

Abstract
Regulatory elements in the 3′ untranslated regions (UTRs) of eukaryotic mRNAs influence mRNA localization, translation, and stability. 3′-UTR length is determined by the location at which mRNAs are cleaved and polyadenylated. The use of alternative polyadenylation sites is common, and can be regulated in different situations. I present a new method to identify cleavage and polyadenylation sites (CSs) at the genome-wide level. The approach is strand-specific, avoids RNA enzymatic modification steps that can introduce sequence-specific biases, and uses unique molecular identifiers to ensure that all identified CS originates from individual RNA molecules. I applied this method to create the first comprehensive genome-wide map of polyadenylation sites of the fission yeast Schizosaccharomyces pombe, comprising the analysis of 2,021,000 individual mRNAs that defined 8,883 CSs. CSs were identified for 90% of coding genes and 50% of ncRNAs. Alternative polyadenylation was prevalent in both groups, with 41% and 45% of all detected genes, respectively, displaying more than one CS. The specificity of the cleavage reaction was gene-specific, resulting in highly variable levels of heterogeneity in 3′-UTR lengths. Finally, I show that for both coding and non-coding genes, the most common regulatory motif associated with CSs in fission yeast is the canonical human AAUAAA sequence.
Keywords: S. pombe, Illumina sequencing, alternative polyadenylation, fission yeast, mRNA processing, unique molecular identifiers
Introduction
Most eukaryotic mRNAs carry a non-encoded tail of adenosine residues at their 3′ end [poly(A) tail]. Poly(A) tails are added in two separate steps: in the first one, nascent mRNAs are cleaved; subsequently, the polyA tail is added to the cleaved substrate. Both reactions are coupled and require the function of a complex multisubunit machinery, which is recruited cotranslationally to pre-mRNAs.1 Cleavage and polyadenylation in mammals are regulated by three major cis-acting sequences:2 the AAUAAA sequence or a closely related motif, located 15‒30 nucleotides upstream of the CS, a uridine-rich upstream sequence element (USE), situated 0‒20 nucleotides upstream of the AAUAAA motif, and a downstream sequence element (DSE), found 0‒20 nucleotides downstream of the cleavage site. In budding yeast, the AAUAAA motif is less conserved, the DSE is not present, and there is a poorly conserved efficiency element (EE) upstream of AAUAAA.3
Most mRNAs have untranslated regions after their coding sequences (3′ UTRs).2 3′ UTRs contain regulatory elements that are recognized by trans factors such as RNA-binding proteins and micro-RNAs, which regulate mRNA localization, stability, and translation. The position of the cleavage and polyadenylation site (CS) determines the length of the 3′ UTR, and, thus, the regulatory elements that will be included in the mature mRNA. The use of multiple polyadenylation sites for a specific mRNA (alternative polyadenylation, or APA) is widespread,4 and appears to be regulated in tissue- and developmental-specific manners. For example, there is a switch to the use of proximal CSs during cell proliferation,5 early development,6,7 and cancer.8 This leads to the production of shorter mRNA isoforms, which lack key binding sites for miRNAs and are therefore differentially regulated.
CSs can be identified from standard RNA-seq data by examining reads that span the boundary between the CS and the poly(A) tail. However, this approach is very inefficient because only a very small fraction of reads the can be used. Recently, a number of studies have employed next-generation sequencing-based approaches specifically targeted to the identification of CSs. These methods have been applied to budding yeast, worms, mammals, and plants (reviewed in ref. 9). I present here a novel technique for the systematic mapping and quantification of CSs. The approach is based on the isolation and sequencing of mRNA ends. It is easy to implement, strand-specific, avoids RNA enzymatic modifications that can cause sequence-specific biases, and uses unique molecular identifiers (UMIs) to eliminate PCR amplification artifacts. I used this approach to create the first genome-wide map of polyadenylation sites in the fission yeast Schizosaccharomyces pombe. The results revealed widespread use of APA in both coding genes and long non-coding RNAs, as well as highly variable levels of specificity in the choice of CSs. In addition, they showed that the most common polyadenylation signal used in fission yeast is the canonical human AAUAAA.
