

Abstract
Disease-onset time (DOT) and disease trajectory concepts were applied to derive an Alzheimer's disease (AD) progression population model using the clinical dementia rating scale—sum of boxes (CDR-SOB) from the AD neuroimaging initiative (ADNI) database. The model enabled the estimation of a DOT and a disease trajectory for each patient. The model also allowed distinguishing fast and slow-progressing subpopulations according to the functional assessment questionnaire, normalized hippocampal volume, and CDR-SOB score at study entry. On the basis of these prognostic factors, 81% of the mild cognitive impairment (MCI) subjects could correctly be assigned to slow or fast progressers, and 77% of MCI to AD conversions could be predicted whereas the model described correctly 84% of the conversions. Finally, synchronization of the biomarker-time profiles on estimated individual DOT virtually expanded the population observation period from 3 to 8 years. DOT-disease trajectory model is a powerful approach that could be applied to many progressive diseases.
Alzheimer's disease (AD), the most common dementing illness in the elderly, is a major growing public health issue as life expectancy increases. AD is characterized by a slow decline in cognitive and functional ability assessed by various clinical, biochemical, imaging, and genetic biomarkers. However, the large variability in disease progression among individuals, from normal to prodromal (predementia), mild cognitive impairment (MCI), and dementia,1 hinders a correct prognosis of the disease and associated assessment of treatment effects of new disease-modifying drugs. An efficient way to comprehensively integrate and use the available information is through population disease progression modeling. In this context, the AD neuroimaging initiative (ADNI),2,3 an on-going longitudinal natural long-term history study of elderly designed to collect validated data such as MRI and PET images, cerebral spinal fluid and blood biomarkers together with clinical/cognitive measurements in normal subjects (NL), MCI, and AD is a first and important step in the early detection and tracking of AD. Different groups have already published disease progression models for cognitive deterioration based on ADNI data.4,5,6,7 These models, briefly summarized hereunder, are mainly based on the longitudinal response in AD assessment scale-cognitive subscale (ADAS-cog).
On the basis of the mixed effects modeling approach, Ito3 developed a linear AD progression model throughout all populations (i.e., NL, MCI, and AD), in which ADAS-cog evolved over time with a constant progression rate. In this model, the progression rate was shown to be influenced by baseline ADAS-cog, age, apolipoprotein (APOE) ε4 genotype, and sex. Samtani built separate nonlinear mixed effect models for AD4 or MCI5 populations using logistic models. In those models, ADAS-cog score deteriorated slowly during the early stage of the disease and more rapidly during the middle stage. Disease progression rates were shown to be mainly influenced by Trail B test, APOE ε4 genotype and high cholesterol (or high p-tau181P/Aβ1-42 ratio), whereas high ADAS-cog baseline values were associated with atrophy of hippocampal volumes (HIPV). Moreover, based on ADAS-cog baseline levels and progression rates, Samtani's model identified two subpopulations of MCI patients (MCI progressers and nonprogressers) which correlated rather well with pathologic cerebrospinal fluid biomarker signature (high p-tau181p and low Aβ1-42) as reported by Meyer.8 Finally, Yang,6 being aware that the duration of clinical trials are too short to show significant changes in biomarkers, judiciously proposed to calculate a time shift for observed ADAS-cog scores across subjects and populations leading to an optimal fit of resulting scores to a theoretical curve of the disease progression. Subsequently, they mapped changes in biomarker data according to their new disease timeline.
However, even if these analyses have brought significant advancement in quantitative understanding of disease progression and in the impact of important covariates, a better understanding of the evolution of individuals at the prodromal phase of AD, i.e., with presymptomatic or mild signs of dementia, remains crucial as they are a high-risk group likely to benefit from effective treatments. In that context, it appears that the clinical dementia rating scale sum of boxes (CDR-SOB) score could be a valuable candidate as outcome indicator in prodromal/MCI populations and facilitate assessment of active vs. placebo treatment.
