

Abstract
Intestinal alkaline phosphatase (IAP) is a brush-border membrane ectoenzyme (BBM-IAP) that is released into the lumen (L-IAP) after a high-fat diet. We examined the effects of oil feeding and the addition of mixed-lipid micelles on the formation of L-IAP in oil-fed rat intestine, Caco-2 cell monolayers, and mouse intestinal loops. We localized IAP in the duodenum of rats fed corn oil using fluorescence microscopy with enzyme-labeled fluorescence-97 as substrate. Four hours after oil feeding, L-IAP increased ∼10-fold accompanied by the loss of BBM-IAP, consistent with BBM-IAP release. Rat IAP isozyme mRNAs progressively increased 4–6 h after oil feeding, followed by the increase of IAP activity in the subapical location at 6 h, consistent with the restoration of IAP protein. Postprandial lipid-micelle components, sodium taurocholate with or without oleic acid, mono-oleylglycerol, cholesterol, or lysophosphatidylcholine (lysoPC) were applied singly or as mixed-lipid micelles to the apical surface of polarized Caco-2 cell monolayers. LysoPC increased L-IAP >10-fold over basal release. LysoPC released IAP into the apical medium more than other intestinal brush-border enzymes, 5′-nucleotidase, sucrase, aminopeptidase N, and lactase, without comparable lactate dehydrogenase release or cell injury. LysoPC increased human IAP mRNA levels by 1.5-fold in Caco-2 cells. Luminally applied lysoPC also increased release of IAP preferentially in mouse intestinal loops. These data show that lysoPC accelerates the formation of L-IAP from BBM-IAP, followed by enhanced IAP synthesis, suggesting the role that lysoPC might play in the turnover of brush-border proteins.
Keywords: Caco-2 cells, lipid absorption, small intestine, enzyme-labeled fluorescence-97
mammalian alkaline phosphatases (APs; orthophosphoric monoester phosphohydrolase, EC 3.1.3.1) are ectoenzymes anchored in the outer leaflet of the plasma membrane by glycosyl phosphatidylinositol (GPI) (15). Human intestine expresses one intestinal type of AP (IAP) encoded by the IAPI gene, whereas rats have two IAP isoenzymes, named IAP-I and IAP-II, which are encoded by the ALPi and ALPii genes, respectively (18). IAP expression is largely restricted to the gut, especially to the brush border of the enterocytes, and its expression and activity are highest in the duodenum (4).
The nonhydrolytic functions of IAP still remain speculative. There has been emerging evidence for the functional role of IAP as a detoxifying enzyme for lipopolysaccharide (LPS) (3, 9). IAP activity is associated with the reduction of inflammatory conditions in the bowel (9): exposure of the intestinal wall to LPS induces IAP gene expression (3). IAP detoxifies LPS by removing its phosphate esters in vitro (30). IAP deficiency is associated with increased LPS toxicity in zebrafish and in Caco-2 cells (3, 9). In vivo, however, the localization of IAP and LPS is mismatched because IAP is expressed mostly in the upper small intestine, whereas the exposure of LPS produced by the microflora to the intestinal mucosa occurs in the ileum and colon.
Many studies suggest that IAP plays a role in the process of lipid absorption and transport. First, an IAP inhibitor l-phenylalanine inhibits fat absorption in the small intestine (14, 19). Second, after ingestion of a high-fat diet, IAP activity in the intestinal luminal content and in the enterocytes increases by more than 10-fold and by two- to threefold, respectively (12, 32). Moreover, there is a parallel increase of IAP activity and triacylglycerol concentration in the thoracic duct during lipid transport (8). Third, alimentary fatty acids may mediate changes in the activity and localization of IAP in the small intestine because the effect of a high-fat diet on the increase of IAP in the luminal contents of the small intestine (L-IAP) is also induced by ingestion of edible oils (12). In addition, oleic acid enhanced rat ALPii gene promoter-mediated expression of luciferase (31). The mechanism by which fatty acids regulate an increase of L-IAP has been studied over the past few decades (12, 16). The process by which ingested fatty acids mediate the IAP release into the intestinal lumen involves production of lipoprotein particles secreted from the enterocytes (5, 16). However, this mechanism does not explain all the changes that occur after ingestion of a fat meal. Such a meal stimulates the secretion of bile and pancreatic juice containing taurocholate and lipases, leading to lipid digestion and micelle formation to enhance fat absorption. Lysophosphatidylcholine (lysoPC) concentration is also increased in the intestinal lumen after fat feeding attributable to the rapid hydrolysis of bile phosphatidylcholine (PC) by pancreatic phospholipase A2 (7). Although lysoPC is a potent detergent molecule, its role in fat absorption or IAP release is unclear.
The aims of the present study were 1) to evaluate the cellular distribution of IAP after corn oil feeding in vivo, 2) to elucidate in Caco-2 cell monolayer the mechanism(s) regulating the dynamics of IAP activity and localization after fat feeding, and 3) to examine the applicability of the IAP release to mouse intestinal loop model. Our results suggest that lysoPC enhances the release of IAP on the brush-border membrane (BBM-IAP) into the intestinal lumen followed by increased IAP synthesis, presumably for BBM-IAP restoration. The roles of lysoPC in the release and restoration of BBM-IAP may be implicated in the turnover of other brush-border proteins as well.
MATERIALS AND METHODS
Animals.
Male Wistar rats (150–200 g) and 8–10-wk-old C57BL male mice were purchased from Clea Japan (Tokyo, Japan). The rodents were kept at 23°C under a 12-h light-dark cycle with free access to water and standard chow. All experiments were approved by the Animal Care Committee of Saitama Medical University.