Results
A method for high-throughput mapping of cleavage and polyadenylation sites
Below I refer to the method as 3PC (for 3’ Poly(A) site mapping using cDNA Circularization). The strategy is summarized in Figure 1. After chemical fragmentation of total RNA, poly(A)-containing RNAs are purified using oligo(dT) magnetic beads. Isolated RNA fragments are reverse-transcribed using a primer (RT primer, Table S1) that contains an anchored oligo(dT) sequence10 and two primer binding sites for nested PCR amplification. The resulting cDNAs are circularized and used as templates for PCR amplification. A spacer that cannot be traversed by DNA polymerases is located between both primer binding sites and allows the use of the circularized cDNA as a template without the need for linearization.11 The nested PCR primers add Illumina-specific adaptors compatible with single or paired-end sequencing. 3PC is strand-specific, an essential feature for the analysis of the highly compact S. pombe genome, where overlapping transcripts are very common.12 The circularization strategy allows the bypass of RNA ligations, which are known to be sequence-dependent.11 The RT primers also contain a random sequence that serves as a unique molecular identifier (UMI).13,14 Therefore, each original cDNA is individually tagged and can be distinguished after PCR amplification. Single-end sequencing is performed on an Illumina platform starting from the side containing the oligo(dT) sequence, thus ensuring that the junction between the mRNA and the poly(A) sequence is reached in every clone. The data are analyzed using a straightforward and stringent bioinformatic pipeline (Fig. S1). First, identical sequences containing the same UMI are discarded, as they are derived from a single RNA fragment. This step removes PCR amplification artifacts and allows better quantification of poly(A) site usage. Second, UMIs and oligo(dT) sequences are removed from the reads. Third, reads are mapped to the S. pombe genome using the Bowtie aligner.15 Finally, to remove sequences that may originate from internal poly(A) sites, reads that map upstream of adenosyl stretches are discarded (see Materials and Methods for details). I applied this strategy to map cleavage and polyadenylation sites in exponentially growing S. pombe cells. Three libraries were generated from independent biological samples. The libraries were sequenced on either an Illumina Genome Analyzer II or a HiSeq 2000 platform using standard Illumina primers, producing a total of 15,626,208 reads (Table S2). 45.6% of the sequences were identified as redundant using UMIs, leaving 8,489,700 unique reads. After all processing steps (Fig. S1), 2,021,000 reads were mapped to unique locations in the S. pombe genome (Table S2). As a result of the use of UMIs, each of these reads corresponds to the CS of an individual mRNA molecule. To monitor the reproducibility of the protocol, the number of reads mapping to the annotated 3′ UTR of every fission yeast gene was quantified for each of the three independent experiments. The data showed very high reproducibility (Fig. S2), with all pair-wise Pearson correlation coefficients above 0.9. To validate the accuracy of the approach, I compared these data with 30 published CSs from three different laboratories mapped using low-throughput methods (Fig. S3).16-18 In every case, CSs mapped using the high-throughput approach overlapped exactly or were within 2 nucleotides of the published ones, demonstrating the ability of this strategy to determine CSs at high resolution.

Figure 1. Outline of 3PC protocol. Total RNA is chemically fragmented and purified using oligo(dT) magnetic beads. Poly(A)-containing fragments are used as substrates for reverse transcription, and the resulting cDNAs are circularized and used as templates for PCR amplification. Poly(A) and poly(T) sequences are indicated. “R” represents the random sequence of the unique molecular identifier, and “B” a multiplexing barcode. Sequences from the RT primer that are used for PCR amplification are represented in cyan and green. See Materials and Methods for more details.
Cleavage and polyadenylation site usage in S. pombe
99.7% of the mapped reads corresponded to annotated genes or non-coding RNAs (Table 1). As expected, there was a very strong bias toward 3′ UTRs (95.8% of all reads). The data provide a comprehensive survey of the CS landscape of S. pombe with an average of 360 reads per 3′ UTR, and 90% of 3′ UTRs with 10 reads or more. Genes for which no CS were detected were enriched in meiotically induced genes,19 in agreement with the low expression levels of these genes in vegetative cells.20 Coverage of ncRNAs was lower, with a mean number of reads of 34 and only 15% of genes with 10 reads or more. This is consistent with previous data showing that ncRNAs tend to be expressed at low levels in S. pombe.21
Table 1. Distribution of CSs among different genomic features.
| Poly(A) site mapping | Reads | Fraction |
|---|---|---|
| Non-coding RNAs |
47,594 |
2.4 |
| CDS |
18,725 |
0.9 |
| 3′-UTR |
1,935,301 |
95.8 |
| 5′-UTR |
13,131 |
0.6 |
| Others |
6,249 |
0.3 |
| Total | 2,021,000 | 100 |
The number of reads mapped to non-coding RNAs, coding sequences (CDS), 5′ UTRs and 3′ UTRs are presented. “Others” represent reads mapping to intergenic sequences and introns, as well as reads mapping to overlapping features (for example, the 3′-UTR and the 5′-UTR of two tandem genes).
Visual inspection of the data revealed variable numbers of CSs. For example, the act1 gene displays three major CSs, while the adh1 gene contains a single one (Fig. 2A‒C). In addition, most CSs displayed some degree of microheterogeneity (Fig. 2B), in which CSs did not map to a single nucleotide (although they tended to cluster in defined regions of the 3′ UTR). A similar phenomenon has been reported in other eukaryotes, and is thought to be caused by the imprecise nature of the cleavage reaction (ref. 4 and references therein).

Figure 2. Examples of polyadenylation sites. The y-axes show the number of reads and x-axes the position of the read along the gene. Only the nucleotide immediately before the poly(A) tail (cleavage site) is plotted. Grey boxes represent coding sequences and the arrows show the direction of transcription. Introns are represented by broken lines. (A) act1, which displays three major CSs in its 3′ UTR. (B) High-resolution view of the first cluster of CSs in the act1 3′ UTR. (C) adh1, which contains a single CS in its 3′ UTR. (D) SPBP4H10.15, which has major CSs at both the coding sequence and the 3′ UTR.
