

Abstract
The calibre of the upper airway is thought to be dependant upon its passive anatomy/collapsibility and the activation of pharyngeal dilator muscles. During awake periods, the more collapsible upper airway in obstructive sleep apnoea (OSA) increases the dilator muscle activity through a negative-pressure reflex.
A direct correlation between the critical closing pressure (Pcrit), as a measure of anatomy/collapsability and electromyogram (EMG) activity of genioglossus EMG (GG-EMG) and tensor palatini EMG (TP-EMG), was hypothesised. The relationship between these indices and pharyngeal resistance (Rphar) was also examined.
The study involved eight males with a mean age of 48 (interquartile range 46–52) yrs with OSA, and an apnoea/hypopnoea index of 75 (65–101)·hr−1 on two nights breathing normally and on nasal continuous positive airway pressure (nCPAP). The Pcrit was measured during nonrapid eye movement sleep on nCPAP using brief, incremental reductions in mask pressure. GG-EMG and TP-EMG were measured breath-by-breath, awake, during sleep onset and on nCPAP. Rphar was measured using airway pressures and flow.
Wakeful GG-EMG, early sleep TP-EMG and the sleep decrement in TP-EMG were directly related to Pcrit. Muscle activation was negatively correlated with Rphar for TP-EMG awake and GGEMG early in sleep.
In conclusion these results confirm that dilator muscle activation is directly related to airway narrowing and reduces resistance across patients with obstructive sleep apnoea.
Keywords: Airway mechanics, airways resistance, muscle function, obstructive sleep apnoea, pharynx
The most vulnerable part of the upper airway is the part behind the base of the tongue and the soft palate, where rigid skeletal support is absent. Here, airway patency is determined by a balance of forces as determined by the interplay of anatomical factors and dilator muscle activation, as shown in figure 1 [1]. The neural drive to the upper airway dilator muscles is integrated at the level of brainstem motor nuclei where multiple neural inputs combine to produce a unitary output to the muscles. For example, the hypoglossal nucleus, which innervates genioglossus (GG), receives: multiple inputs from the pre-motor pacemaker neurones of the respiratory pattern generator [2]; reflex inputs from the upper airway negative-pressure receptors (NPR) [3–7]; chemoreceptor inputs [8, 9]; vagal input, due to change in lung volume [10–12]; and a wakefulness component from the brainstem reticular formation and other sources [13]. These various neural inputs have been studied in animals [14–16] as well as in humans.
FIGURE 1.
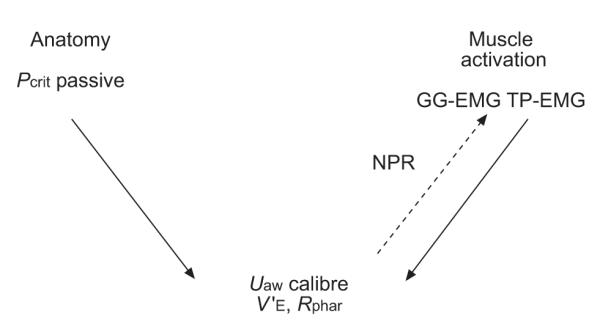
Hypothetical relationships between the determinants of upper airway calibre. In this model, airway anatomy/collapsibility and dilator muscle activation are the two principal determinants of airway calibre. Upper airway (Uaw) dilator muscle activation is partly driven (- - -) by the negative pressure reflex (NPR) and responds to Uaw narrowing by increasing its activation. Pcrit: critical closing pressure; GG-EMG: genioglossus electromyogram; TP: tensor palatini; V’: flow; Rphar: pharyngeal resistance.
The relative magnitude of each of the components of upper airway muscle activation may vary amongst the sets of upper airway muscles involved, and typically some dilators, e.g. GG, have more marked phasic inspiratory activity than others, e.g. tensor palatini (TP), the activity of which is less phasically modulated by the respiratory cycle and, in normal subjects at least, is predominantly tonic [17]. Wasicko et al. [18] have also shown that added resistive loads lead to differential muscle activation in normal subjects and that these responses are deficient in obstructive sleep apnoea (OSA) subjects.
In patients with OSA, anatomical factors resulting in narrowing of the upper airway have been characterised on magnetic resonance imaging (MRI) [19]. These include an increase in lateral wall thickness and fat pad size with reduction in lateral diameter and cross-sectional area of the retro-pharyngeal airway. During wakefulness, patency of the narrowed upper airway in OSA is maintained by augmented dilator muscle activation [20, 21]. The current authors have recently shown that this increased activity in OSA patients is largely due to a heightened NPR component, as it is markedly reduced by the application of continuous positive airways pressure (CPAP) [22]. However, even on CPAP, GG electromyogram (GG-EMG) activity in OSA remains higher during wakefulness and falls further at sleep onset (α to Θ transitions) than that in normal sleep, suggesting that in OSA sleep there is also augmentation of the wakefulness or respiratory pattern generator drives. Furthermore, there is recruitment of dilator muscle activation in OSA subjects early on after sleep onset, suggesting that the NPR remains active, to some extent, during sleep in this group.
The passive anatomy of the upper airway relates to its structure and collapsibility. This may be assessed during sleep using the passive method for the measurement of critical closing pressure (Pcrit) described by Schwartz et al. [23]. The upper airway dilator muscles remain relatively inactive for several breaths after sudden reduction of CPAP during sleep and the measured collapsibility reflects the passive anatomical characteristics of the upper airway. This method differs from the earlier method [24], in which airway pressure is gradually reduced and measures airway collapsibility during sleep, with the dilator muscles partially activated by negative pressure. Other methods, which have included using imaging techniques for assessing the size, cross-sectional and three-dimensional shape of the upper airway with MRI [19] and computed tomography (CT) [25, 26], have proven useful in identifying differences between OSA patients and normal subjects. However, these methods have not been systematically applied during sleep, because of the difficulty in achieving sleep in the scanners, or on a breath-by-breath basis, because of the time constraints of image acquisition. In addition, these methods measure anatomical factors with concomitant muscle activation. Philip-Joet et al. [27] have demonstrated a linear correlation between muscle activation during sleep and Pcrit measured in the negative-pressure activated upper airway of normal subjects. However, the relationship between muscle activation and passive collapsibility across sleep onset in OSA has not previously been evaluated.