CDR-SOB is commonly used to asses both cognitive and functional impairment of AD. It has been shown to be less variable than ADAS-cog, to have excellent 2-year responsiveness and to be appropriate in distinguishing between MCI and AD patients.9
The objective of this work is to develop an original natural history population disease progression model based on CDR-SOB scores from ADNI by integrating the experiences from previous ADAS-cog modeling. The model presented herein, similarly to Yang's model,6 is based on the concept that study entry time does not correspond to the start of the disease as illustrated in Figure 1. Analyzing the data at the population level permits derivation of the most likely common disease trajectory using time scale adjustment and back- and forth extrapolation between AD and MCI patients. In addition, the different approaches developed by Samtani4,5 have been adapted to the CDR-SOB score.
Figure 1.

Scale synchronization on disease onset time. (a) Typical clinical dementia rating scale—sum of boxes score profiles in normal (NL), mild cognitive impairment (MCI), and Alzheimer's disease (AD) subjects from AD neuroimaging initiative. (b) Subpopulation time scale adjustment based on the hypothesis of common disease trajectory. (c) Individual back (AD to MCI) and forward (MCI to AD) scale adjustments by estimation of the disease onset time (DOT). (d) Synchronization on DOT instead of time of study entry.
Results
Available data
Model building was performed on data obtained from the ADNI database (www.adni.loni.ucla.edu).10 The final analysis dataset consisted of 2,700 CDR-SOB values collected from 380 MCI and 180 AD patients for up to 4 years. It excluded control subjects for which, contrary to ADAS-cog scores, most CDR-SOB scores were equal or close to zero (Figure 2a,b), as well as subjects with a single CDR-SOB record. Guided by prior disease modeling in this area, a total of 19 covariates were included (see Methods). As cerebrospinal fluid (CSF) biomarkers were only available in about half of the patients, their impact could not be fully assessed.
Figure 2.

Clinical dementia rating scale–sum of boxes and hippocampal volume vs. status at study entry. (a) Relationship between clinical dementia rating scale–sum of boxes (CDR-SOB) and Alzheimer's disease assessment scale-cognitive subscale at study entry and (b) CDR-SOB change from baseline at 2 years in function of baseline values at study entry for the different diagnostic groups. Lines are smooth through all the data. (c) Distribution of hippocampal volumes at study entry centered to median value of controls or (d) normalized to healthy values for same age and head size (RHPNM) in function of Alzheimer's disease status.
Special attention has been given to the HIPV which is known to be an important covariate in AD.11,12 HIPV has been shown to be affected by head size, age, and sex13 and to significantly drop in the elderly.14 Therefore, its standardization to individual characteristics should improve its disease predictive value.15 So, in addition to the observed HIPV centered to the median value of healthy population, a HIPV normalized to that of a healthy subject with the same age and head size (RHPNM) has been derived for each MCI and AD patient. The normalization was based on the relationship between HIPVs, individual estimated intracranial volumes,16 and ages in healthy subjects (see Methods) and was used to compare the HIPV atrophy between patients. It should be noted that sex was no longer necessary in this normalization because it was already included in estimated intracranial volumes. As expected and shown in Figure 2c,d, this transformation decreased the variance of the HIPV measurement by about one-third.
Structural base model development
As previously described for ADAS-cog,4,5 graphical analysis of CDR-SOB progressions indicated a continuous increase in disease progression rate over time in most patients, the higher the CDR-SOB score the faster the progression rate. Like ADAS-cog, CDR-SOB score is also constrained between lower and upper limits (here, 0–18) and in particular, left censored (Figure 2a,b).