The amount of CS heterogeneity varied widely among different genes (Fig. 3). For instance, the two genes in Figure 3A and B are expressed at similar levels and have UTRs of almost identical lengths, but display very different CS profiles. To quantify this effect, a heterogeneity score was defined as the minimal number of CS positions in a given region required to account for at least 90% of all observed CSs (Fig. 3C). The average number for all 3′ UTRs was 13.7, with a standard deviation of 6.2. The genes displayed in Figure 3A and B had scores of 3 and 31, respectively. The heterogeneity score did not correlate with expression levels (Pearson correlation -0.04), although it showed a weak correlation with 3′ UTR length (Pearson correlation 0.43). ncRNAs had a slightly higher score (mean 16.6, standard deviation 8.0). Although microheterogeneity is a well-known phenomenon, the reasons for the specific behavior of different CSs are not clear.
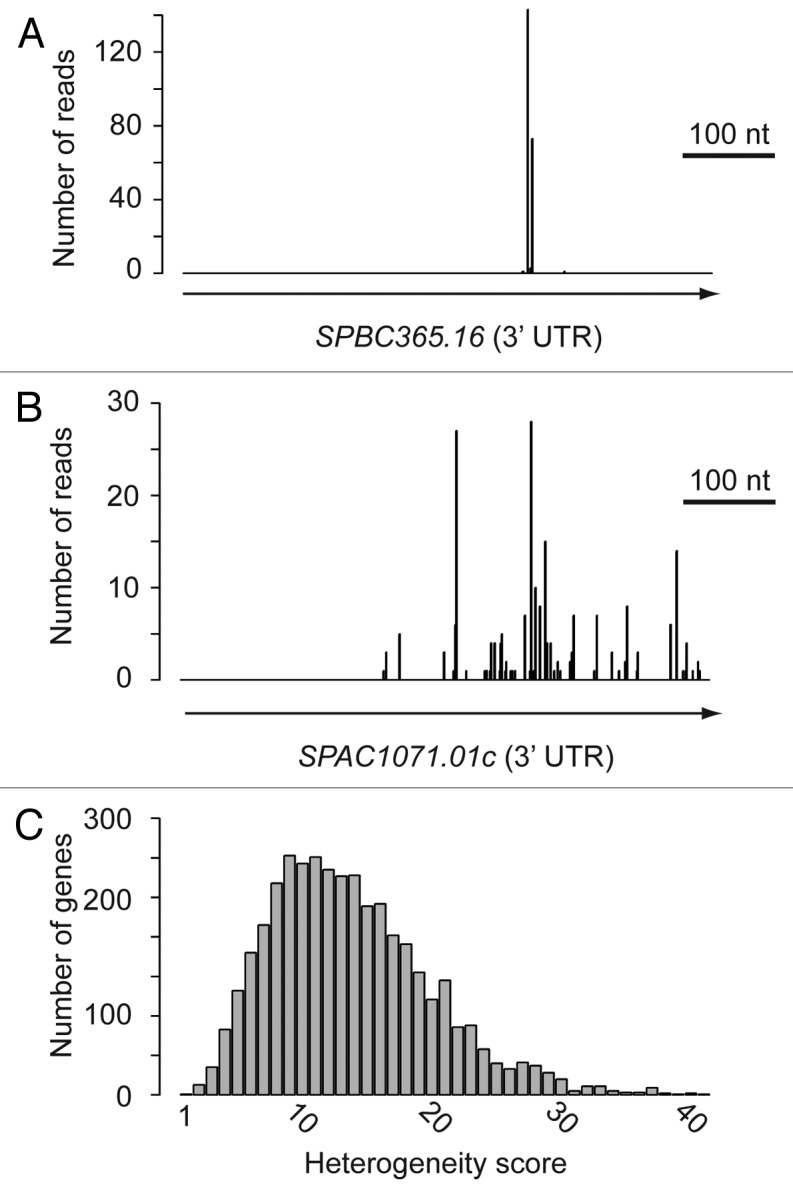
Figure 3. Analysis of cleavage site heterogeneity. In (A and B) the y-axes show the number of reads and the x-axes the position of the read along the gene. Only the nucleotide immediately before the poly(A) tail (cleavage site) is plotted. Only the 3′ UTRs are depicted, with the arrows showing the direction of transcription. (A) SPBC365.16, which has a heterogeneity score of 3. (B) SPA1071.01c, which has a heterogeneity score of 31. (C) Histogram displaying the heterogeneity score for all 3′ UTRs.