The primary aim of the current study was to investigate the relationships between passive upper airway collapsibility, dilator muscle activation and airway calibre (estimated as pharyngeal resistance: Rphar), across a group of subjects with OSA. It was hypothesised that the dilator muscle activation is strongly related to passive anatomical calibre (estimated as Pcrit) and that subjects whose upper airway is narrower than normal will have greater activation of upper airway muscles. Furthermore, this relationship may be stronger during wakefulness than early after sleep onset, when the NPR is less active. A relationship between dilator muscle activation and airway calibre (measured as Rphar) was also anticipated. This was predicted by the present authors to be stronger during wakefulness and to weaken during sleep with the removal of the wakefulness drive and reduction of the NPR components of muscle activation. The current authors subsequently aimed to measure passive anatomy (Pcrit), wakeful and sleep muscle activation (GG-EMG and TP-EMG) and airway calibre as Rphar and to assess the relationships between these variables across a group of patients with OSA.
METHODS
Subjects
Eight male subjects with OSA were recruited from the sleep clinic. Median (interquartile range) patient characteristics were: age 48 (46–52) yrs; body mass index (BMI) 32.8 (29.6–41.2) kg·m−2; and apnoea/hypopnoea index (AHI) 75 (65–101) events·h−1. The eight subjects undergoing detailed assessment of upper airway mechanics and collapsibility in this report were part of a larger group of OSA subjects in whom the sleep-onset effects on dilator muscle activation have recently been published [22]. All eight subjects in the current study had been on nasal CPAP therapy for a minimum of 3 months and had demonstrated compliance (≥6 h·night−1 by history). Nasal CPAP therapy was continued until the night before the studies took place. Patients on regular medication, other than stable antihypertensive therapy, were excluded from the study. Informed consent was obtained from each subject, with the protocol conforming to the principles outlined in the declaration of Helsinki and having the prior approval of the human subjects ethics committee of the Brigham and Women’s Hospital (Boston, MA, USA).
Protocol
Each subject was studied for 2 nights separated by at least 1 week. On one night the patient was studied on nasal CPAP for the purpose of measuring airway collapsibility (Pcrit) during sleep. On the other, spontaneous breathing measurements of dilator muscle activation (GG-EMG/TP-EMG) and airway calibre (Rphar) during wakefulness and sleep were obtained. The order of the study nights (CPAP or basal breathing (BB)) was randomised. Subjects reported to the laboratory at ~21:00 h, having fasted for a minimum 4 h. After obtaining informed consent, the sleep staging electrodes, pressure catheters and intramuscular EMG wires were positioned. Subjects then assumed a supine posture in bed and the nasal mask was attached. Subjects subsequently lay with opened eyes in this posture and were allowed to acclimatise to the equipment.
Data recording
The laboratory procedures, EMG recordings and measurement of ventilation and resistance were conducted as previously described [5, 17]. In order to assess sleep–wake state, subjects were instrumented with two channels of electroencephalography (EEG), two channels of electro-oculography (EOG) and chin EMG.
All signals (GG-EMG and TP-EMG, raw and an electronically derived moving time average (MTA); airway pressures mask, choanal and epiglottic; end-tidal carbon dioxide (ETCO2); ECG; sleep staging; and inspiratory flow) were recorded on a 16-channel Grass model 78 polygraph (Grass Instruments Astro-Med Inc., West Warwick, RI, USA). Certain signals (GG-EMG, TP-EMG, airway pressures, EEG, inspiratory flow and ECG) were also recorded on a computer using data acquisition software (Spike 2, version 3.17; Cambridge Electronic Design Ltd, Cambridge, UK).
For each individual, the occipital EEG during each breath was assessed as being predominantly α or Θ, as previously described [28, 29].
Airway mechanics
Subjects wore a nasal mask (Respironics Inc., Murraysville, PA, USA) connected to a non-rebreathing valve. Inspiratory and expiratory airflow were determined with a pneumotachometer (model 3700A; Hans Rudolph Inc., Kansas City, MO, USA) and differential pressure transducer (Validyne Corp., Northridge, CA, USA), calibrated with a rotameter. Subjects were instructed to breathe exclusively through the nose and were carefully monitored by video camera to ensure that the mouth was completely closed. The mouth was taped shut during sleep, in order to minimise the subject breathing through the mouth. Mask leak was detected from a perforated catheter surrounding the mask-face interface, which was continuously sampled for CO2. In addition, end-tidal CO2 tension (PET,CO2) was monitored from the mask using an infrared analyser (Capnograph Monitor; BCI, Waukesha, WI, USA).
Pressures were monitored in the mask with an open catheter attached to a pressure transducer (Validyne Corp.) and in the airway at the levels of the posterior choanae and the epiglottis using two pressure-tipped catheters (MPC-500; Millar, Houston, TX, USA). One nostril was decongested (oxymetazalone HCl; Taro Pharmaceuticals USA Inc., Mawthorne, NY, USA) and anesthetised (lidocaine HCl; Roxane Laboratories Inc., Columbus, OH, USA), and the Millar catheters were inserted through this nostril and localised at the posterior choanae and the epiglottis. Prior to insertion, the pressure signals were calibrated simultaneously in a rigid cylinder using a standard water manometer (Dwyer Instruments Inc., Michigan City, IN, USA). These pressure signals plus flow were demonstrated to be without amplitude or phase lags at up to 2 Hz. Any drift in the pressure catheters was corrected on a breath-by-breath basis by an automated computer program that defined end inspiration and end expiration by identifying the point of zero flow and then correcting any offset in the pressure signals. Minute ventilation and Rphar (measured as peak inspiratory resistance and at a fixed flow rate (0.2 L·s−1)), were calculated on a breath-by-breath basis, Rphar being the pressure difference between the choanae and the epiglottis related to flow. The automated analysis computer program was modified to allow analysis of breaths with complete airway obstruction (as occurred in some OSA patients on the normal breathing night). The algorithm initially used the flow signal to define the beginning and end of each inspiration. However, if respiratory-related variations in pressure were detected in the absence of variations in flow, the computer then used the epiglottic pressure signal to define the beginning and end of inspiration. In addition, on breaths with extremely low flow rates, resistance values were set at a maximum of 99 cm H2O−1·L−1·s−1.
