcriptional and post-transcriptional silencing have failed. We thus conclude that the inhibition of UPB1b protein production by the Athila-derived 21‒22 nt siRNA854 acts as a suppressor of a host TE silencing mechanism, demonstrating a derived function for an individual epigenetically regulated TE siRNA.

Figure 5. UBP1b is a stress granule protein that acts to repress Athila protein production. (A) Sub-cellular GFP localization of the UPB1b protein in dark-grown stressed root meristem cells. Plants also contained a transgene with RFP fused to either the DCP2 P-body protein, or the G3BP stress granule marker. (B) Western analysis of Athila6 GAG protein levels in double and triple mutant plants. Actin is shown as a protein loading control. The highest level of GAG protein is detected in ddm1/rdr6/ubp1b triple mutant plants. (C) Statistical analysis of GAG protein accumulation in three independent westerns as in (B). For each of the three analyses, GAG protein is highest in the ddm1/rdr6/ubp1b triple mutant plants. GAG protein levels in each western and for each sample were normalized to the level of Actin protein accumulation.
Discussion
TEs are appreciated as regulators of gene expression in cases where a TE or TE fragment neighbors the gene being regulated (reviewed in ref. 15). This type of regulation can occur due to a TE being in close proximity to a gene’s protein coding region, or at a larger distance if the TE influences a cis-regulatory element such as an enhancer element. Recently, two individual examples demonstrated that a TE can regulate a gene through a small RNA in trans from an unlinked position, without the gene containing any annotated TE fragment or homology to the TE outside of partial complementarity over the length of the small RNA.16,22 Here, we have demonstrated that gene regulation by TE siRNAs is far more pervasive than the previous two examples. We have gone from two experimentally validated targets of TE small RNAs (nanos from Drosophila and UPB1b from Arabidopsis) to seven in Arabidopsis (Table 1), with an additional 24 likely targets informatically predicted by our analysis (Table S4). We are now able to estimate the total number of genes targeted by TE siRNAs, and we estimate that this number lies between 20‒300 (see below). In addition, we have demonstrated that one genic target of TE siRNAs plays a direct role in repressing TE activity, and this data provides evidence that these TE siRNA-target mRNA interactions provide the TE with a replicative advantage that may be evolutionarily selected for. In this case, the siRNA (siRNA854) comes from a non-protein coding region of the Athila6 retrotransposon, and acts as a suppressor of a host mechanism to repress TE activity.
Number of target genes
We have experimentally validated seven genes (0.03% of the tested Arabidopsis genes) that are regulated by TE siRNAs. Although this is a low number, it is significant, as we have demonstrated a methodology that can be used to identify additional genes regulated by TE siRNAs, and this number serves as a lower bound for the number of genes regulated by TE siRNAs. In addition to these seven confirmed targets, an additional 24 targets are excellent bioinformatic candidates based on their complementarity to a TE siRNA, their decreased expression in ddm1 F6 mutants, and their lack of a neighboring TE (Table S4). Overall, we conservatively estimate that at least 20 genes are targeted by TE siRNAs, and this number may easily be more than 10-fold higher. Our upper estimate of the number of genes regulated by TE 21‒22 nt siRNAs in Arabidopsis is 300, as this is the number of genes with decreased expression in ddm1 F6 plants without a neighboring TE. This is likely an overestimation, as not all of these genes will be regulated by this one mechanism. For example, some of these genes may be regulated by neighboring TEs that lie over 1 kb from the gene’s start and stop sites that we used as a cutoff in our analysis. This overestimation may account for why the overlap is not greater between the genes with decreased expression in ddm1 F6 mutants and the predicted targets of AGO1-incorporated ddm1 TE siRNAs.