To quantify APA, neighboring CSs were grouped into clusters (see Materials and Methods). The number of identified clusters were 7,253 for 3′ UTRs (Data Sets S1 and S2), 1,277 for ncRNAs (Data Sets S3 and S4), and 353 for CDSs (Data Sets S5 and S6). Most reads could be assigned to clusters (Fig. 4A), which had an average length of 55.4 nucleotides. The median distance between the edges of adjacent clusters was 73 nucleotides, and the mean separation was 160, strongly suggesting that the clusters represent independent CSs. The position of the 3′ UTR CS with most reads within a cluster (peak) was used to estimate the length of the 3' UTR (Fig. 4B). Based on these data, the median length of a 3′ UTR in S. pombe is 203 nucleotides, and the mean is 284. This is close to the medians of 166 nucleotides of the budding yeast Saccharomyces cerevisiae22 and the 130 of Caenorhabditis elegans.7
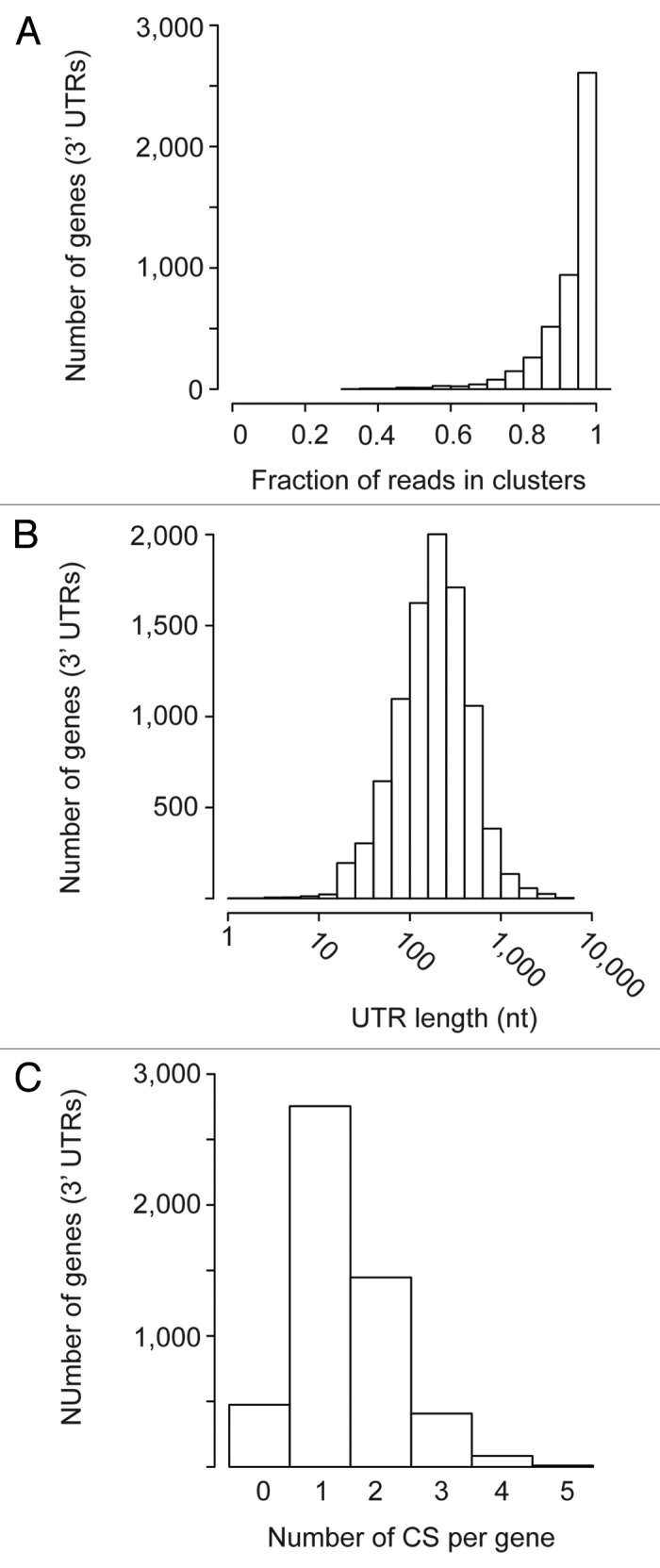
Figure 4. Analysis of poly(A) site usage in 3′ UTRs. The y-axes show the number of genes in each category. (A) Histogram showing the fraction of reads that could be assigned to a cluster within a 3′ UTR. (B) Histogram of 3′ UTR lengths, calculated using the peak of each cluster. (C) Histogram displaying the number of identified CSs per 3′ UTR.
The positions of 3′ UTR CSs, which define UTR lengths, were compared with the lengths of annotated UTRs (Fig. S4 and Data Set S7). The comparison was performed in two ways: first, by using the most distal single CS identified by 3PC; second, by considering the peak of the most distal cluster. In the first case, the correlation was very high (Pearson correlation = 0.94), although the UTRs defined by 3PC tended to be longer (Fig. S4A). In the second case, the correlation was poorer (Pearson = 0.64, Fig. S4B). This is consistent with the fact that annotated UTRs are usually defined by the longest observed transcript, while the position of the peak of the cluster represents the length of a “typical” UTR.
Analysis of the data for 3′ UTRs also revealed a high prevalence of APA, with the number of clusters ranging between one and five, and a mean number of 1.54 clusters per gene (Fig. 4C; Data Set S2). 41% of all detected S. pombe 3′ UTRs contained more than one cluster. In ncRNAs, there was a similar trend to the presence of multiple CSs, with 45% of all detected ncRNAs having more than one cluster, and a mean of 1.63 clusters per gene (Fig. S5 and Data Set S4). These results demonstrate that APA is widespread in S. pombe. The extent of APA is comparable to that of multicellular eukaryotes. For example, 47% of genes expressed in HeLa cells display APA, with an average of 1.9 isoforms per gene.23
Analysis of APA in several organisms has revealed preferences for the use of proximal or distal sites, which are often dynamically regulated.5-8 To investigate if S. pombe displays a positional bias in the use of CS, I concentrated on those genes with exactly two CSs. The distributions of the number of reads that map to the proximal and the distal sites for each 3′ UTR were compared with each other (Fig. S6). The distributions for both sites were very similar, demonstrating that S. pombe vegetative cells do not show a global positional preference in the use of CSs.