Muscle activation
The GG-EMG was measured with a pair of unipolar intramuscular electrodes referenced to a single ground, producing a bipolar recording. Two stainless steel Teflon-coated 30-gauge wire electrodes were inserted 15–20 mm into the body of the GG muscle, 3 mm lateral to the frenulum on each side, using a 25-gauge needle that was quickly removed, leaving the wires in place. This technique has been used previously [5, 28–32]. TPEMG was measured in a similar manner to that of the GG muscle, using a pair of referenced unipolar intramuscular electrodes producing a bipolar recording. The tip of the pterygoid hamulus was located at the junction of the hard and soft palate, on each side of the palate. A 25-gauge needle with a 30-gauge stainless steel Teflon-coated wire was then inserted at a 45-degree angle along the lateral surface of the medial pterygoid plate, to a depth of ~10–15 mm into the palate. The needle was then removed, leaving the electrode in place. To establish maximal EMG activation and to confirm GG and TP electrode placement, the following respiratory manoeuvres, which have been documented previously [33], were performed: maximal tongue protrusion, sucking, blowing and swallowing. These manoeuvres were performed in triplicate until reproducible EMG activation by each was obtained.
The raw EMG signal was amplified to provide an easily visible phasic inspiratory signal during quiet breathing (Grass Instruments, Quincy, MA, USA), band pass filtered (between 30–1,000 Hz) and stored for subsequent data analysis. Sections of the recording containing movement or other artefacts were removed before analysis. The raw EMG signals for the muscles were then integrated using a 100-ms MTA. Several values were calculated on a breath-by-breath basis for each muscle. The first was the tonic level. Activity in the expiratory phase was divided into 10 equal segments and the level of tonic activity of the muscle was defined as the activity of the lowest segment. The second was peak activity, which was defined as the highest value that occurred during inspiration. The third was phasic activity, which was defined as the difference between peak activity and tonic activity. In order to allow comparison between subjects and between the CPAP and control nights, the GG-EMG and TP-EMG were quantified as a per cent of the maximal total activity observed during forced tongue protrusion, as previously described [33]. The tonic, phasic, and peak EMG activity for each muscle for each breath were then expressed as a percentage of the maximal values.
Comparison of the magnitude of EMG activation between swallows, which remained stable across the night, was used to establish that changes in EMG electrode impedance had not occurred.
Basal breathing night
After recording data during stable wakefulness, subjects were allowed to fall asleep (remaining in the supine posture). On the BB night, in order to obtain multiple sleep onsets (α–Θ transitions) during normal breathing, subjects were awoken if they slept for 5 mins consecutively without spontaneous awakenings, and were thereafter allowed to fall asleep again. This procedure was repeated until ~4 h of data had been collected.
On the BB night, once each breath had been classified as α or Θ, computer software was used to identify sets of consecutive breaths occurring on either side of an α–Θ transition. An adequate α–Θ transition was defined by having at least three consecutive α breaths followed by at least two consecutive Θ breaths. Each breath in the transition was then assigned a position relative to the transition from −5– +3 as previously described [28]. For each subject, the following parameters were averaged at each breath position: minute ventilation (V’E), Rphar, GG-EMG and TP-EMG (as percentage of maximal activity). Waking values for each variable were determined from the initial stable wakeful period at the start of the night and also from the mean α level (−5– -1) immediately before sleep onset. Sleep values were measured for each Θ breath (1–3) following sleep onset and compared with the mean α level preceding sleep onset.
Determination of airway collapsibility
On the CPAP night, subjects were studied using the same nasal mask with CPAP applied. Initially 5–10 mins of data were recorded during quiet wakefulness, lying supine without CPAP in order to estimate baseline GG-EMG/TP-EMG. Nasal CPAP was then applied. Subjects breathed through a nasal mask connected to a modified CPAP device incorporating a valve for rapidly switching between two preset levels of positive or negative pressure (Respironics Inc.). CPAP was initially at the patient’s previously prescribed pressure and, if flow limitation was noted during sleep, the pressure was increased until this was abolished and the minimum level of GG-EMG was obtained.
Passive Pcrit was measured using the method of Schwartz et al. [23]. After the patient had been in stable stage 2 sleep for ≥10 mins without flow limitation, transient decrements in pressure for three breaths were applied by switching between breaths to a reduced level of CPAP. Pressure decrements started at 2 cmH2O below baseline CPAP and increased progressively by a further 2 cmH2O from baseline. These were applied repeatedly after intervals ensuring return of EMG activity to stable baseline until flow limitation to complete cessation of airflow or arousal occurred. When arousal occurred, that pressure decrement series was terminated and the patient was allowed to return to stable stage 2 sleep for ≥5 mins before another series of progressive pressure decrements was applied. In each patient, data from at least four such series was collected.
To measure critical airway closing pressure by the passive method, nasal pressure and maximal flow values for each pressure decrement were plotted for flow-limited breaths in each patient. The Pcrit value was taken as the intercept on the nasal pressure axis at zero maximal flow established as a best fit for all pressure decrement series for that patient.
Statistical analyses
Correlation between Pcrit, dilator muscle EMG (GG-EMG/TP-EMG) and Rphar was performed using Spearman’s rank test. For all analyses, α was set at 0.05. Results are presented as Spearman correlation coefficients and p-values with lines of best fit determined by linear correlation. The Mann–Whitney U-test was used to compare wake with sleep data. Only BB night measures of EMG and Rphar were used to assess correlation between collapsibility, muscle activation and resistance.
RESULTS
Patient demographics and study variables are presented in tables 1 and 2, respectively. Raw data from one subject’s CPAP/Pcrit night showing a run of progressive pressure decrements resulting in flow limitation is shown in figure 2. Note the relatively stable GG-EMG recording demonstrating little muscle recruitment during the first few breaths following the pressure decrements.
TABLE 1.