Detection of alkaline phosphatase activity in situ.
Rats were fasted overnight and fed 2 ml of corn oil by gavage. Then the rats were euthanized by terminal exsanguination under pentobarbital sodium anesthesia (115 mg/kg) at 0, 2, 4, and 6 h after corn oil gavage. The whole small intestine was rapidly excised and cut into the duodenum, jejunum, and ileum. The luminal side of the ileum segments was washed with 10 ml of cold PBS (pH 7.4) (12), and the luminal washings were assayed for AP activity and protein concentration as described below. A duodenal portion was fixed in 4% paraformaldehyde and embedded in optimal cutting temperature compound (Sakura Finetek, Tokyo, Japan). Frozen sections were cut with a cryostat at 4-μm thickness. To visualize AP activity, the serial sections were incubated with a fluorogenic AP substrate, 2-(5′chloro-2′-phosphoryloxyphenyl)-6-chloro-4(3H)-quinazolinone (enzyme-labeled fluorescence-97, ELF-97; Invitrogen, Carlsbad, CA) as previously described (1). The sections were counterstained by AlexaFluor 488-phalloidin (Invitrogen) for brush border and 7-aminoactinomycin D (Invitrogen) for nuclei. Briefly, the sections were first stained with phalloidin for 20 min, followed by the incubation with 125 μM of ELF-97 for 1 min, and then treated with 7-aminoactinomycin D for 1 h. All reactions were performed at room temperature in a dark room. All fluorescence detections with ELF-97 were performed with an exposure time of 100 ms. The images of sections were observed with a fluorescent microscopy (Zeiss Axioplan 2 imaging; MOT, Carl Zeiss, Jena, Germany) and captured using AxioVision (Carl Zeiss). Densitometric analysis was performed for ELF-97 and phalloidin staining with an image analyzing software Image-J (http://rsb.info.nih.gov/ij/).
Caco-2 cell culture.
Caco-2 cells were obtained from Dr. Terrence Riehl (Washington University School of Medicine), which were originally cloned by Dr. Jeffrey Field (Department of Internal Medicine, University of Iowa). This cell line possesses a marked induction of IAP expression after confluence (28). Cell culture reagents were purchased from Invitrogen (Tokyo, Japan), FCS from Biowest (Nuaille, France), and microporous PET membrane inserts (1-μm pore size, 23-mm diameter) from BD Biosciences (Franklin Lakes, NJ).
Caco-2 cells (between passages 37 and 42) were seeded at a density of 5 × 104 cells/cm2 on a porous filter membrane and grown in DMEM containing 25 mM glucose and GlutaMax (Invitrogen) containing 20% heat-inactivated FCS under a humidified atmosphere (5% CO2-95% atmosphere) at 37°C. Cells were grown to confluence for 1 wk and then cultured in the same medium containing 5 mM glucose in the upper compartment and 20% FCS in the lower compartment for the following 4 days. Serum-free low-glucose medium containing 1% insulin, transferrin, and selenium-G (Invitrogen) was used for the last 3 days. All media were changed daily. Penicillin/streptomycin (100 IU/ml and 100 μg/ml, respectively) and 1% nonessential amino acids were added to all the media. Transepithelial resistance (TER) of Caco-2 cells grown on filters was measured by using a Millicell-ERS instrument (Millipore, Billerica, MA).
Preparation of mixed-lipid micelles.
Oleic acid, mono-oleylglycerol, cholesterol, and lysoPC (Sigma Chemical, St. Louis, MO) were dried under nitrogen gas individually. The residue obtained was dissolved in 2 mM taurocholate (Nacalai Tesque, Kyoto, Japan) in serum-free medium and mixed to generate micelles. Final lipid concentrations in the Caco-2 culture media (in mM) were 0.6 for oleic acid, 0.05 for cholesterol, 0.2 for oleylglycerol, and 0.2 for lysoPC. Each freshly prepared and sterile preparation of single lipid component (e.g., lysoPC plus taurocholate) or mixed-lipid micelles were added to the apical side of cell monolayer to mimic luminal contents in the small intestine after ingestion of a meal.
AP activity assay.
We measured AP activity at 37°C using 1 M ethylaminoethanol buffer (pH 10.5) containing 15 mM p-nitrophenyl-phosphate and 5 mM MgCl2 as previously described (17). Absorbance for p-nitrophenol was measured at 405 nm with a microplate reader (Sunrise; Tecan Group, Männedorf, Switzerland). IAP activity was expressed in units (U), which defines the enzyme activity that catalyzes the hydrolysis of 1 μmol of p-nitrophenyl-phosphate/min. BBM-IAP activity was estimated as described previously in the cell monolayer (21). Briefly, the cells grown on filters were fixed with 1% glutaraldehyde in 10 mM Tris·HCl buffer (pH 8.0) for 15 min at 4°C and washed with cold Tris-buffered saline. BBM-IAP activity was then detected at 37°C using 0.2 M carbonate buffer (pH 10.5) containing 10 mM p-nitrophenyl-phosphate and 5 mM MgCl2. The latter method was also used to measure AP activity in the luminal washings.
Caco-2 cell preparation and toluidine blue staining.
Caco-2 cell monolayers were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) for an hour and postfixed with 1% OsO4 solution buffered with Sorenson's phosphate at pH 7.4 for another hour. Then the monolayers were dehydrated in alcohol series and embedded in epoxy resin (Epok 812; Oken Shoji, Tokyo, Japan). Semi-thin sections were cut and stained with 0.3% toluidine blue.