As a starting model, we selected a nonlinear model adapted from Samtani4 based on differential equations with an evolving disease progression rate. As subjects entered the study at different stages of the disease, a disease-onset time (DOT) similar to the time shift introduced by Yang6 was also applied:
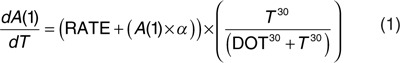 |
(1)
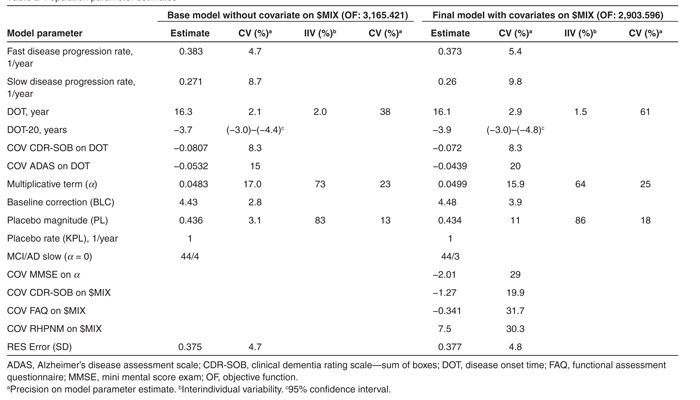
When time (T) equaled DOT, an increase in A(1), a continuous variable related to CDR-SOB, was triggered (i.e., by (T30/(DOT30+T30)) which due to the power arbitrary set to 30 approximated a step function from 0 to 1). The change in A(1) was described by a constant disease progression rate (RATE) plus an additive term (A(1) × α) in which α was a parameter for the individual change in progression rate. To deal with floor and ceiling CDR-SOB scores, the modeling was performed in the logit domain and additive residual error was used. Therefore, A(1) represents the time course of CDR-SOB in the logit domain. Inherent to the nature of the model, at least one covariate indicative of the patient's status was required. The most appropriate one was the cognitive CDR-SOB score at study entry since it carried most of the information on the patient disease status and DOT (i.e., the higher the score the earlier the disease started). It should also be noted that due to numerical difficulties, log normally distributed interindividual variability (IIV) could only be implemented on one of the two parameters, RATE or DOT, forcing either the same RATE or the same DOT for every subject. The model was stable when implementing IIV on parameter DOT.
Therefore, for a CDR-SOB score at study entry and its progression as a function of time, the modeling exercise essentially consisted of identifying a DOT for each individual that minimized the objective function given the population disease trajectory. The combination of DOT synchronization and population analysis—information from one individual enriched by the information coming from the other individuals—allowed the estimation of the most likely disease trajectory of the CDR-SOB score.
However, when applying this model, two major observations were noted. First, observed disease scores evolved more slowly in a significant number of subjects, mainly belonging to the group classified as MCI. Second, CDR-SOB scores following study entry showed an early drop or delay before progression of the disease. These two observations were in agreement with the previous findings5,17 when modeling ADAS-cog scores.
To allow the model to capture the difference in the disease progression rate, a mixture model was implemented allowing to assign subjects to one out of the two subpopulations ($MIX).18 For the first subpopulation, RATE was estimated without IIV and α set to 0, whereas for the second subpopulation, RATE without IIV and α with IIV were estimated together. Subjects were assigned to either subpopulation independently of their disease status (i.e., MCI and AD status was not used as a covariate). The second observation was accounted for by adding a ‘placebo' effect at the time the patient entered the study as given by the following equation where CDR_SOB represents the predicted CDR-SOB score, PL the magnitude of the ‘placebo' effect, and KPL the equilibration rate constant of the effect:
(2)
Finally, in addition to the IIV on DOT and α, the score-time profile was best captured by log normally distributed IIV on PL; the estimation of IIV on KPL was not supported by the data.
Covariate analysis
Covariate analyses presented hereunder pursued two aims. The first one was to find among available biomarkers those that could best explain the variability in disease progression, described in this model by DOT and α parameters, and the second aim was to identify biomarkers that could allow the discrimination between slow and fast MCI progressers.