Demographics for the eight obstructive sleep apnoea patients
| Age yrs | 48 (46–52) |
| BMI kg·m−2 | 32.8 (29.6–41.2) |
| AHI·h−1 | 75 (65–101) |
| Pcrit cmH2O | 2.4 (0.6–3.6) |
Data are presented as median (25th–75th percentiles). BMI: body mass index; AHI: apnoea/hypoapnoea index; Pcrit: critical closing pressure.
TABLE 2.
Study variables for the eight obstructive sleep apnoea patients
| Awake | S2 | S3 | |
|---|---|---|---|
| GG-EMG % max | |||
| Peak | 7.6 (5.6–9.8) | 8.4 (7.6–10.7) | 10.9 (8.2–13.2) |
| Phasic | 4.0 (2.9–4.5) | 5.4 (4.6–5.8) | 6.3 (4.9–7.8) |
| Tonic | 3.9 (3.0–5.2) | 3.0 (2.8–5.5) | 3.7 (3.5–5.7) |
| TP-EMG % max | |||
| Peak | 9.4 (4.5–21.4) | 6.0 (4.3–10.4) | 5.9 (4.1–10.2) |
| Phasic | 1.5 (0.7–3.9) | 3.4 (1.8–6.5) | 3.7 (1.9–5.8) |
| Tonic | 6.8 (3.8–14.5) | 2.6 (2.1– 4.1) | 2.6 (2.1–4.4) |
| Rphar | 1.7 (0.9–2.6) | 5.6 (3.2–7.1)* | 4.1 (3.4–6.2)* |
Data are presented as median (25th–75th percentiles). S2: second Θ breath; S3: third Θ breath; GG-EMG: genioglossus electromyogram; TP-EMG: tensor palatini electromyogram; Rphar: pharyngeal resistance.
: p<0.05, Mann–Whitney U-test versus basal breathing awake.
FIGURE 2.

– An example of raw data in one obstructive sleep apnoea subject on the continuous positive airway pressure/critical closing pressure night. Note that with progressively increasing brief decrements in nasal pressure, increasing flow limitation and eventually apnoea occur. Note the lack of recruitment in genioglossus electromyogram (GG-EMG) during the brief pressure drops during sleep. EEG: electroencephalography; TP-EMG: tensor palatini electromyogram; Pepi: epiglottic pressure; Pmask: mask pressure.
The determination of the Pcrit is shown in figure 3 for one of the subjects. The intercept on the nasal pressure axis at zero flow determined by the line of best fit is taken as that patient’s value of Pcrit.
FIGURE 3.
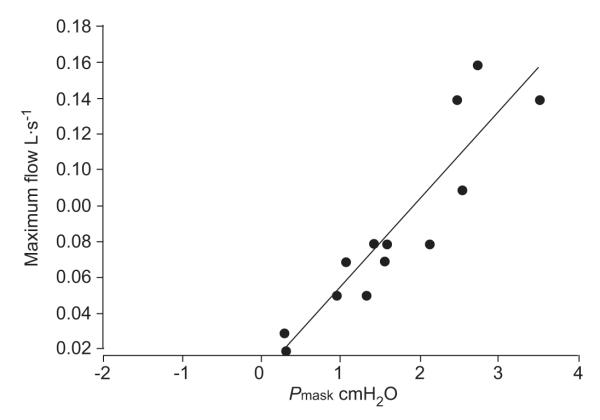
Measurement of critical closing pressure (Pcrit) in one obstructive sleep apnoea (OSA) subject. Plot of nasal pressure versus maximal flow for flow-limited breaths without arousal during pressure decrements in one (OSA) subject on the continuous positive airway pressure/Pcrit night. Data points are collected during multiple pressure drop runs. Pcrit is determined from the pressure intercept at zero flow from the line of best fit, following Schwartz and co-workers [23]. Pmask: mask pressure.
Median (25th–75th percentiles) values for EMG and Rphar study variables awake and for the second (S2) and third Θ breaths (S3) during BB are shown for the eight obstructive sleep apnoea subjects in table 2.
Sleep onset effects
On the BB night, Rphar increased dramatically and significantly between wake and sleep breaths two and three (both p<0.05) during BB. Typically, apnoea then arousal occurred within three to four breaths after sleep onset in patients with severe OSA. In seven of the eight patients, GG-EMG and TP-EMG either decreased or changed little between wake and the second and third sleep breaths, consistent with previous studies of normal subjects [28, 29] and in a larger group of OSA patients, which included those in the present study [22]. Tonic activity of both muscles but particularly TP-EMG fell at sleep onset (wake versus sleep breaths two and three on the BB night) but none of these differences between wake and sleep EMG reached statistical significance for either muscle. In one subject there was an unexpected and substantial increase in both GG-EMG and TP-EMG from wakefulness to sleep, although the subject’s Rphar increased from wake to sleep, as did the other subjects. The current authors could find no explanation to account for the increase in muscle activity in this subject. Correlation analyses were performed both excluding (n=7) and including (n=8) this subject. The latter yielded slightly lower r-values for most of the correlations.
Relationship between critical closing pressure and waking upper airway muscle activation
The results of linear correlation analyses (Spearman rank test) between Pcrit and BB-EMG activation during stable wakefulness, expressed as percentage maximal activation, are shown for the seven subjects in whom muscle activation fell at sleep onset in table 3.
TABLE 3.