Cytotoxicity assays for lysoPC.
To examine the effect of lysoPC on cell viability, lactate dehydrogenase (LDH) release and propidium iodide (PI) staining were examined in Caco-2 cell monolayers. The apical side of Caco-2 cell monolayers was washed with 2 ml Krebs buffer, and we added 1 ml Krebs buffer containing 5 mM glucose. LysoPC (0.2 or 2 mM) was added to the apical solution, and the monolayers were incubated for 1 h at 37°C. LDH activity in the apical solution and whole cell lysate was measured with LDH cytotoxicity detection kit (Roche Applied Science, Mannheim, Germany). For PI staining to detect the damaged cells, PI (1 μM; Molecular Probes, Eugene, OR) was added to the apical Krebs buffer with or without lysoPC. After 1 h of incubation, PI-positive damaged cells were visualized at 535-nm excitation and 590-nm emission using a Zeiss microscope with a ×10 objective lens. Images were recorded with a cooled charge-coupled device video camera (Hamamatsu Orca-EN; Hamamatsu USA, Bridgewater, NJ), captured, and digitized using an image analyzing software (OpenLab; Improvision, Lexington, MA). The cell monolayers fixed with 4% paraformaldehyde overnight at 4°C were used to count the total number of the cells in the observed area. The number of PI-positive cells in each image was counted and expressed as a percentage of total cell number.
Enzyme release experiments.
The jejunum was removed from mice after an overnight fasting, and the luminal contents were flushed out with 5 ml cold saline. After the proximal end was ligated, the lumen was filled with PBS with or without 4 mM lysoPC. After the other end was ligated, the loops were bathed in warm PBS, incubated for 20 min at 37°C, and then washed with 2 ml PBS. All of the fluid in the lumen was collected. After the segments were opened longitudinally, the mucosal surface was scraped lightly with Whatman no. 3 filter papers, and then the papers were immediately soaked in cold 10 mM Tris·HCl buffer containing 1% Triton X-100, 0.1 mM phenylmethylsulfonyl fluoride, and 2 mM benzamidine (16) and were sonicated for 1 min. The luminal washings and the scrapings were assayed for enzyme activities.
We assayed aminopeptidase N, sucrase, and lactase as previously described (6) with some modifications. Aminopeptidase N (EC 3.4.11.2) activity was assayed with a substrate buffer containing 50 mM Na2HPO4 and 18 mM l-alanine-p-nitroanilide hydrochloride (pH 7.0). The sample solutions obtained from the mouse jejunal loops or from Caco-2 cells treated with or without lysoPC were added to the buffer and incubated at 37°C for 20 min. The reactions were stopped by heating at 98°C for 2 min. The absorbance for p-nitroaniline was measured at 405 nm. Sucrose and lactose (300 mM each) were used as substrates for sucrase (EC 3.2.1.48) and lactase (EC 3.2.1.23–62), respectively. Each enzyme activity was measured in 50 mM sodium maleate buffer (pH 6.0). The same assay condition for aminopeptidase N was used. The end product of both enzyme reactions, d-glucose, was measured using a Glucose C2 kit (mutarotase-GOD method; Wako, Osaka, Japan). 5′-Nucleotidase was assayed with a commercial kit (Diazyme Laboratories, San Diego, CA).
The percentage of enzyme released from the Caco-2 cell culture was calculated by means of the following formula: enzyme release (%) = enzyme activity in the apical media/total enzyme activity (cell extract and media) × 100.
Real-time RT-PCR.
Total RNA was extracted from the cells or intestinal segments (duodenum, jejunum, and ileum) with ISOGEN (Nippon Gene, Toyama, Japan) according to the manufacturer's instructions. The samples were treated with RNase-free DNase I (Invitrogen) for 30 min at 37°C. First-strand cDNA synthesis was performed with a Superscript-II reverse transcription kit (Qiagen, Tokyo, Japan) after denaturation of total RNA (10 μg, 15 min, 65°C). Reactions were performed in a total volume of 25 μl containing 2.5 μl of cDNA solution, 12.5 μl of qPCR Master Mix (Applied Biosystems, Tokyo, Japan), and 1.25 μl of TaqMan probe. Reactions were run on an ABI PRISM 7900 Sequence Detector (Applied Biosystems). The cycle threshold (Ct), which corresponds to the number of cycles after which the target-DNA concentration increase becomes exponential, was monitored. Results were analyzed using SDS 2.1 Software (Applied Biosystems). Each expression level was normalized against a housekeeping gene, the gene coding ribosomal protein L19. All reactions were done in duplicate.
Statistical analysis.
Data are shown as means ± SE. For parametric data, means were compared with Student's t-test. For nonparametric data, Mann-Whitney U-test was used. The statistical analyses were performed with StatView (version 5.0 for Win software) (SAS Institute).
RESULTS
Dynamics in the activity and localization of alkaline phosphatase in rat duodenum.