The upper bound estimate of Arabidopsis genes regulated by TE 21‒22 nt siRNAs is difficult to estimate for the following reasons. First, we have used stringent arbitrary cutoffs, and, therefore, many TE siRNAs and genic mRNAs were not considered. For example, we both limited our scope to the TE-derived siRNAs that accumulated to at least 100 RPM and to only the 403 genes that we have experimentally identified as having at least a 2-fold decrease (and P < 0.05) in expression in ddm1 F6 plants. Although limiting our scope to those genes with at least a 2-fold decrease makes verification of these targets easier, it likely does not take into consideration any TE-regulated genes that experience a more subtle change in expression, underestimating the total number of genes that experience regulation by TE siRNAs. Second, like UBP1b, many of the genes targeted by TE siRNAs may not show decreased mRNA accumulation, as both plant and animal siRNAs can inhibit protein production on the translational level.5 Using only mRNA expression data, the number of genes regulated by translational repression of TE siRNAs is difficult to gauge, and since translational inhibition cannot be assayed on a high-throughput genome-wide level, translational repression is overlooked in our analysis. For example, we predicted 826 genes to be targeted on the translational level by TE 21‒22 nt siRNAs that are incorporated into AGO1 (Table S2), but these predicted targets were not investigated further. Third, we investigated only one tissue at one developmental stage, and additional regulation will likely take place as the genic transcriptome changes from tissue to tissue. Lastly, we have chosen analysis with STTM transgenes as the gold standard to validate TE siRNA/genic mRNA targeting (Fig. 4). This method is scientifically accurate, but laborious and, therefore, has limited the scope and number of target genes we have experimentally validated.
Decrease in target gene expression
The level of decrease of the genes regulated by TE siRNAs is relatively minor compared to either knockout mutations or targeting by a microRNA. Knockout mutations occur on the genetic level and affect all copies of the potential transcript, and gene regulation by microRNAs stems from thousands of RPM of the microRNA accumulating in AGO1-IPs (Fig. 1F). We observe that a range of 126‒816 RPM TE siRNA accumulation is sufficient for regulation of their respective target genes (Table 1). However, this is a 15‒100-fold decrease in levels of the TE siRNAs accumulating in AGO1 compared to the accumulation of the average microRNA. Therefore, the regulation by TE siRNAs is likely more of a 2‒5-fold modulation of mRNA levels rather than a destruction of all mRNA and absolute lack of protein production. However, in the case of the target gene AMS, enough gene expression change is generated by the TE siRNAs to see a visible though subtle phenotype (Fig. 4C).
Types of targeted genes
Two observations stem from the identities of the genes regulated by TE siRNAs. First, we demonstrated that one target mRNA, UPB1b, encodes a protein that specifically acts as a host cell defense mechanism to inhibit TE activity (Fig. 5). This suggests that one set of TE siRNA target genes likely includes the host proteins responsible for TE surveillance and repression, and that the ability to regulate a gene via a TE siRNA has evolved from the evolutionary arms race between the TE and host genome. TE siRNAs may represent suppressors of host silencing and repression systems, allowing the TE to gain activity when some of its TE transcripts are degraded by the host small RNA defense system. The second observation is that the gene ontology category of “primary metabolic processes” is statistically enriched for the genes regulated by TE siRNAs. However, the purpose of these common-function targets is not clear.
TEs that participate in siRNA-mediated gene regulation
In ddm1 F6 mutants, nearly all TEs are transcriptionally reactivated.26 Of these, we previously identified 15 TE sub-families that are targeted by endogenous RNAi and produce 21‒22 nt siRNAs.31 Here, we have identified only five TE families whose TE siRNAs are incorporated into AGO1 (Athila, AtGP1, AtENSPM, Vandal, and Helitron). Of these, we find that only two families (Athila and AtENSPM) are able to regulate gene expression. Overall, we find no correlation between those TEs that produce 21‒22 nt siRNAs and genome position, copy number, or expression level of reactivated TEs in ddm1.31 After production of 21‒22 nt siRNAs, why some are incorporated into AGO1 and others are not is also currently unknown. We cannot determine if the most abundantly produced TE siRNAs are those that get incorporated into AGO1, because AGO1 protects these siRNAs from degradation, resulting in their high accumulation levels. However, we believe that the siRNAs that accumulate to the highest levels in AGO1 are most likely to regulate genes. We also must consider that the identification of only two TE families that can regulate genes may be a bias generated from the 100 RPM cutoff of TE siRNAs in AGO1 we used. Although other TE families do not produce many TE siRNAs that are incorporated into AGO1, we cannot definitively say that they have no regulatory effect on the genic transcriptome.