Spearman rank correlations between study variables
|
Pcrit |
Rphar |
BMI |
||||
|---|---|---|---|---|---|---|
| r-values | p-values | r-values | p-values | r-values | p-values | |
| Awake GG-EMG | ||||||
| Peak | 0.79 | 0.02 | 0.00 | 0.97 | 0.66 | 0.07 |
| Phasic | 0.82 | 0.01 | 0.00 | 0.97 | 0.68 | 0.07 |
| Tonic | 0.68 | 0.07 | −0.29 | 0.49 | 0.39 | 0.34 |
| Awake TP-EMG | ||||||
| Peak | −0.04 | 0.91 | −0.78 | 0.03 | 0.10 | 0.78 |
| Phasic | 0.46 | 0.25 | 0.18 | 0.66 | 0.50 | 0.21 |
| Tonic | −0.14 | 0.72 | −0.64 | 0.09 | 0.17 | 0.66 |
| Sleep breath 2 | ||||||
| GG-EMG | ||||||
| Peak | 0.07 | 0.84 | 0.43 | 0.30 | −0.29 | 0.49 |
| Phasic | 0.32 | 0.43 | 0.11 | 0.78 | −0.07 | 0.84 |
| Tonic | 0.001 | 0.97 | 0.34 | 0.39 | −0.21 | 0.60 |
| Sleep breath 2 | ||||||
| TP-EMG | ||||||
| Peak | 0.75 | 0.04 | −0.50 | 0.21 | 0.53 | 0.18 |
| Phasic | 0.64 | 0.09 | −0.39 | 0.34 | 0.50 | 0.21 |
| Tonic | 0.42 | 0.30 | −0.54 | 0.18 | 0.64 | 0.09 |
| Sleep breath 3 | ||||||
| GG-EMG | ||||||
| Peak | 0.32 | 0.44 | −0.82 | 0.05 | 0.17 | 0.66 |
| Phasic | 0.64 | 0.09 | −0.82 | 0.05 | 0.11 | 0.78 |
| Tonic | −0.14 | 0.72 | −0.60 | 0.24 | −0.14 | 0.72 |
| Sleep breath 3 | ||||||
| TP-EMG | ||||||
| Peak | 0.64 | 0.09 | −0.02 | 1.00 | 0.55 | 0.22 |
| Phasic | 0.61 | 0.12 | −0.02 | 1.00 | 0.39 | 0.34 |
| Tonic | 0.42 | 0.30 | 0.14 | 0.80 | 0.64 | 0.09 |
Data are presented as percentage maximum. Pcrit: critical closing pressure; Rphar: pharyngeal resistance; BMI: body mass index; GG-EMG: genioglossus electromyogram; TP-EMG: tensor palatini electromyogram.
Waking peak GG-EMG as percentage maximal showed a significant relationship with Pcrit (r=0.79, p=0.02) as shown in figure 4a. This relationship was also seen for phasic GG-EMG (r=0.82, p=0.01) and a trend for tonic GG-EMG (r=0.68, p=0.07). The relationships of waking TP-EMG with Pcrit were not significant.
FIGURE 4.
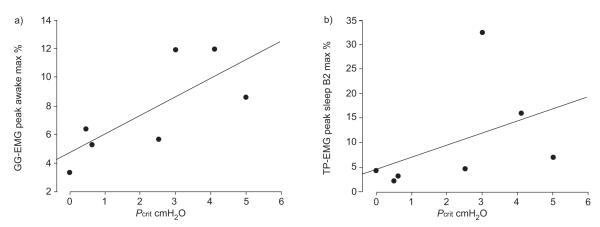
Relationships of genioglossus electromyogram (GG-EMG) and tensor palatini electromyogram (TP-EMG) activation to critical closing pressure (Pcrit) in obstructive sleep apnoea. max %: per cent maximum; B2: second sleep Θ breath. a) r=0.79; p=0.02. b) r=0.75; p=0.04.
Relationship between critical closing pressure and upper airway muscle activation after sleep onset
For GGEMG, the relationship to Pcrit weakened, becoming nonsignificant during sleep. For TPEMG, activity at the second and third breaths following onset of Θ activity became more strongly related to Pcrit than during wakefulness and, for peak activity, the relationship was significant at the second breath after sleep onset (r=0.75, p=0.04) as shown in figure 4b.
Relationship between critical closing pressure and the fall in muscle activation at sleep onset
The fall in TP-EMG activity between wakefulness (breaths −5– −1) and sleep (breaths +2, +3) was also significantly and positively related to Pcrit. Correlation coefficients for TPEMG (wake–S3) versus Pcrit were significant for peak (r=0.71, p=0.05) and phasic (r=0.89, p=0.01) components. These relationships did not reach significance for GG-EMG.
Relationship between upper airway muscle activation, pharangeal resistance and their changes at sleep onset
Waking TP-EMG peak (% maximum) activity was significantly and negatively related to wakeful Rphar (r= −0.78, p=0.03) during BB as shown in figure 5a. This relationship was not significant for GG-EMG.
FIGURE 5.
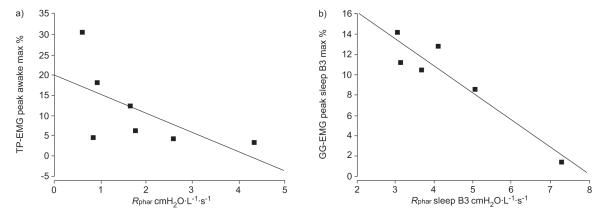
Relationships of tensor palatini electromyogram (TP-EMG) and genioglossus electromyogram (GG-EMG) activation to pharyngeal resistance (Rphar) in obstructive sleep apnoea. Pharyngeal resistance during sleep breath 3 was available in only six patients. max %: per cent maximum; B3: third sleep Θ breath. a) r= −0.78, p=0.03. b) r= −0.82, p=0.05.
During sleep (S3) GG-EMG was significantly related to sleep Rphar (r= −0.82, p=0.05) as shown in figure 5b, whereas for TP-EMG these relationships were not significant.
The rise in Rphar at sleep onset (wake–S3) was strongly related to wakeful baseline TP-EMG for both peak and tonic activity (r=0.94, p=0.01). It was not significantly related to the fall in muscle activation at sleep onset.
Relationship between muscle activation and other factors
The results of the correlations between GG-EMG and TP-EMG with BMI are also shown in table 3. Generally, waking GGEMG showed a weaker relationship with BMI than with Pcrit. Waking TP-EMG was not significantly related to BMI and, for GG-EMG, the relationship with BMI weakened during sleep.
Other relationships
Rphar measured during BB was not significantly related to Pcrit either awake (r= −0.21, p=0.60) or after sleep onset (S2: r= −0.46; p=0.25; S3: r= −0.60; p=0.24). Similarly, Rphar was not significantly related to BMI in wakefulness or sleep across the group.