A newly developed fluorescence microscopy technique for AP activity detection (1) was used to better observe the dynamics of IAP activity and localization in enterocytes in rats that were fed 2 ml of corn oil as previously described (34). Fluorescence microscopy analysis with ELF-97 showed that, at t = 0, all IAP activity was located external to the internal margin of the brush border, identified with phalloidin binding to F-actin (Fig. 1A, right). IAP staining on brush border was gradually decreased by 4 h (Fig. 1A), corresponding to the increase of IAP activity in the luminal washings in the ileum, where L-IAP was gradually increased by ∼10-fold at 6 h after the oil feeding compared with those at t = 0 (Fig. 1C). The BBM-IAP activity became nearly undetectable at 4 h and 6 h (Fig. 1A). Intracellular IAP activity was detected close to the brush border at 4 h after oil feeding as seen by the orange band containing both green IAP and red phalloidin fluorescence, and it became stronger and more obvious at 6 h (Fig. 1A), suggesting the increase of IAP activity in the subapical pool. At 6 h, IAP was also located external to the band of phalloidin staining, representing IAP restoration in the BBM. Densitometric analysis, however, showed that most of the IAP activity was still localized within the cytosol at 6 h (Fig. 1B). The rat duodenum showed high mRNA expression levels of both IAP isoenzymes, as expected (Fig. 1D). In particular, mRNA coding ALPii is dominantly expressed in the duodenum. The mRNA level of the gene encoding IAP-I (ALPi) increased gradually more than twice after oil feeding, whereas the mRNA encoding IAP-II (ALPii) increased by approximately threefold.
Fig. 1.
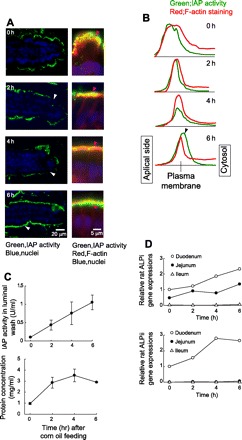
Effect of corn oil feeding on dynamics of intestinal alkaline phosphatase (IAP) in the rat duodenum. A: fluorescence microscopic images of the rat duodenal villi after corn oil feeding up to 6 h. Rats (n = 3 for each period of time) were fed with 2 ml of corn oil by gavage, and the duodenum was removed at the time point indicated. Duodenal frozen sections were stained with enzyme-labeled fluorescence (ELF)-97 for IAP activity (green), phalloidin for F-actin (red), and 7-aminoactinomycin D (7-AAD) for nuclei (blue). Left: lower magnification of ELF-97 and 7-AAD staining. IAP activity (white arrow heads) was gradually decreased up to 4 h after oil feeding but was recovered at 6 h. Right: high magnification of the corresponding images in the left panels with phalloidin staining. IAP activity (purple arrow heads) was observed in the surface of the villi above phalloidin at 0 h. The surface IAP activity was decreased at 2 and 4 h. At 6 h, IAP activity was detected beneath phalloidin. B: densitometric analysis of ELF-97 and phalloidin staining was vertically performed for the images of A, right. Densitometric curves for ELF-97 (green lines) and for phalloidin staining (red lines) were shown from apical side (left) to the cytosol (right). Y-axis represents relative fluorescent intensity. The red peak positions are vertically adjusted and indicated as “Plasma membrane.” The distribution of IAP activity became short and narrow up to 4 h with its peak positioned to the right of the red peak at 6 h (black arrow head). C: time course changes of IAP activity (top) and protein concentration (bottom) in the luminal contents of the rat ileum after oil feeding. Each plot indicates means ± SE (n = 3). D: time course changes of rat IAP (ALPi and ALPii) mRNA levels in the rat small intestine after oil feeding. mRNA was extracted from each two 5-mm segments of the duodenum (○), jejunum (•), and ileum (▵) up to 6 h. Real-time PCR was performed in triplicate for each extract. Data were normalized against L19, a housekeeping gene, and are shown as the means of the values obtained from 2 segments.
IAP activity after adding mixed-lipid micelles and lysoPC to Caco-2 cell cultures.
Human ALPI mRNA expression, IAP secretion, and cellular IAP activity were progressively increased in the Caco-2 cells cultured with serum-containing medium (Fig. 2, A and B). After 2 wk of culture (serum-free medium in the last 4 days), Caco-2 cells demonstrated a complete monolayer with morphological differentiation as manifested by polarized IAP localization (data not shown). Thus the culture conditions as described in materials and methods were used in the following studies. Most IAP secretion was apically directed in all cultures, and basolateral secretion was either undetectable in the presence of serum (Fig. 2A) or limited when induced by the addition of mixed-lipid micelles (Fig. 2C).
Fig. 2.
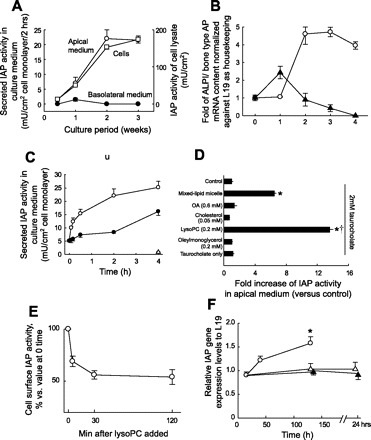
IAP activity and expression in Caco-2 cell cultures. A: AP activity in cell lysate and culture medium during the cell culture period. The cell monolayers were incubated with fresh media for 2 h, followed by collection of the cells and culture media for AP activity assay. AP activities in the apical medium (○), in the basolateral medium (•), and in cell lysate (□) are shown. Each data point represents mean ± SE (n = 3). B: human AP mRNA expression levels in Caco-2 cell cultures. Caco-2 monolayers were harvested as noted in A, with mRNA isolated and quantitated as outlined in materials and methods. ○, IAP; ▴, bone/liver/kidney AP. Each point represents the mean ± SE for n = 3. C: AP activity in apical and basolateral medium in response to mixed-lipid micelles. Mixed micelles were added to a final concentration of lipids as described in materials and methods. ○, apical medium; •, basolateral medium. Each point represents the mean ± SE of 3 determinations. D: effect of lipid micelle or each lipid component on IAP release to the apical medium of Caco-2 cell culture. Each point represents the mean ± SE (n = 3). OA, oleic acid; LysoPC, lysophosphatidylcholine. *P < 0.001 vs. control, †P < 0.001 vs. mixed-lipid micelle. E: effect of lysoPC on IAP activity in the brush-border membrane. Each point represents the mean ± SE (n = 3). F: effect of lysoPC on human IAP mRNA expression levels. Time course changes of relative IAP mRNA levels in lysoPC-treated group (0.2 mM, ○), oleic acid-treated group (0.6 mM, ▵), or untreated control (▴) are shown. Each point represents the mean of duplicate assay. *P < 0.05 vs. control.