A small number of CSs mapped within coding sequences (see Fig. 2D for an example). In contrast to 3′ UTRs and ncRNAs, the number of CSs per gene within a coding sequence was almost always one (Fig. S5 and Data Sets S5 and S6), with a mean of 1.14. These mRNAs tended to have additional CSs in the 3′ UTR (93.0%, with an average of 1.42). CSs within coding sequences would generate mRNAs without stop codons, which are likely to be degraded by the non-stop decay pathway.24
Cis-sequences regulating cleavage and polyadenylation
To identify cis-regulatory regions, the sequence distribution in the regions around CSs was examined (Fig. 5A). For this purpose, the peak of each CS cluster was used. A very strong enrichment in adenosines was present in nucleotides 1 and 2 after the CS. Note that the drop in adenosines immediately before the CS is due to the way in which the sequences are processed (see Materials and Methods for details), as it is not possible to unambiguously map cases in which cleavage occurs after an adenosine. In addition, there was an enrichment in adenosines upstream of the CS, extending approximately between -29 and -13, and with a peak at position -20. Finally, uridines were enriched before and around the CS, showing peaks at positions -8 and 1. A very similar pattern was found for ncRNAs (Fig. S7A). Comparable patterns have been observed in budding yeast and multicellular organisms.7,22,25
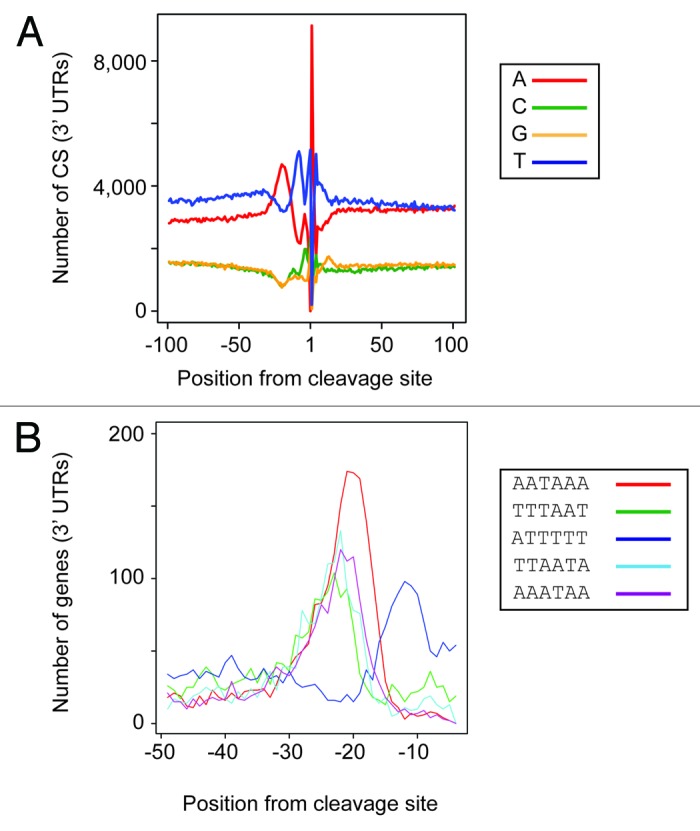
Figure 5. Cleavage and polyadenylation regulatory sequences in 3′ UTRs. The y-axes show the number of CSs in 3′ UTRs. Only the peak of each cluster was used for the analyses. (A) Nucleotide composition of sequences around the CS. (B) Location of the most enriched motifs relative to the position of the CS. The numbers refer to the location of the first nucleotide of the hexamer.
I then searched for over-represented sequence motifs in the regions surrounding the CSs. As above, the peaks of each cluster were chosen for this analysis. For upstream motifs, the sequences between -5 and -50 were scanned. The search revealed 40 enriched hexamers with compositions high in adenosines and/or uridines (Table S3). The most abundant hexamer was the canonical human AAUAAA (present in 20.1% of all CSs), while several other motifs had sequences that partly overlapped with it. The majority of A-rich motifs were located around positions -23 to -26 (Fig. 5B; Table S3). For example, AAUAAA peaked at position -24. By contrast, some U-rich motifs, such as UUUUUU, were located closer to the CS (position -14). This is consistent with the overall enrichment in U-rich sequences observed at this distance from the CS (Fig. 5A). The specific location of these motifs suggests that they may be of functional importance. Examination of ncRNAs revealed related motifs (Table S4), with AAUAAA having the smallest P value and being the most abundant (25.1%). AAUAAA was also located at a similar position to that of 3′ UTRs, peaking at -22 from the CS (Fig. S7B). These results suggest that the signals controlling the polyadenylation of coding and non-coding genes are similar. By contrast, no over-represented sequences were detected downstream of the CS. This was unexpected, given that elements located after CSs have been shown to be important for efficient cleavage and polyadenylation in S. pombe.26
Discussion
I present here a new method to map cleavage and polyadenylation sites and its application to the fission yeast S. pombe. 3PC is based on the use of an oligodT primer to enrich mRNA ends, followed by sequencing of the library across the oligo(dT) sequence. The protocol is straightforward, highly reproducible, strand-specific, compatible with multiplexing, and avoids the use of enzymatic modifications of RNA, especially RNA ligations, which suffer from sequence biases.11 Moreover, 3PC uses UMIs to avoid PCR amplification artifacts. A number of similar protocols have been published recently (see ref. 27 for a comparison), but none of them involves the use of UMIs.