DISCUSSION
In this study, the current authors set out to determine the relationship between upper airway calibre evaluated as Rphar and its two principal determinants: 1) the passive anatomical structure/collapsibility of the upper airway; and 2) the activation of upper airway dilator muscles across a group of patients with OSA. The current authors observed a significant, direct relationship with Pcrit for both waking GG-EMG and early sleep TP-EMG. Waking TP-EMG was also inversely related to Rphar across the group. Rphar itself was not correlated with Pcrit. These findings taken together are consistent with the hypothesis that narrowing of a more collapsible upper airway augments dilator muscle activation via the negative pressure reflex to widen the upper airway and that this upper airway muscle response reduces Rphar most in those with greatest dilator activation.
The present authors have previously shown in OSA patients [31] and normal subjects [31, 34] that increasing negative pressure in the upper airway, with the addition of an inspiratory resistive load or using a negative pressure ventilator, causes an increase in GG-EMG within the individual that is proportional to both the increased epiglottic pressure and the Rphar that develops. The current findings suggest that this negative pressure reflex is consistent between patients with sleep apnoea as well as within the individual patient.
The correlation between GG muscle activation and upper airway collapsability (Pcrit) during wakefulness is explained by a narrower, more collapsible upper airway (high Pcrit) experiencing more negative inspiratory pressure and activation of the NPR to augment dilator muscle activation. This relationship weakens in the early breaths after sleep onset in this study. The current authors previously demonstrated a loss of dilator muscle activation for GG (and for TP) at sleep-onset in normal subjects and in patients with OSA [28, 29, 32, 35]. These findings are consistent with diminution of both the augmented wakeful NPR and the phasic central pattern generator components of GG activation at the point of sleep onset when the wakefulness drive is lost.
In the current study of patients with OSA, the tonic activation of both muscles, but particularly that of the predominantly tonic TP-EMG, tends to diminish at sleep onset. This diminution in tone at sleep onset has been observed in previous studies of normal subjects [29] and OSA patients [22] and would have been likely to reach significance in this study given a larger number of subjects. The relationship between TP muscle activation and Pcrit is weak before but becomes more strongly positive after sleep onset and is significant by the second sleep breath. This suggests that the NPR is still present during early sleep. Reduction of phasic GG activity at sleep onset may result in the relationship between activation of the more tonic TP and airway collapsibility (via negative pressure in the airway lumen) becoming more clearly apparent.
The direct influence of upper-airway dilator muscle activation on airway calibre is seen in the negative correlations between wakeful TP-EMG and Rphar, and between GG-EMG after sleep onset (S3) and Rphar. These are consistent with NPR-augmented activation of TP during wakefulness in OSA subjects serving to decrease upper-airway resistance in compensation for the airway narrowing. If this compensatory mechanism were completely efficient, no correlation across subjects between these two variables would be expected. The presence of a significant inter-individual relationship between GG-EMG and Rphar during the early sleep breaths is also consistent with higher activation of GG early after sleep onset in those subjects with a narrower upper airway. The current authors have previously documented the NPR as an intra-individual muscle activation/epiglottic pressure relationship in normal subjects [31, 34] and OSA patients [31]. The current study suggests that amongst OSA patients, higher muscle activation results in better preserved airway calibre.
The magnitude of the increase in Rphar at sleep onset was also positively correlated with wakeful TP-EMG activity. This is consistent with the greatest sleep onset-related rise in airway resistance occurring in those subjects with the most NPR-activated tonic muscle activity during wakefulness. The lack of a significant relationship between TP-EMG and Rphar during sleep in this study is also consistent with a previous finding of lesser recruitment of TP than of GG by the NPR during sleep in OSA [35] and in normals [28]. The lack of a correlation between Pcrit and Rphar during wakefulness is not unexpected given the intermediate influence of dilator muscle activation to reduce Rphar below its passive value during wakefulness.
This study is the first to document the relationship between muscle activation and passive airway collapsibility across sleep onset in patients with OSA. One previous study in a group of normal subjects by Philip-Joet et al. [27] demonstrated that GG activation during stable sleep is positively correlated with Pcrit measured with the upper airway muscles activated by continuous negative airway pressure. However, there have been no previous studies relating muscle activation and resistance both awake and at sleep onset to measures of the passive upper airway.
The present authors have also demonstrated that dilator muscle activation correlates less well with BMI than with passive Pcrit. The strength of this correlation would not be expected to be as high as that between muscle activation and Pcrit because BMI is a general anthropometric measure rather than a specific measure of upper airway calibre. Craniofacial anatomical factors independent of body weight and body fat distribution are also important in determining upper airway anatomy. The influence of craniofacial characteristics on closing pressure and site of collapse of the passive upper airway in OSA has previously been demonstrated in anaesthetised humans [36]. This is separate from the lateral pharyngeal wall fat deposition effect of obesity and the lung volume effect of reduction in functional residual capacity leading to reduced caudal traction on the upper airway, thus increasing its resistance [37]. All of the subjects in this study were overweight or obese with their mean BMI clearly in the obese range at 32.8. The current study’s findings may thus not be generalisable to nonobese OSA subjects in whom other mechanisms, such as craniofacial abnormalities or respiratory control factors, are primarily responsible for sleep disordered breathing.
One limitation of the current study is that Rphar increases substantially in the first few breaths after sleep onset, often precipitating apnoea, which then results in arousal, making it difficult to measure sleep effects on Rphar in severe OSA patients. In the current study, there was frequent rapid cycling between wakefulness, and sleep with flow limitation or apnoea repeatedly resulting in arousal within three to four breaths of sleep onset. It was thus not possible to study muscle activation and upper airway mechanics deeper than three to four breaths into sleep. Also, when airflow limitation develops, inspiratory flow becomes independent of downstream (epiglottic) pressure upon which the Rphar measurement depends. In the present study, correlations with Rphar measured at low flow (200 mL·s−1) were of similar strength to those with peak inspiratory resistance (Rphar), suggesting that the current study’s data is not seriously affected by this consideration. The use of rank correlation analysis is also appropriate in this regard.