Feeding a meal rich in triacylglycerol to animals increases IAP activity in the luminal contents of the small intestine as much as >10-fold (12). Because fatty acids may mediate the IAP release in the small intestine after fat feeding, we examined the effect of fatty acid-containing mixed micelles on IAP activity in Caco-2 cell monolayers. Mixed-lipid micelles resembling the lipid composition of the postprandial period were added to the apical medium of Caco-2 cell monolayers. The addition of mixed-lipid micelles rapidly increased IAP activity in the apical medium (Fig. 2C). By contrast, little IAP activity was detected in the basolateral medium at 2 h although after that time basolateral secretion did increase somewhat. Dot blot analysis confirmed that the IAP protein concentration increased two- to threefold, consistent with increased IAP activity (data not shown). Choline moieties and lysoPC were reported to affect BBM-AP activity in the cultured cells (11, 29), but we found that neither PC nor lysoPC had any direct effect on AP enzyme activity (data not shown). These findings suggested that the increased IAP activity in the apical medium after mixed-lipid micelle treatment was attributable to IAP release derived from BBM-IAP.
Next, we attempted to identify which component in the mixed-lipid micelles increased IAP release into the apical medium from the Caco-2 cell monolayer. LysoPC alone increased IAP activity in the apical medium, ∼14-fold greater than control, and twofold more than the mixed-lipid micelles, whereas other components had no effect (Fig. 2D). The addition of lysoPC rapidly reduced BBM-IAP activity by about half within 30 min (Fig. 2E). Because fatty acids increase IAP activity in the intestinal tissues after fat feeding (20, 31), we also compared the effects of oleic acid and lysoPC on IAP gene (ALPI) expression in Caco-2 cells. Real-time PCR showed that oleic acid had no significant effect on the level of IAP gene expression (Fig. 2F). In contrast, lysoPC significantly increased IAP gene expression ∼1.5-fold at 2 h after the treatment.
The detergent-like action of lysoPC suggested that the release of IAP from the BBM could be attributable to the nonspecific shedding of the membranes or cells and/or attributable to a cytotoxic effect of lysoPC. LysoPC (0.2 mM) at the concentration used in Caco-2 cell study had no effect on TER, whereas a higher concentration (2 mM) decreased TER (Fig. 3A). The polarized monolayers were morphologically intact with or without exposure to 0.2 mM lysoPC (Fig. 3, B and C). LysoPC (0.2 mM) had a little effect on LDH release into the apical medium (Fig. 3D), suggesting that the large increase of IAP release (∼50%) induced by lysoPC was attributable to an effect on the apical membrane distinct from cell lysis. In addition, exposure of Caco-2 cells to 0.2 mM lysoPC had no significant effect on the number of damaged cells as determined by PI staining (Fig. 3E).
Fig. 3.
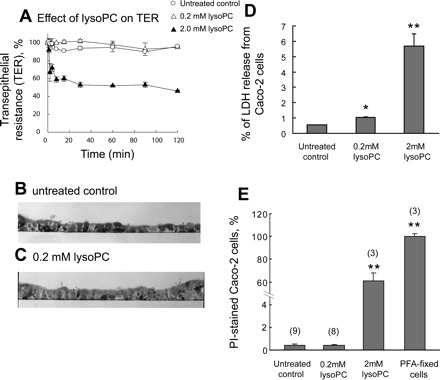
Effect of lysoPC application in the apical medium on the integrity of Caco-2 cell monolayers. A: effect of lysoPC in the apical medium on transepithelial resistance (TER) of Caco-2 cell monolayer. TER of the differentiated Caco-2 cell monolayers on the filter membrane support was measured at the indicated periods. The addition of 0.2 mM lysoPC (▵) had no effect on TER compared with the untreated control (○), whereas 2.0 mM lysoPC rapidly decreased TER. Each data point represents mean ± SE (n = 3). B and C: semi-thin vertical sections of Caco-2 cell monolayers with or without lysoPC treatment. The sections were counterstained with toluidine blue. B: untreated control. C: lysoPC (0.2 mM) treatment for 2 h. D: effect of lysoPC on lactate dehydrogenase (LDH) release from Caco-2 cells. Caco-2 cells were treated with 0.2 or 2 mM lysoPC for 2 h. LDH content in the apical medium and cell lysate was assayed. Data points are shown as LDH release from the cells (percentage of total cellular LDH). Each data point represents mean ± SE (n = 3). *P < 0.05; **P < 0.01 vs. untreated control. E: effect of lysoPC on cell damage. Caco-2 cell monolayers were incubated with 0.2 mM or 2 mM lysoPC in the presence of propidium iodide (PI) for 1 h, and the number of PI-positive cells was counted and expressed as a percentage of total cell number (mean ± SE). Total cell number in the examined area was obtained by PI staining after the cells were fixed. The total cell number in the area was 754 ± 20 (n = 3, SE). The parentheses indicate the number of replications. **P < 0.01 vs. untreated control. PFA, paraformaldehyde.