Of particular interest is the ability of the 3′ region of Athila LTR retrotransposons to produce large amounts of TE 21‒22 nt siRNAs (Fig. 2) and regulate multiple genes (Table 1; Table S4). The Athila 3′ region of siRNA production overlaps with the ancient envelope protein coding domain and the non-protein coding 1.6 kb region adjacent to the 3′ LTR (Fig. 2D). Therefore, this well-conserved region of Athila acts as a suppressed repository for gene regulatory siRNAs and has the potential to regulate hundreds of genes (Fig. 2E; Fig. S4, Table S2). Although the RDR6-dependent siRNA processing of Athila is a cellular response to inhibit the element’s proliferation, we speculate that Athila TEs may have retained this 3′ non-protein coding region to regulate a diverse set of genes as a secondary function of these siRNAs, similar to how tasiRNA-producing loci regulate suites of target genes. In addition, TE 21‒22 nt siRNA generating loci, such as the 3′ end of Athila, may themselves be conserved epigenetically regulated tasiRNA loci, and possibly represent the evolutionary origin of the other developmentally regulated tasiRNA-generating loci.
Our inability to detect similarity between the gene target and TE outside of the siRNA binding site suggests that specific genes are not targeted because of an unannotated TE fragment inserted within their gene space. Instead, we favor a stochastic model in which TE siRNAs are produced, and regulate somewhat random targets. If this targeting is beneficial to either the TE or host, this targeting will be selected and maintained.23 Another possibility is that short gene sequences are incorporated into TEs, a process referred to as trans-duplication.45 Since different organisms, and perhaps different strains of the same organism, have vastly different complements of TEs, the possibility exists that the genes regulated by TE siRNAs differ widely and contribute to the generation of species- or strain-specific variability.
TE siRNA accumulation in AGO1
When TE 21‒22 nt siRNAs are incorporated into the AGO1 protein in ddm1 mutants, the level of AGO1 protein does not increase; rather, the accumulation of specific microRNAs is reduced to accommodate the new TE siRNAs (Fig. 1; Fig. S2). We do not believe that the AGO1 protein is selectively binding TE siRNAs, but instead, the high level of TE 21‒22 nt siRNAs produced when TEs are transcriptionally activated in ddm1 mutants results in a flooding of AGO1 with TE siRNAs due to their sheer abundance at the expense of microRNAs. Why specific microRNAs experience reduced levels in AGO1 when TEs are active is currently unknown. We observe that the microRNAs that experience changes in AGO1 incorporation do so without consequence to the regulation of their target mRNAs (Fig. S3), possibly due to an excess of microRNA accumulation in AGO1 when TEs are epigenetically silenced. We speculate that there is a feedback mechanism that buffers the levels of particular microRNAs in AGO1 against drastic changes due to the critical mass of microRNA required for proper gene regulation, particularly for low-abundance microRNAs.
Many different TE siRNAs are produced in ddm1 plants; however, only some of them are incorporated to high levels in AGO1 protein complexes (Fig. 1; Table S1). Individual TE siRNAs are not as selectively enriched in AGO1 protein complexes (relative to the total TE small RNA pool) compared to the enrichment of individual microRNAs. The primary function of these TE siRNAs is likely the post-transcriptional degradation of TE mRNAs. However, we have found that particular TE siRNAs can regulate genic mRNAs due to their partial complementarityy and incorporation into AGO1. Why particular TE siRNAs are incorporated into AGO1 is likely due to their specific size generated by DCL4 and DCL2 (21 and 22 nt, respectively), their 5′ nucleotide,32 and their subcellular proximity to AGO1 protein.