It is also important to note that the OSA subjects in this study had all been on nasal CPAP therapy for ≥3 months with good compliance. It is possible that treatment may have improved upper airway muscle function and reflexes so that the resultsmay not be fully applicable to untreated OSA patients. Another potential limitation of this study is the confounding effect of the instrumentation required to measure airway mechanics (Millar catheters at posterior choanae and epiglottis via the nasal cavity) and muscle activation (intramuscular wire electrodes) on arousal threshold. These may make arousal more likely to occur during the airway pressure decrements used to measure the passive Pcrit (breaths in which arousal is observed need to be excluded from analysis). This effect appears to be less of a problem in OSA patients than in similarly instrumented normal subjects in whom it was more difficult to reliably obtain consistent flow limitation without arousal as needed for Pcrit determination. This is clearly less of a problem when Pcrit measurements are made using nasal pressure without instrumentation of the upper airway, as originally described by Schwartz and co-workers [23, 24].
In summary, the present authors have shown that dilator muscle activation relates both to anatomical airway narrowing/collapsibility (positive end-expiratory pressure) and to airway patency (pharangeal resistance) across a group of subjects with obstructive sleep apnoea. These findings are consistent with the model of passive anatomical factors and muscle activation (in part driven by the negative-pressure reflex) being the principal determinants of pharyngeal patency. The relationship between critical closing pressure and genioglossus muscle activation weakens when dilator tone diminishes with loss of the wakefulness drive at sleep-onset but, for the activity of tensor palatine it becomes stronger suggesting persistence of the negative-pressure reflex during early sleep. Upper airway muscle activation is more strongly related to airway collapsibility than to body mass index, reflecting the fact that obesity is only one of several factors narrowing the airway in patients with obstructive sleep apnoea.
Footnotes
STATEMET OF INTEREST Statements of interest for D. White and A. Malhotra can be found at www.erj.ersjournals.com/misc/statements.shtml
REFERENCES
- 1.Remmers JE, DeGroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol. 1978;44:931–938. doi: 10.1152/jappl.1978.44.6.931. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi A, Denavit-Saubie M, Champagnat J. Central control of breathing in mammals: neuronal circuitry, membrane properties, and neurotransmitters. Physiological Reviews. 1995;75:1–31. doi: 10.1152/physrev.1995.75.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Horner RL, Innes JA, Holden HB, Guz A. Afferent pathway(s) for pharyngeal dilator reflex to negative pressure in man: a study using upper airway anaesthesia. J Physiol (Lond) 1991;436:31–44. doi: 10.1113/jphysiol.1991.sp018537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horner RL, Innes JA, Murphy K, Guz A. Evidence for reflex upper airway dilator muscle activation by sudden negative airway pressure in man. J Physiol (Lond) 1991;436:15–29. doi: 10.1113/jphysiol.1991.sp018536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wheatley J, Mezzanotte W, Tangel D, White D. Influence of sleep on genioglossus muscle activation by negative pressure in normal men. Am Rev Respir Dis. 1993;148:597–605. doi: 10.1164/ajrccm/148.3.597. [DOI] [PubMed] [Google Scholar]
- 6.Mathew OP, Abu-Osba YK, Thach BT. Genioglossus muscle response to upper airway pressure changes: afferent pathways. J Appl Physiol. 1982;52:445. doi: 10.1152/jappl.1982.52.2.445. [DOI] [PubMed] [Google Scholar]
- 7.Mathew OP, Abu-Osba YK, Thach BT. Influence of upper airway pressure changes on genioglossus muscle and respiratory activity. J Appl Physiol. 1982;52:438. doi: 10.1152/jappl.1982.52.2.438. [DOI] [PubMed] [Google Scholar]
- 8.Onal E, Lopata M, O’ Connor TD. Diaphragmatic and genioglossal electromyogram responses to CO2 rebreathing in humans. J Appl Physiol. 1981;50:1052–1055. doi: 10.1152/jappl.1981.50.5.1052. [DOI] [PubMed] [Google Scholar]
- 9.Onal E, Lopata M, O’Connor T. Diaphragmatic and genioglossal electromyogram responses to isocapnic hypoxia in humans. Am Rev Respir Dis. 1981;124:215–217. doi: 10.1164/arrd.1981.124.3.215. [DOI] [PubMed] [Google Scholar]
- 10.Bartlett D, Jr, St John WM. Influence of lung volume on phrenic, hypoglossal and mylohyoid nerve activities. Respir Physiol. 1988;73:97–109. doi: 10.1016/0034-5687(88)90130-2. [DOI] [PubMed] [Google Scholar]
- 11.Begle RL, Badr S, Skatrud JB, Dempsey JA. Effect of lung inflation on pulmonary resistance during NREM sleep. Am Rev Respir Dis. 1990;141:854–860. doi: 10.1164/ajrccm/141.4_Pt_1.854. [DOI] [PubMed] [Google Scholar]
- 12.Akahoshi T, White DP, Edwards JK, Beauregard J, Shea SA. Phasic mechanoreceptor stimuli can induce phasic activation of upper airway muscles in humans. J Physiol (Lond) 2001;531:677–691. doi: 10.1111/j.1469-7793.2001.0677h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orem J. The nature of the wakefulness stimulus for breathing. Prog Clin Biol Res. 1990;345:23–30. discussion 31. [PubMed] [Google Scholar]
- 14.Peever JH, Mateika JH, Duffin J. Respiratory control of hypoglossal motoneurones in the rat. Pflugers Arch. 2001;442:78–86. doi: 10.1007/s004240000502. [DOI] [PubMed] [Google Scholar]
- 15.Peever JH, Necakov A, Duffin J. Nucleus raphe obscurus modulates hypoglossal output of neonatal rat in vitro transverse brain stem slices. J Appl Physiol. 2001b;90:269–279. doi: 10.1152/jappl.2001.90.1.269. [DOI] [PubMed] [Google Scholar]