Release of brush-border enzymes from Caco-2 cells and mouse jejunum.
The effect of lysoPC for the release of IAP and other brush-border enzymes was examined in Caco-2 cells and mouse jejunal loops. The IAP release into the apical medium from Caco-2 cells (10-fold increase vs. untreated control) and into the luminal contents from the mouse jejunum (24-fold increase) was the greatest of the enzymes tested (Fig. 4, A and B). LysoPC also increased the release of other enzymes both from Caco-2 cells and mouse jejunum but to a lesser extent than the release of IAP. Thus lysoPC has a nonselective effect on increasing brush-border enzyme release, but IAP release predominates. There was no significant change in any of the enzyme levels measured in the light scrapings from mouse jejunum (Fig. 4C). The effect of lysoPC thus appeared to be restricted to the BBM and did not affect the overlying surfactant-like particles that are found in the light scrapings (5, 16).
Fig. 4.
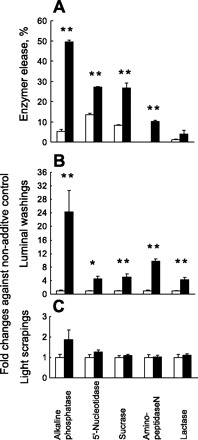
Effect of lysoPC application on brush-border enzyme release from Caco-2 cells and from mouse jejunum. A: 5 intestinal brush-border enzyme activities in the apical media and in Caco-2 cell lysate were assayed after 2-h incubation with 0.2 mM lysoPC (solid bar) or with the untreated control (open bar). Each bar represents the mean ± SE of enzyme activity (n = 3) released into the apical media as a percentage of the total cellular activity. **P < 0.01 vs. untreated control. B and C: 5 intestinal brush-border enzyme activities in the luminal washings (B) or light mucosal scrapings (C) of mouse jejunal loops were assayed after 30-min exposure to 4 mM lysoPC (solid bar) or to the untreated control (open bar). Each bar represents the mean ± SE (n = 3) of changes in each enzyme activity vs. untreated control. *P < 0.05, **P < 0.01 vs. untreated control.
DISCUSSION
The concentration of lysoPC is rapidly increased after a meal intake and reaches 2–3 mM in the upper small intestine (22) because of the rapid hydrolysis of bile PC (11–12 g/day) by pancreatic phospholipase A2 (7). In the present study, we have demonstrated that lysoPC mediated the release of IAP from the BBMs of Caco-2 cells and increased mRNA encoding IAP. These data are consistent with the increase of IAP release into the small intestinal lumen after corn oil feeding, accompanied by the rapid loss of BBM-IAP with the increased IAP mRNA expression, followed by BBM-IAP restoration demonstrated in rat duodenum. Our results suggest that lysoPC is a major factor that mediates the release of BBM-IAP and its restoration in the upper small intestine after corn oil feeding. Surfactant-like particles (SLPs) provide one mechanism whereby an IAP-enriched membrane is secreted to the luminal surface of the enterocytes following corn oil feeding (5). Since lysoPC, but not oleic acid, increased IAP mRNA expression, lysoPC may be an additional factor in inducing secretion of SLPs in the intestinal mucosa during the postprandial period (28), a process associated with increased expression of IAP. The direct action of lysoPC on the BBM appears to be a separate mechanism from the effect of corn oil stimulating SLP production after fat feeding because lysoPC does not lead to an enrichment in IAP over other brush-border enzymes in the light scrapings of the intestinal mucosa, the compartment in which SLP are concentrated (Fig. 4C).
Cholecystokinin-induced increases of L-IAP as well as other brush-border enzymes (5, 10) can be explained by this effect of lysoPC because cholecystokinin stimulates the secretion of pancreatic juice and gallbladder contraction, producing luminal contents containing phospholipid lipases and lecithin as substrate. LysoPC had little cytotoxicity and had no obvious effect on TER or cellular morphology at the concentration used (0.2 mM), inconsistent with IAP release by detergent-based shedding or cell lysis. Hung and Melnykovych (11) showed that lysoPC at 2 mM (calculated as palmitoyl) solubilizes the cellular membranes and releases membrane-bound tissue-nonspecific AP from HeLa cells. However, the predominant lysoPC-mediated IAP release compared with other hydrolases indicates that the enzyme release from the BBM is not attributable solely to nonspecific membrane shedding. Absorption of long-chain fatty acids and larger lipids is limited by diffusion through the unstirred water layer adjacent to the apical enterocyte membrane (27). Thus the effective lysoPC concentration at the surface of the BBM may not be predicted by the total concentration in luminal fluid (2–3 mM) and may be low enough to release IAP in preference to other hydrolases.
The preferential release of IAP may be related to the fact that it is a GPI-anchored and lipid raft-associated protein. Interestingly, Staneva et al. (24, 25) show that lysoPC induces the fission and budding of lipid-raft domains from artificial membranes. We have observed that methyl-β-cyclodextrin, which disrupts membrane rafts by depleting cholesterol, completely inhibited lysoPC-induced release of IAP (T. Nakano, unpublished observations). Although lysoPC increased the release of various brush-border enzymes from Caco-2 cells and the mouse jejunum, lysoPC released IAP more than the other ectoenzymes. The heterogeneity of the lipid and protein composition of lipid raft and GPI domains may affect the liability to enzyme release by lysoPC. LysoPC rapidly diffuses into the outer leaflet of the apical membrane, thereby disturbing the structure and properties of a variety of microdomains (2, 11). The modestly greater release of IAP by lysoPC compared with other enzymes (Fig. 4, A and B) suggests that there is not a large margin between concentrations that produce selective IAP release and those that lead to generalized enzyme shedding. LysoPC in concentrations as low as 0.1 mM produced evidence of increased mucosal and cellular permeability in the rat enterocyte (26). The factors that protect the intestine in vivo from the potentially toxic effects of lysoPC remain to be determined.