Current protocols for the preparation of libraries for Illumina sequencing involve PCR amplification. This can lead to biases if the efficiency of amplification of individual clones is different. Moreover, random sampling of clones within the library means that the number of times a clone is sequenced may not reflect its original abundance in the library. A key advantage of 3PC is the use of UMIs, which ensures that every sequenced poly(A) site corresponds to that of an individual RNA molecule, thus providing more accurate quantification of relative CS usage. In addition, given the number of sequence reads and the fraction of unique sequenced clones (obtained using UMIs), it is possible to estimate the complexity of the original library (Fig. S8). This information can be used to guide future experiments: for example, if the complexity of a library is low, additional sequencing will provide little new information, and efforts should concentrate on generating a new library.
3PC requires sequencing through the oligo(dT) sequence. This ensures that every read reaches a CS site, but can lead to loss of accuracy and efficiency during sequencing.27 A number of solutions have been proposed to this problem, including filling in the oligo(dA) sequencing with thymidines immediately before sequencing,27 and the use of custom sequencing primers.28 These procedures are incompatible with the use of UMIs as implemented here, which requires that they be located 5′ relative to the oligo(dT) in the RT oligo [and thus that they be sequenced before the poly(A) tail]. To avoid problems caused by accuracy loss, a very stringent analysis protocol was implemented, in which only reads that contain exactly the expected number of Ts in their sequence followed by the expected anchor were retained (see Materials and Methods). This resulted in the removal of ~38% of sequences. As using the methods discussed above would involve losing the ability to employ UMIs, and given that with current technologies sequencing depth is rarely limiting, I consider this lower efficiency is acceptable.
This is the first genome-wide analysis of CS usage in the fission yeast S. pombe. Over 2 million individual non-redundant CSs were mapped, identifying 8,883 major CSs. This resulted in good coverage for more than 90% of coding sequences and 15% of annotated ncRNAs. Although 3′ UTRs have been annotated in S. pombe,29-32 the data were based on standard RNA-seq experiments and could not distinguish between different isoforms generated by APA.
A very interesting finding is the widespread use of APA in fission yeast. Even in vegetative conditions, it appears that many mRNAs exist as different isoforms, with the length of their UTRs differing in some cases by hundreds of nucleotides (as an example, see act1, a highly abundant mRNA, Fig. 2). APA can affect mRNA fate; for example, there are two forms of the brain-derived neurotrophic factor BDNF that differ in the lengths of their 3′ UTRs that differentially localized and translated (reviewed in ref. 33). It has also been shown that longer UTRs can have a negative effect on gene expression through the increased number of miRNA binding sites.8 However, in most cases, the physiological importance of the existence of multiple forms, especially in unicellular eukaryotes, is unknown.
The comprehensive data set presented in this manuscript will be of great value for S. pombe researchers, by assisting in the annotation of the fission yeast genome, and as the basis for hypothesis-driven experiments into gene expression control in this model organism. In addition, I expect that these data will provide the foundation for comparative studies of among various organisms.
Materials and Methods
Fission yeast methods
Standard fission yeast methods were used for all experiments. 972 h-cells were grown in Edinburgh Minimal Medium (EMM) at 32 °C.