Environmental and species-specific modulation of gene expression via TE siRNAs
We chose the ddm1 mutant for our analysis because it has a very high level of global TE transcriptional activity,28 and it produces very high levels of TE 21‒22 nt siRNAs.16 However, gene regulation in this mutant context was a proof-of-principle to determine how many and which genes are regulated in this manner. Now that we have rough answers to the questions of the number of genes regulated, the identity of those genes, and if TEs can purposely encode a siRNA to regulate a gene, our attention turns to biologically relevant cases of TE activation to determine the role of TE regulation of non-neighboring genes. First, in contrast to the wt Col Arabidopsis genome, other plants and animals have genomes with naturally active TEs and accumulate TE siRNAs as a product of the post-transcriptional host defense against TE activity.46-48 It will be interesting to determine if and how genomes with naturally active TEs deal with constant gene regulation produced from ever-present gene-regulating TE small RNAs. Second, environmental stresses such as large swings in temperature are known to epigenetically activate TEs,20,21 and we hypothesize that this transient TE activation will lead to transient gene regulation, potentially contributing an epigenetic effect on an organism’s response to stress.
Materials and Methods
Plant material
The mutant alleles used in this study are described in Table S5. Plants were grown under standard long day conditions at 22 °C. Inflorescence tissue was used in each experiment unless otherwise noted.
AGO1 immunopurification
The AGO1 protein was immunoprecipitated as in McCue et al.16 For the AGO1 western blot, protein was obtained by grinding inflorescence tissue with liquid nitrogen and homogenizing in 2 mL extraction buffer [100 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, and 5 mM DTT containing one tablet/10 mL protease inhibitor cocktail (Roche)] per gram of tissue. Protein was run on an SDS-PAGE gel, transferred by Trans Blot SD Semi-Dry transfer system (BioRad), and immunoblotted with α-AGO1 and α-ACT11 (loading control) antibodies (Agrisera AB).
Small RNA deep sequencing and analysis
Libraries were produced using the TruSeq Small RNA Sample Preparation Kit (Illumina). Each library was barcoded and sequenced in one lane of an Illumina Genome Analyzer IIx. The resulting sequences were de-multiplexed, adapter trimmed, and filtered on length and quality, and tRNA/rRNA and low complexity reads were removed. Small RNAs were matched to the Arabidopsis genome, and sequences that did not perfectly align were discarded. Library size was normalized by calculating reads per million of 18‒28 nt genome-matched small RNAs. Small RNAs were also matched to the TAIR10 (www.arabidopsis.org) and Repbase (www.girinst.org) annotation of the TE portion of the Arabidopsis genome using bowtie. To best handle multi-mapping sequences generated from repetitive regions of the genome, the bowtie modifiers “-best -M1 -strata” were employed.49 If more than one genome perfect match for a TE siRNA exists, only one random match is assigned per small RNA read. The raw sequencing and genome-matched small RNAs analyzed are available from NCBI GEO repository number GSE45990. Small RNA tracks and display of the data in Figure 2 were performed using the SiLoMa small RNA locus mapper of the UEA small RNA toolkit.50 Predictions of small RNA target genes were made using the psRNA-Target analysis tool.34 We used a psRNA-Target mRNA/small RNA pair scoring cutoff of E ≤ 5 points, which allows for some G:U wobble pairings (0.5 points each), insertions/deletions (2.0 points each), and non-canonical Watson-Crick pairings (1 point each), with 0.5 points added if the mismatch occurs in the small RNA 5′ seed region.51
Small RNA northern analysis
Total RNA was isolated using TRIzol reagent (Life Technologies), and small RNAs were concentrated with polyethylene glycol. Twenty-eight micrograms of small RNA-enriched RNA were loaded in each lane. Immunoprecipitated small RNA northern analysis was performed by directly isolating RNA from immunoprecipitations by TRIzol (Life Technologies) with no further small RNA size enrichment. Eight µg of RNA was loaded in each lane and the MicroRNA Marker (New England Biolabs) was used for size comparison. Gel electrophoresis, blotting, and cross-linking were performed as described in Pall et al.52 Athila probes were generated by randomly degrading a P32-labeled in vitro RNA transcript. PCR primers used to generate the probes are listed in Table S5.