LysoPC may facilitate the turnover of IAP as well as other membrane-bound proteins during the postprandial period when extensive membrane reorganization is required to process the large amount of nutrients rapidly entering the cells (13). Our finding that BBM-IAP was rapidly restored after the loss of BBM-IAP because of its release induced by oil feeding is consistent with the rapid turnover of IAP facilitated by lysoPC. Brush-border turnover as measured by double-labeled isotope techniques in fasted rats demonstrated increased turnover of brush-border proteins with molecular weight of more than 200 mediated by pancreatic proteases. The released enzymes included the disaccharidases, which were released only modestly in the present experiments. Proteins having a molecular weight from 70,000 to 100,000 did not demonstrate an increased turnover rate, but IAP was not specifically measured (2). IAP comprises only a small percent of brush-border proteins, so it is possible that the turnover of IAP following fat feeding might have been missed or that the turnover of IAP might be mediated by a mechanism different from that related to the pancreatic proteases. In fact, lumenal proteases added in vitro to human brush borders did not lead to IAP release (33).
Recent findings suggest that IAP functions as a host-defense molecule by detoxifying LPS (3, 9). IAP expression is highest in the duodenum, intermediate in the jejunum, and low in the ileum and the large intestine (1, 19), where most LPS is produced by bacteria. IAP sloughed from the duodenum and jejunum after a meal might provide a LPS-detoxifying enzyme in the lumen throughout the length of the intestine. Feeding dietary oils to rats leads to a marked increase in the accumulation of luminal AP activity (12). We demonstrated that IAP secretion into the duodenal lumen increased in the rat after corn oil feeding and into the jejunal lumen of the mouse after lysoPC exposure. These findings suggest that duodenal and jejunal BBM-IAP released into the lumen during fat digestion might act within the lumen of the ileum and large intestine to detoxify LPS. IAP is relatively resistant to pancreatic proteases, consistent with the possibility that some IAP might survive transit to the colonic lumen in an active form (23).
In summary, lysoPC increased IAP expression in the epithelium and IAP release to the small intestinal lumen and changed the activity and localization of IAP. The replenishment of IAP within 6 h after the loss of BBM-IAP suggests that lysoPC may be one factor that mediates brush-border protein turnover. Furthermore, the released IAP may have a functional role in host defense throughout the gut, even as far as the distal small intestine and colon.
GRANTS
This work was supported in part by a grant from the Japan Foundation of Applied Enzymology for T. Nakano, the CNRU grant from NIH, DK56341, for partial support for D. H. Alpers, and a Department of Veterans Affairs Merit Review Award and NIH-NIDDK R01 DK54221 for J. D. Kaunitz.
Acknowledgments
The authors thank Dr. Sylvie Demignot (INSERM, Paris, France) for advice on Caco-2 cell culture and for providing a Caco-2 subclone and Dr. Terrence Riehl (Washington University School of Medicine) for providing us with the second Caco-2 subclone. We also express our gratitude to Dr. Kayoko Tanaka for help in histochemical analysis.
REFERENCES
- 1.Akiba Y, Mizumori M, Guth PH, Engel E, Kaunitz JD. Duodenal brush border intestinal alkaline phosphatase activity affects bicarbonate secretion in rats. Am J Physiol Gastrointest Liver Physiol 293: G1223–G1233, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Alpers DH, Tedesco FJ. The possible role of pancreatic proteases in the turnover of intestinal brush border proteins. Biochim Biophys Acta 401: 28–40, 1975 [DOI] [PubMed] [Google Scholar]
- 3.Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2: 371–382, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calhau C, Hipolito-Reis C, Azevedo I. Alkaline phosphatase and exchange surfaces. Clin Biochem 32: 153–154, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Eliakim R, Mahmood A, Alpers DH. Rat intestinal alkaline phosphatase secretion into lumen and serum is coordinately regulated. Biochim Biophys Acta 1091: 1–8, 1991 [DOI] [PubMed] [Google Scholar]
- 6.Fan MZ, Stoll B, Jiang R, Burrin DG. Enterocyte digestive enzyme activity along the crypt-villus and longitudinal axes in the neonatal pig small intestine. J Anim Sci 79: 371–381, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Froehlich F, Gonvers JJ, Fried M. Role of nutrient fat and cholecystokinin in regulation of gallbladder emptying in man. Dig Dis Sci 40: 529–533, 1995 [DOI] [PubMed] [Google Scholar]
- 8.Glickman RM, Alpers DH, Drummey GD, Isselbacher KJ. Increased lymph alkaline phosphatase after fat feeding: effects of medium chain triglycerides and inhibition of protein synthesis. Biochim Biophys Acta 201: 226–235, 1970 [DOI] [PubMed] [Google Scholar]
- 9.Goldberg RF, Austen WG Jr, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, Warren HS, Narisawa S, Millan JL, Hodin RA. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci USA 105: 3551–3556, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gotze H, Adelson JW, Hadorn HB, Portmann R, Troesch V. Hormone-elicited enzyme release by the small intestinal wall. Gut 13: 471–476, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hung SC, Melnykovych G. Alkaline phosphatase in HeLa cells. Stimulation by phospholipase A2 and lysophosphatidylcholine. Biochim Biophys Acta 429: 409–420, 1976 [DOI] [PubMed] [Google Scholar]