Library preparation and sequencing
All primers used are described in Table S1 and the structure of the RT primer is presented in Figure S9. Total RNA was prepared using a hot-phenol extraction protocol. 100 μg of total mRNA were incubated with 1/9 volumes of RNA fragmentation reagents (Ambion, Life Technologies) at 70 °C for 4.5 min. The reaction was stopped by adding 1/9 volumes of stop solution (Ambion, Life Technologies) and transferring the samples to 4 °C. Polyadenylated fragments were purified using oligo(dT) magnetic beads (Life Technologies). 200 μl of beads were washed in binding buffer (20 mM TRIS-HCl pH 7.0, 2 mM EDTA, 1 M LiCl). 50 μl of binding buffer was added to 50 μl of fragmented RNA, and the mix was incubated at 65 °C for 2 min and then transferred to ice. The RNA was then added to the beads, and incubated at room temperature for 5 min. The beads were washed twice in wash buffer (10 mM TRIS-HCl pH 7.0, 1 mM EDTA, 0.15 M LiCl). RNA was eluted by resuspending the beads in 22 μl of 10 mM Tris pH 8.0 and incubating at 80 °C for 2 min. The remaining RNA in lysis buffer was incubated with the beads once more and washed as described above, and the two eluates combined. For reverse transcription, 11 μl of purified RNA fragments were mixed with 1 μl of dNTPs (10 mM each) and 1 μl of 50 μM RT primer, incubated at 65 °C for 5 min and transferred to ice. Four µl of 5X FFS buffer (Invitrogen, Life Technologies), 1 µl of SuperaseIN (Ambion, Life Technologies), 1 µl of 0.1 M DTT, and 1 µl of Superscript III (Invitrogen, Life Technologies) were added, and the reaction incubated at 48 °C for 40 min. RNA was hydrolyzed by adding 2.3 µl of 1 M NaOH and incubating at 95 °C for 15 min, and the solution neutralized with 2.3 µl of 1 M HCl. The cDNA was purified using Agencourt AMPure magnetic beads (Beckman Coulter). 46 μl of beads were added to the cDNA solution (~1.85 volumes), and the beads washed twice with 180 μl of freshly prepared 70% ethanol. The cDNA was eluted by resuspending the beads in 40 μl of water. A second round of purification was performed as above, except that a lower ratio of beads to sample (1.6 volumes) was used. This second round is essential for the complete removal of RT primer, which can otherwise circularize and be amplified by PCR. Eight μl of cDNA were circularized using Circligase II (Epicenter) as follows: 0.5 µl of 50 mM MnCl2, 1 µl of 10X Reaction buffer and 0.5 µl of enzyme were added, and the reaction incubated at 60 °C for 60 min and at 80 °C for 10 min. PCR amplification was performed using Phusion High Fidelity Polymerase (Thermo Scientific). Two µl of circularised cDNA were used as a template, and amplified in a 50 µl volume as described by the manufacturer. Primers P3 and P5 were used at a final concentration of 0.2 µM. PCR amplification was performed in two stages: four cycles using an annealing temperature of 66°C, followed by a variable number of cycles at 72°C. Small scale reactions were performed to determine the optimal number of cycles, which ranged between 12 and 18 (including both stages). Samples were sequenced in either an Illumina Genome Analyzer II or a HiSeq 2000 platform. As low diversity sequences [such as oligo(dT) stretches] can affect sequencing performance on Illumina platforms, samples were mixed with unrelated high-diversity libraries, and/or mixed with 20% of a PhiX control.
Bioinformatics analyses
All data were processed using custom-made scripts written in Perl and visualized using the Integrated Genome Viewer.34 Downstream statistical analyses were performed with R. Reads were first demultiplexed to separate different libraries. To remove redundant sequences, a single copy was retained of identical reads containing the same random UMI. Reads that did not contain only Ts between positions 7‒21, followed by A/C/G at position 22, were discarded (see Fig. S9). After that, sequences between bases 1‒21 were removed (including random UMIs, multiplexing barcodes, and dT sequences from the RT primer, Fig. S9), and all sequences were reverse complemented. For all analyses, S. pombe annotations and sequences available from GeneDB (http://old.genedb.org/), now PomBase (http://www.pombase.org/), on May 9, 2011 were used. Processed sequences were aligned to the S. pombe genome using Bowtie15 with the following parameters: -S -v 2 -m 1 (two mismatches allowed, and reporting only reads that map to a single location in the genome). Aligned reads were then filtered to remove sequences potentially generated from internal poly(A) sequences, by discarding reads followed by either four or more consecutive As, or by seven or more As in the 10 downstream bases. Finally, reads that contained a mismatch in the last base were also discarded. The most downstream base within the mapped sequence was defined as the CS. 3′ UTRs were extended by 200 nucleotides from the annotated transcription termination site. Clusters were defined iteratively, by merging reads separated by less than a fixed number of nucleotides from each other. As the distance used to define clusters will influence their length and number, we performed the analysis using distances from 1‒30 (Fig. S10). As expected, the fraction of genes containing more than one cluster decreased with length. However, the conclusion that APA is widespread remained robust for all numbers tested. A conservative distance of 25 nucleotides was used for all the analyses, which resulted in a median separation between cluster edges of 73 nucleotides. Only clusters containing at least six reads and at least 10% of the reads assigned to the corresponding gene were considered. To quantify heterogeneity, the minimal number of positions required to account for 90% of all observed CSs within a 3′ UTR was calculated. To avoid biases created by differential read coverage,4 a fixed number of reads was set at 200. For genes with less reads, the data were expanded by sampling with replacement from the original data set until 200 was reached. For genes with more, 200 reads were randomly selected. Only genes with more than 30 reads were used to calculate the heterogeneity score. To identify regulatory elements, sequences between -50 and -5 from the CS, or between +2 and +50, were scanned for the presence of over-represented hexamers. The background model was generated from the frequency of all hexamers in the regions -200 to -100 from the CS. Statistical significance was calculated using a normal approximation to the cumulative binomial distribution. For this analysis, a single event from each cluster of poly(A) sites was used, corresponding to the nucleotide with the highest number of reads. All raw data files have been deposited in the ArrayExpress database with accession number E-MTAB-1642.