Microarray and analysis
Wild-type Col and ddm1 F6 plants were grown side-by-side, and three non-overlapping pools of inflorescence tissue were collected. RNA isolation was performed using TRIzol reagent (Life Technologies) followed by RNeasy MinElute Cleanup Kit (Qiagen). Oligo-dT primed cDNA was labeled and hybridized to the Affymetrix ATH1-121501 gene expression microarray. Three biological replicates were performed for each genotype. Analysis of the microarray data was performed using the GeneSpring software suite (Agilent Technologies). Relative expression levels for the genes selected in Table S3 were calculated using gcRMA normalization. Figure 3 utilized MAS5.0 normalized data for improved data display. Gene Ontology categorization and statistical over-enrichment tests were performed using the Panther Classification System and Bonferroni correction for multiple testing.53 The raw microarray files, normalized dataset, and further experimental details are available from NCBI GEO repository number GSE46050.
Transgene construction
The 35S-driven short tandem target mimic (STTM) transgenes were generated by cloning the STTM sequence specific to each TE siRNA of interest into the BsrGI/SpeI-digested binary plasmid pB2GW7. The STTM sequences were generated as a 96 nt long DNA oligo (Sigma), amplified using primers with homology to the ends of the linearized plasmid, and recombined into the vector using In Fusion HD (Clontech).
The 35S:UBP1b-GFP transgene was produced by Gateway (Life Technologies) cloning the coding sequence (no introns) of UBP1b (At1g17370) into the binary plasmid pK7FWG2, resulting in a C-terminal protein fusion to eGFP. The 35S:DCP2-RFP and 35S:G3BP-RFP transgenes were produced by Gateway cloning the coding sequences (no introns) of DCP2 (At5g13570) and G3BP (At5g60980) into the binary plasmid pH7RWG2, resulting in a C-terminal protein fusion to RFP. PCR primer sequences are listed in Table S5.
Microscopy
Dark-grown etiolated seedlings were grown on 1/2X MS media for 7 d and their roots were analyzed using a C1 confocal microscope and NIS-Elements software (Nikon Corporation). The pattern of fluorescence accumulation was verified in 10 or more cells from individual plants.
Expression analysis by qRT-PCR
Three biological replicates were performed for each genotype. Each replicate consisted of a non-overlapping pool of individuals. qRT-PCR was performed and analyzed as in reference 31: cDNA was generated using an oligo-dT primer and Tetro Reverse Transcriptase (Bioline). qPCR was performed using SensiMix (Bioline) on a Mastercycler ep realplex thermocycler (Eppendorf). The At1g08200 gene was used as a reference in all qRT-PCR reactions. qRT-PCR primers are listed in Table S5. Relative expression was calculated using the “delta-delta method” formula 2-(ΔCP sample - ΔCP control), where 2 represents perfect PCR efficiency. All values were normalized relative to wt Col, where wt Col was set to 1. Statistical significance was calculated with an unpaired t-test using Welch’s correction. PCR primer sequences are listed in Table S5.
Cleavage RACE-PCR
Cleavage RACE-PCR was performed with the GeneRacer kit (Life Technologies). To select for cleaved products with a 5′-OH, the protocol was modified to begin with ligation of the RNA adapter oligo to 5 µg of the starting RNA sample. Subsequent RNA extraction, cDNA synthesis, and PCR amplification followed. Nested PCR was performed using the PCR primer sequences listed in Table S5.
Analysis of pollen sterility
Pollen sterility was analyzed by Alexander’s staining as in Peterson et al,54 except that the tissue was not fixed. Live anthers were subject to Alexander’s stain for 20 min at room temperature before being analyzed by light microscopy.