- 12.Kaur J, Madan S, Hamid A, Singla A, Mahmood A. Intestinal alkaline phosphatase secretion in oil-fed rats. Dig Dis Sci 52: 665–670, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Ladman AJ, Padykula HA, Strauss EW. A morphological study of fat transport in the normal human jejunum. Am J Anat 112: 389–419, 1963 [DOI] [PubMed] [Google Scholar]
- 14.Linscheer WG, Malagelada JR, Fishman WH. Diminished oleic acid absorption in man by L-phenylalanine inhibition of an intestinal phosphohydrolase. Nat New Biol 231: 116–117, 1971 [DOI] [PubMed] [Google Scholar]
- 15.Low MG, Prasad AR. A phospholipase D specific for the phosphatidylinositol anchor of cell-surface proteins is abundant in plasma. Proc Natl Acad Sci USA 85: 980–984, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahmood A, Shao JS, Alpers DH. Rat enterocytes secrete SLPs containing alkaline phosphatase and cubilin in response to corn oil feeding. Am J Physiol Gastrointest Liver Physiol 285: G433–G441, 2003 [DOI] [PubMed] [Google Scholar]
- 17.McComb RB, Bowers GN Jr. Study of optimum buffer conditions for measuring alkaline phosphatase activity in human serum. Clin Chem 18: 97–104, 1972 [PubMed] [Google Scholar]
- 18.Millan JL. Mammalian alkaline phosphatases. In: Biology to Applications in Medicine and Biotechnology. Weinheim, Germany: Wiley-VCH Verlag, 2006, p. 1–322.
- 19.Nakano T, Inoue I, Koyama I, Kanazawa K, Nakamura K, Narisawa S, Tanaka K, Akita M, Masuyama T, Seo M, Hokari S, Katayama S, Alpers DH, Millan JL, Komoda T. Disruption of the murine intestinal alkaline phosphatase gene (Akp3) impairs lipid transcytosis and induces visceral fat accumulation and hepatic steatosis. Am J Physiol Gastrointest Liver Physiol 292: G1439–G1449, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Narisawa S, Hoylaerts MF, Doctor KS, Fukuda MN, Alpers DH, Millan JL. A novel phosphatase upregulated in Akp3 knockout mice. Am J Physiol Gastrointest Liver Physiol 293: G1068–G1077, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J Cell Biol 127: 1199–1215, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Persson EM, Nilsson RG, Hansson GI, Lofgren LJ, Liback F, Knutson L, Abrahamsson B, Lennernas H. A clinical single-pass perfusion investigation of the dynamic in vivo secretory response to a dietary meal in human proximal small intestine. Pharm Res 23: 742–751, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Riepe SP, Goldstein J, Alpers DH. Effect of secreted Bacteroides proteases on human intestinal brush border hydrolases. J Clin Invest 66: 314–322, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staneva G, Angelova MI, Koumanov K. Phospholipase A2 promotes raft budding and fission from giant liposomes. Chem Phys Lipids 129: 53–62, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Staneva G, Seigneuret M, Koumanov K, Trugnan G, Angelova MI. Detergents induce raft-like domains budding and fission from giant unilamellar heterogeneous vesicles: a direct microscopy observation. Chem Phys Lipids 136: 55–66, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Tagesson C, Franzen L, Dahl G, Westrom B. Lysophosphatidylcholine increases rat ileal permeability to macromolecules. Gut 26: 369–377, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomson AB, Schoeller C, Keelan M, Smith L, Clandinin MT. Lipid absorption: passing through the unstirred layers, brush-border membrane, and beyond. Can J Physiol Pharmacol 71: 531–555, 1993 [DOI] [PubMed] [Google Scholar]
- 28.Tietze CC, Becich MJ, Engle M, Stenson WF, Mahmood A, Eliakim R, Alpers DH. Caco-2 cell transfection by rat intestinal alkaline phosphatase cDNA increases surfactant-like particles. Am J Physiol Gastrointest Liver Physiol 263: G756–G766, 1992 [DOI] [PubMed] [Google Scholar]
- 29.Tiruppathi C, Alpers DH, Seetharam B. The role of choline on the activity-temperature relationship of brush-border alkaline phosphatase. Biochim Biophys Acta 898: 283–292, 1987 [DOI] [PubMed] [Google Scholar]
- 30.Tuin A, Huizinga-Van der Vlag A, Van Loenen-Weemaes AM, Meijer DK, Poelstra K. On the role and fate of LPS-dephosphorylating activity in the rat liver. Am J Physiol Gastrointest Liver Physiol 290: G377–G385, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Xie Q, Alpers DH. The two isozymes of rat intestinal alkaline phosphatase are products of two distinct genes. Physiol Genomics 3: 1–8, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Yamagishi F, Komoda T, Alpers DH. Secretion and distribution of rat intestinal surfactant-like particles after fat feeding. Am J Physiol Gastrointest Liver Physiol 266: G944–G952, 1994 [DOI] [PubMed] [Google Scholar]
- 33.Young GP, Das L. Influence of duodenal secretions and its components on release and activities of human brush-border enzymes. Biochim Biophys Acta 1022: 393–400, 1990 [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Shao JS, Xie QM, Alpers DH. Immunolocalization of alkaline phosphatase and surfactant-like particle proteins in rat duodenum during fat absorption. Gastroenterology 110: 478–488, 1996 [DOI] [PubMed] [Google Scholar]