Base model. As mentioned previously, the primary covariate that needed to be accounted for was CDR-SOB at study entry, put on DOT. Next, covariates such as baseline ADAS-cog, hippocampal volume, mini mental score exam (MMSE), and functional assessment questionnaire (FAQ), preselected mainly by graphical analysis (i.e., correlation with DOT) and clinical significance were also tested on DOT as described in Table 1 (part I). ADAS-cog was retained since it decreased the objective function the most (100 units). None of the other tested covariates were found statistically significant after selection of CDR-SOB and ADAS-cog. This model was considered as the base model (Table 2, left part) and was used to search for potential covariates for slow and fast MCI progressers. At that stage, tentative implementation of covariates on parameter α was explored but always resulted in numerically instable models.
Table 1. Summary of model development.
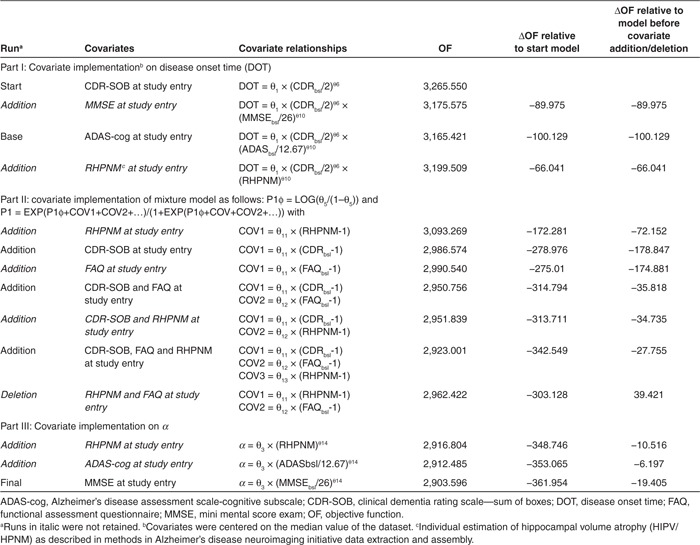
Table 2. Population parameter estimates.
Slow- and fast-progressing MCI populations. Effect of covariates to identify slow- and fast-progressing MCI populations was explored using generalized additive models (GAM) in univariate and multivariate steps. First, all covariates (i.e., in total 19) were tested separately. As shown in Figure 3a, sex and APOE ε4 carrier status were not identified by univariate analysis. Conversion from MCI to AD appeared indicative of fast progression since 84% (136 of 162) of fast progressers had converted although 33% (69 of 207) of not-yet-converters were also identified as fast progressers by the model. Interestingly but not surprisingly, use of co-medication was also indicative of fast disease progression. Second, an automated multivariate step-GAM analysis with forward selection and backward deletion was performed using relevant covariates from the univariate tests that were available in almost all MCI patients, i.e., MMSE, ADAS-cog, FAQ, Trail B test, CDR-SOB, ventricular, and hippocampal volumes. Sex and APOE ε4 carrier status were also tested again. From this multivariate step-GAM analysis, the most significant covariates to differentiate between slow and fast MCI progressers were CDR-SOB (P = 8.7 × 10−6), FAQ (P = 1.85 × 10−5), and RHPNM (P = 4 × 10−4). Sex was also identified but was less significant (P = 0.026). APOE ε4 carrier status was not identified.
Figure 3.

Probability of fast progression. (a) Univariate generalized additive models (GAM) analysis on mild cognitive impairment (MCI) slow (bottom side) of and fast (top side) progressers for, from left to right, sex (1 = male; 2 = female), apolipoprotein ε4 (0 = noncarrier; 1 = carrier), MCI to Alzheimer's disease (AD) conversion (0 = no; 3 = yes), comedication (0 = no anticholinesterase; 1 = with anticholinesterase), age, cholesterol, cerebrospinal fluid Aβ1-42, Tau (log scale), p-tau181P (log scale), and p-tau181P/Aβ1-42(log scale). (b) Probability of fast disease progression based on MCI slow/fast GAM analyses according to base or (c) final model. In this latter, the five slow AD disease patients are represented by green-dotted lines.