- 16.Peever JH, Shen L, Duffin J. Respiratory pre-motor control of hypoglossal motoneurons in the rat. Neuroscience. 2002;110:711–722. doi: 10.1016/s0306-4522(01)00594-2. [DOI] [PubMed] [Google Scholar]
- 17.Fogel RB, White DP, Pierce RJ, et al. Control of upper airway muscle activity in younger versus older men during sleep onset. J Physiol. 2003;553:533–544. doi: 10.1113/jphysiol.2003.045708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wasicko M, Erlichman J, Leiter J. Control of segmental upper airway resistance in patients with obstructive sleep apnea. J Appl Physiol. 1993;74:2694–2703. doi: 10.1152/jappl.1993.74.6.2694. [DOI] [PubMed] [Google Scholar]
- 19.Schwab RJ, Gupta KB, Gefter WB, Metzger LJ, Hoffman EA, Pack AI. Upper airway and soft tissue anatomy in normal subjects and patients with sleep-disordered breathing. Significance of the lateral pharyngeal walls. Am J Respir Crit Care Med. 1995;152:1673–1689. doi: 10.1164/ajrccm.152.5.7582313. [DOI] [PubMed] [Google Scholar]
- 20.Mezzanotte WS, Tangel DJ, White DP. Waking genioglossal EMG in sleep apnoea patients versus normal controls (a neuromuscular compensatory mechanisms) J Clin Inves. 1992;89:1571–1579. doi: 10.1172/JCI115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mezzanotte WS, Tangel DJ, White DP. Influence of sleep onset on upper-airway muscle activity in apnea patients versus normal controls. Am J Respir Crit Care Med. 1996;153:1880–1887. doi: 10.1164/ajrccm.153.6.8665050. [DOI] [PubMed] [Google Scholar]
- 22.Fogel RB, Trinder J, Malhotra A, et al. The effect of sleep onset on upper airway muscle activity in patients with sleep apnea versus controls. J Physiol. 2004;156:95–240. doi: 10.1113/jphysiol.2005.083659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz AR, O’Donnell CP, Baron J, et al. The hypotonic upper airway in obstructive sleep apnea. Am Rev Respir Crit Care Med. 1998;157:1051–1057. doi: 10.1164/ajrccm.157.4.9706067. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz AR, Smith PL, Wise RA, Gold AR, Permutt S. Induction of upper airway occlusion in sleeping individuals with subatmospheric nasal pressure. J Appl Physiol. 1988;64:535–542. doi: 10.1152/jappl.1988.64.2.535. [DOI] [PubMed] [Google Scholar]
- 25.Haponik E, Smith P, Bohlman M, Allan R, Goldman S, Bleecker E. Computerized tomography in obstructive sleep apnea: correlation of airway size with physiology during sleep and wakefulness. Am Rev Respir Dis. 1983;127:221–226. doi: 10.1164/arrd.1983.127.2.221. [DOI] [PubMed] [Google Scholar]
- 26.Burger CD, Stanson AW, Sheedy PFD, Daniels BK, Shepard JW., Jr Fast-computed tomography evaluation of age-related changes in upper airway structure and function in normal men. Am Rev Respir Dis. 1992;145:846–852. doi: 10.1164/ajrccm/145.4_Pt_1.846. [DOI] [PubMed] [Google Scholar]
- 27.Philip-Joet F, Marc I, Series F. Effects of genioglossal response to negative airway pressure on upper airway collapsibility during sleep. J Appl Physiol. 1996;80:1466–1474. doi: 10.1152/jappl.1996.80.5.1466. [DOI] [PubMed] [Google Scholar]
- 28.Worsnop C, Kay A, Kim Y, Trinder J, Pierce R. Effect of age on sleep onset-related changes in respiratory pump and upper airway muscle function. J Appl Physiol. 2000;88:1831–1839. doi: 10.1152/jappl.2000.88.5.1831. [DOI] [PubMed] [Google Scholar]
- 29.Worsnop C, Kay A, Pierce R, Kim Y, Trinder J. Activity of respiratory pump and upper airway muscles during sleep onset. J Appl Physiol. 1998;85:908–920. doi: 10.1152/jappl.1998.85.3.908. [DOI] [PubMed] [Google Scholar]
- 30.Malhotra A, Fogel RB, Edwards JK, Shea SA, White DP. Local mechanisms drive genioglossus activation in obstructive sleep apnoea. Am J Respir Crit Care Med. 2000;161:1746–1749. doi: 10.1164/ajrccm.161.5.9907109. [DOI] [PubMed] [Google Scholar]
- 31.Fogel RB, Malhotra A, Pillar G, et al. Genioglossal activation in patients with obstructive sleep apnoea versus control subjects. Mechanisms of muscle control. Am J Respir Crit Care Med. 2001;164:2025–2030. doi: 10.1164/ajrccm.164.11.2102048. [DOI] [PubMed] [Google Scholar]
- 32.Fogel RB, Trinder J, Malhotra A, et al. Within-breath control of genioglossal muscle activation in humans: effect of sleep-wake state. J Physiol. 2003a;550:899–910. doi: 10.1113/jphysiol.2003.038810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malhotra A, Pillar G, Fogel RB, et al. Genioglossal but not palatal muscle activity relates closely to pharyngeal pressure. Am J Respir Crit Care Med. 2000;162:1058–1062. doi: 10.1164/ajrccm.162.3.9912067. [DOI] [PubMed] [Google Scholar]
- 34.Malhotra A, Pillar G, Edwards JK, et al. Pharyngeal pressure and flow effects on genioglossus activation in normal subjects. Am J Respir Crit Care Med. 2002;165:71–77. doi: 10.1164/ajrccm.165.1.2011065. [DOI] [PubMed] [Google Scholar]
- 35.Fogel RB, Malhotra A, White DP. Sleep. 2: pathophysiology of obstructive sleep apnoea/hypopnoea syndrome. Thorax. 2004;59:159–163. doi: 10.1136/thorax.2003.015859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe T, Isono S, Tanaka A, Tanzawa H, Nishino T. Contribution of body habitus and craniofacial characteristics to segmental closing pressures of the passsive pharynx in patients with sleep-disordered breathing. Am J Respir Crit Care Med. 2002;165:260–265. doi: 10.1164/ajrccm.165.2.2009032. [DOI] [PubMed] [Google Scholar]
- 37.Stanchina ML, Malhotra A, Fogel RB, et al. The influence of lung volume on pharyngeal mechanics, collapsibility, and genioglossus muscle activation during sleep. Sleep. 2003;26:851–856. doi: 10.1093/sleep/26.7.851. [DOI] [PubMed] [Google Scholar]