Supplementary Material
Acknowledgments
I thank members of the Mata Lab and Samuel Marguerat for helpful discussions, and SM for comments on the manuscript. This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) (grant number BB/J007153/1).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: http://www.landesbioscience.com/journals/rnabiology/article/25758/
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/25758
References
- 1.Mandel CR, Bai Y, Tong L. Protein factors in pre-mRNA 3′-end processing. Cell Mol Life Sci. 2008;65:1099–122. doi: 10.1007/s00018-007-7474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Proudfoot NJ. Ending the message: poly(A) signals then and now. Genes Dev. 2011;25:1770–82. doi: 10.1101/gad.17268411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63:405–45. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33:201–12. doi: 10.1093/nar/gki158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–7. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji Z, Lee JY, Pan Z, Jiang B, Tian B. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci USA. 2009;106:7028–33. doi: 10.1073/pnas.0900028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jan CH, Friedman RC, Ruby JG, Bartel DP. Formation, regulation and evolution of Caenorhabditis elegans 3’UTRs. Nature. 2011;469:97–101. doi: 10.1038/nature09616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayr C, Bartel DP. Widespread shortening of 3’UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–84. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y. Alternative polyadenylation: new insights from global analyses. RNA. 2012;18:2105–17. doi: 10.1261/rna.035899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nam DK, Lee S, Zhou G, Cao X, Wang C, Clark T, et al. Oligo(dT) primer generates a high frequency of truncated cDNAs through internal poly(A) priming during reverse transcription. Proc Natl Acad Sci USA. 2002;99:6152–6. doi: 10.1073/pnas.092140899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–43. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 13.Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, et al. iCLIP–transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J Visualized Exps. 2011;50:e2638. doi: 10.3791/2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2012;9:72–4. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 15.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rissland OS, Norbury CJ. Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat Struct Mol Biol. 2009;16:616–23. doi: 10.1038/nsmb.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen K, Birse CE, Proudfoot NJ. Nascent transcription from the nmt1 and nmt2 genes of Schizosaccharomyces pombe overlaps neighbouring genes. EMBO J. 1998;17:3066–77. doi: 10.1093/emboj/17.11.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cremona N, Potter K, Wise JA. A meiotic gene regulatory cascade driven by alternative fates for newly synthesized transcripts. Mol Biol Cell. 2011;22:66–77. doi: 10.1091/mbc.E10-05-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mata J, Lyne R, Burns G, Bähler J. The transcriptional program of meiosis and sporulation in fission yeast. Nat Genet. 2002;32:143–7. doi: 10.1038/ng951. [DOI] [PubMed] [Google Scholar]
- 20.Lackner DH, Beilharz TH, Marguerat S, Mata J, Watt S, Schubert F, et al. A network of multiple regulatory layers shapes gene expression in fission yeast. Mol Cell. 2007;26:145–55. doi: 10.1016/j.molcel.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, Bähler J. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell. 2012;151:671–83. doi: 10.1016/j.cell.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, et al. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell. 2010;143:1018–29. doi: 10.1016/j.cell.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shepard PJ, Choi E-A, Lu J, Flanagan LA, Hertel KJ, Shi Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011;17:761–72. doi: 10.1261/rna.2581711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–56. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- 25.Sherstnev A, Duc C, Cole C, Zacharaki V, Hornyik C, Ozsolak F, et al. Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat Struct Mol Biol. 2012;19:845–52. doi: 10.1038/nsmb.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphrey T, Birse CE, Proudfoot NJ. RNA 3′ end signals of the S. pombe ura4 gene comprise a site determining and efficiency element. EMBO J. 1994;13:2441–51. doi: 10.1002/j.1460-2075.1994.tb06529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkening S, Pelechano V, Järvelin AI, Tekkedil MM, Anders S, Benes V, et al. An efficient method for genome-wide polyadenylation site mapping and RNA quantification. Nucleic Acids Res. 2013;41:e65. doi: 10.1093/nar/gks1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–83. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ, Habib N, et al. Comparative functional genomics of the fission yeasts. Science. 2011;332:930–6. doi: 10.1126/science.1203357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lantermann AB, Straub T, Strålfors A, Yuan GC, Ekwall K, Korber P. Schizosaccharomyces pombe genome-wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae. Nat Struct Mol Biol. 2010;17:251–7. doi: 10.1038/nsmb.1741. [DOI] [PubMed] [Google Scholar]
- 31.Dutrow N, Nix DA, Holt D, Milash B, Dalley B, Westbroek E, et al. Dynamic transcriptome of Schizosaccharomyces pombe shown by RNA-DNA hybrid mapping. Nat Genet. 2008;40:977–86. doi: 10.1038/ng.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bitton DA, Grallert A, Scutt PJ, Yates T, Li Y, Bradford JR, et al. Programmed fluctuations in sense/antisense transcript ratios drive sexual differentiation in S. pombe. Mol Syst Biol. 2011;7:559. doi: 10.1038/msb.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Giammartino DC, Nishida K, Manley JL. Mechanisms and consequences of alternative polyadenylation. Mol Cell. 2011;43:853–66. doi: 10.1016/j.molcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.