GAG protein accumulation by western
Protein was extracted from 0.2 g of fresh inflorescence tissue using 1 ml of extraction buffer (0.2 M Tris-HCl pH 7.0, 20 mM EDTA, 1.5 M Urea, and 2% Triton-X100). Quantification of soluble protein was performed by DC protein assay (BioRad). One hundred and twenty-five µg/ul of protein was mixed with 1:1 suspension buffer (100 mM sodium phosphate pH 7.5, 100 mM NaCl) and 2X SDS dye-loading solution (200 mM Tris-HCl pH 6.8, 4% SDS, 20% Glycerol, 400 mM DTT, 0.2% bromophenol blue) and boiled for 10 min. Protein was run on a 12% SDS-PAGE gel and transferred to PVDF membrane (Millipore). We raised a mouse polyclonal antibody against the Athila GAG epitope GDKAHQWEKS (Abmart), and used this at a 1:500 dilution. The ACT-11 antibody was used as a loading control and is described above.
Supplementary Material
Acknowledgments
The authors thank Christopher DeFraia and Kaushik Panda for assistance and Jay Hollick for critically reviewing this manuscript. ADM is a fellow of The Ohio State Center for RNA Biology. This work was supported by grants MCB-1020499 and MCB-1252370 to RKS from the U.S. National Science Foundation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/25555
References
- 1.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 2.Park W, Li J, Song R, Messing J, Chen X. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol. 2002;12:1484–95. doi: 10.1016/S0960-9822(02)01017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaucheret H, Vazquez F, Crété P, Bartel DP. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004;18:1187–97. doi: 10.1101/gad.1201404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumberger N, Baulcombe DC. Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc Natl Acad Sci USA. 2005;102:11928–33. doi: 10.1073/pnas.0505461102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, et al. Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008;320:1185–90. doi: 10.1126/science.1159151. [DOI] [PubMed] [Google Scholar]
- 6.Vazquez F, Vaucheret H, Rajagopalan R, Lepers C, Gasciolli V, Mallory AC, et al. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell. 2004;16:69–79. doi: 10.1016/j.molcel.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 7.Vaucheret H. MicroRNA-dependent trans-acting siRNA production. Sci STKE. 2005;2005:pe43. doi: 10.1126/stke.3002005pe43. [DOI] [PubMed] [Google Scholar]
- 8.Garcia D, Collier SA, Byrne ME, Martienssen RA. Specification of leaf polarity in Arabidopsis via the trans-acting siRNA pathway. Curr Biol. 2006;16:933–8. doi: 10.1016/j.cub.2006.03.064. [DOI] [PubMed] [Google Scholar]
- 9.Fahlgren N, Montgomery TA, Howell MD, Allen E, Dvorak SK, Alexander AL, et al. Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr Biol. 2006;16:939–44. doi: 10.1016/j.cub.2006.03.065. [DOI] [PubMed] [Google Scholar]
- 10.Hunter C, Willmann MR, Wu G, Yoshikawa M, de la Luz Gutiérrez-Nava M, Poethig SR. Trans-acting siRNA-mediated repression of ETTIN and ARF4 regulates heteroblasty in Arabidopsis. Development. 2006;133:2973–81. doi: 10.1242/dev.02491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tran RK, Zilberman D, de Bustos C, Ditt RF, Henikoff JG, Lindroth AM, et al. Chromatin and siRNA pathways cooperate to maintain DNA methylation of small transposable elements in Arabidopsis. Genome Biol. 2005;6:R90. doi: 10.1186/gb-2005-6-11-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng X, Zhu J, Kapoor A, Zhu J-K. Role of Arabidopsis AGO6 in siRNA accumulation, DNA methylation and transcriptional gene silencing. EMBO J. 2007;26:1691–701. doi: 10.1038/sj.emboj.7601603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Havecker ER, Wallbridge LM, Hardcastle TJ, Bush MS, Kelly KA, Dunn RM, et al. The Arabidopsis RNA-directed DNA methylation argonautes functionally diverge based on their expression and interaction with target loci. Plant Cell. 2010;22:321–34. doi: 10.1105/tpc.109.072199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton A, Voinnet O, Chappell L, Baulcombe D. Two classes of short interfering RNA in RNA silencing. EMBO J. 2002;21:4671–9. doi: 10.1093/emboj/cdf464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–85. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 16.McCue AD, Nuthikattu S, Reeder SH, Slotkin RK. Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 2012;8:e1002474. doi: 10.1371/journal.pgen.1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalmay T, Hamilton A, Rudd S, Angell S, Baulcombe DC. An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell. 2000;101:543–53. doi: 10.1016/S0092-8674(00)80864-8. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki T, Nishihara H, Hirakawa M, Fujimura K, Tanaka M, Kokubo N, et al. Possible involvement of SINEs in mammalian-specific brain formation. Proc Natl Acad Sci USA. 2008;105:4220–5. doi: 10.1073/pnas.0709398105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakanishi A, Kobayashi N, Suzuki-Hirano A, Nishihara H, Sasaki T, Hirakawa M, et al. A SINE-derived element constitutes a unique modular enhancer for mammalian diencephalic Fgf8. PLoS One. 2012;7:e43785. doi: 10.1371/journal.pone.0043785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grandbastien M-A. [Stress activation and genomic impact of plant retrotransposons] J Soc Biol. 2004;198:425–32. [PubMed] [Google Scholar]
- 21.Tittel-Elmer M, Bucher E, Broger L, Mathieu O, Paszkowski J, Vaillant I. Stress-induced activation of heterochromatic transcription. PLoS Genet. 2010;6:e1001175. doi: 10.1371/journal.pgen.1001175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rouget C, Papin C, Boureux A, Meunier AC, Franco B, Robine N, et al. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature. 2010;467:1128–32. doi: 10.1038/nature09465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCue AD, Slotkin RK. Transposable element small RNAs as regulators of gene expression. Trends Genet. 2012;28:616–23. doi: 10.1016/j.tig.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Baumberger N, Tsai C-H, Lie M, Havecker E, Baulcombe DC. The Polerovirus silencing suppressor P0 targets ARGONAUTE proteins for degradation. Curr Biol. 2007;17:1609–14. doi: 10.1016/j.cub.2007.08.039. [DOI] [PubMed] [Google Scholar]
- 25.Jeddeloh JA, Stokes TL, Richards EJ. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat Genet. 1999;22:94–7. doi: 10.1038/8803. [DOI] [PubMed] [Google Scholar]
- 26.Zemach A, Kim MY, Hsieh P-H, Coleman-Derr D, Eshed-Williams L, Thao K, et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193–205. doi: 10.1016/j.cell.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gendrel A-V, Lippman Z, Yordan C, Colot V, Martienssen RA. Dependence of heterochromatic histone H3 methylation patterns on the Arabidopsis gene DDM1. Science. 2002;297:1871–3. doi: 10.1126/science.1074950. [DOI] [PubMed] [Google Scholar]
- 28.Lippman Z, Gendrel A-V, Black M, Vaughn MW, Dedhia N, McCombie WR, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430:471–6. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 29.Kakutani T, Jeddeloh JA, Flowers SK, Munakata K, Richards EJ. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc Natl Acad Sci USA. 1996;93:12406–11. doi: 10.1073/pnas.93.22.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature. 2001;411:212–4. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- 31.Nuthikattu S, McCue AD, Panda K, Fultz D, DeFraia C, Thomas EN, et al. The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21-22 nucleotide small interfering RNAs. Plant Physiol. 2013;162:116–31. doi: 10.1104/pp.113.216481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell. 2008;133:116–27. doi: 10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Zhang X, Liu J, Kiba T, Woo J, Ojo T, et al. Deep sequencing of small RNAs specifically associated with Arabidopsis AGO1 and AGO4 uncovers new AGO functions. Plant J. 2011;67:292–304. doi: 10.1111/j.1365-313X.2011.04594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39(Web Server issue):W155-9. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slotkin RK, Vaughn M, Borges F, Tanurdzić M, Becker JD, Feijó JA, et al. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell. 2009;136:461–72. doi: 10.1016/j.cell.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kines KJ, Belancio VP. Expressing genes do not forget their LINEs: transposable elements and gene expression. Front Biosci. 2012;17:1329–44. doi: 10.2741/3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan J, Gu Y, Jia X, Kang W, Pan S, Tang X, et al. Effective small RNA destruction by the expression of a short tandem target mimic in Arabidopsis. Plant Cell. 2012;24:415–27. doi: 10.1105/tpc.111.094144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sorensen A-M, Kröber S, Unte US, Huijser P, Dekker K, Saedler H. The Arabidopsis ABORTED MICROSPORES (AMS) gene encodes a MYC class transcription factor. Plant J. 2003;33:413–23. doi: 10.1046/j.1365-313X.2003.01644.x. [DOI] [PubMed] [Google Scholar]
- 40.Arteaga-Vázquez M, Caballero-Pérez J, Vielle-Calzada J-P. A family of microRNAs present in plants and animals. Plant Cell. 2006;18:3355–69. doi: 10.1105/tpc.106.044420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber C, Nover L, Fauth M. Plant stress granules and mRNA processing bodies are distinct from heat stress granules. Plant J. 2008;56:517–30. doi: 10.1111/j.1365-313X.2008.03623.x. [DOI] [PubMed] [Google Scholar]
- 42.Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, et al. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–68. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aparicio O, Carnero E, Abad X, Razquin N, Guruceaga E, Segura V, et al. Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res. 2010;38:750–63. doi: 10.1093/nar/gkp1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erickson SL, Lykke-Andersen J. Cytoplasmic mRNP granules at a glance. J Cell Sci. 2011;124:293–7. doi: 10.1242/jcs.072140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoen DR, Park KC, Elrouby N, Yu Z, Mohabir N, Cowan RK, et al. Transposon-mediated expansion and diversification of a family of ULP-like genes. Mol Biol Evol. 2006;23:1254–68. doi: 10.1093/molbev/msk015. [DOI] [PubMed] [Google Scholar]
- 46.Mitra R, Li X, Kapusta A, Mayhew D, Mitra RD, Feschotte C, et al. Functional characterization of piggyBat from the bat Myotis lucifugus unveils an active mammalian DNA transposon. Proc Natl Acad Sci USA. 2013;110:234–9. doi: 10.1073/pnas.1217548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nobuta K, Venu RC, Lu C, Beló A, Vemaraju K, Kulkarni K, et al. An expression atlas of rice mRNAs and small RNAs. Nat Biotechnol. 2007;25:473–7. doi: 10.1038/nbt1291. [DOI] [PubMed] [Google Scholar]
- 48.Nobuta K, Lu C, Shrivastava R, Pillay M, De Paoli E, Accerbi M, et al. Distinct size distribution of endogeneous siRNAs in maize: Evidence from deep sequencing in the mop1-1 mutant. Proc Natl Acad Sci USA. 2008;105:14958–63. doi: 10.1073/pnas.0808066105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moxon S, Schwach F, Dalmay T, Maclean D, Studholme DJ, Moulton V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics. 2008;24:2252–3. doi: 10.1093/bioinformatics/btn428. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y. miRU: an automated plant miRNA target prediction server. Nucleic Acids Res. 2005;33(Web Server issue):W701-4. doi: 10.1093/nar/gki383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pall GS, Codony-Servat C, Byrne J, Ritchie L, Hamilton A. Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piRNA by northern blot. Nucleic Acids Res. 2007;35:e60. doi: 10.1093/nar/gkm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(Database issue):D377–86. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peterson R, Slovin JP, Chen C. A simplified method for differential staining of aborted and non-aborted pollen grains. International Journal of Plant Biology. 2010;1:e13. doi: 10.4081/pb.2010.e13. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.