As mentioned earlier, CSF tau, p-tau181P , and Aβ1-42 levels, biomarkers well known to be altered in AD,19 were only available in a subset of MCI patients (Figure 3a) and therefore were not included in the multivariate GAM analysis. However, in addition to the univariate analysis, a graphical analysis indicated a possible effect of CSF Aβ1-42 in discriminating fast from slow-progressing MCI subjects.
Final model development
The three major covariates selected by the multivariate GAM analysis were implemented in NONMEM by estimating their effect on the modeled fraction of the population in the first (or second) subpopulation ($MIX)16 and further analyzed as described in Table 1 (right part). All three, (CDR-SOB, FAQ, and RHPNM) were found to significantly improve the model objective function of the base model (ΔOF > 200). A final search of covariates for their potential influence on parameter α was undertaken as described in part III of Table 1 and led to the inclusion of the MMSE score. In the resulting final model, all included covariates were statistically significant (P < 0.001); population parameter estimates are given in Table 2 (right part) in comparison with the base model.
Model evaluation
The goodness of fit plots (not shown) were carefully examined and used together with the minimization of the objective function as guides throughout all the model development. Visual predictive checks are shown in Figure 4 for base and final models. As shown in this figure, both models were able to satisfactorily predict the course of the disease progression in the different diagnosed groups. The base model performed slightly better for AD patients and the final model for fast MCI progressers, particularly for the fastest ones.
Figure 4.

Model visual predictive check. Visual predictive checks for the base (left side) and final (right side) Alzheimer's disease (AD) progression models. Red lines are the 10th, 50th, and 90th progressions of clinical dementia rating scale—sum of boxes (CDR-SOB) from the AD neuroimaging initiative trial whereas dots are the individual observations. Green areas represent the 10th, 50th, and 90th percentiles of the predicted CDR-SOB progressions from the 100 simulated datasets.
Results of GAM analyses for the slow and fast MCI populations were also evaluated for both models by applying the logistic models to all subjects from the ADNI database with the appropriate covariates as shown in Figure 3b,c. As shown in this figure, predictions made on GAM-selected covariates allowed to reasonably differentiate subjects relative to their probability of disease progression and were in good agreement with ADNI classification. Indeed, the NL and AD subjects who were not part of the GAM analysis of slow and fast MCI subjects could be well categorized based on their covariates, for their probability of progression.
Discussion
Considering AD as a continuously progressing disease, CDR-SOB scores from MCI and AD patients from the ADNI database have been used in a combined analysis using DOT synchronization in order to reconstruct a coherent trajectory for disease progression. DOT and the disease trajectory were implemented in a differential equation. At the time the disease started in an individual, the estimated DOT activated the increase in the CDR-SOB score following a RATE adjusted to each subject's progression velocity by an additive disease increasing rate term (CDR × α). Initially to deal with the floor and ceiling effect of the score, modeling was performed in the logit domain. However, this approach also implies that the disease progression itself occurs in the logit domain. In fact, a score is only an expression of a disease status and is limited to a time window when it is sensitive to the disease progression. For instance, the disease can have started a while ago, but the score is not yet sensitive enough to detect its progression (e.g., CDR-SOB score below 0.5). On the opposite, the disease can still evolve but the score can no more describe its progression because it has already reached the maximum value (e.g., CDR-SOB score of 18). If we accept that the disease progresses in the logit domain, the different scores (e.g., ADAS-cog, CDR-SOB, MMSE, FAQ, etc) can be positioned at different phase of the disease progression and can translate the disease progression rate through different link functions. Unfortunately, today, the ‘true' or reference disease status and trajectory have not yet been defined. Imaging techniques might help in this quest. This makes that A(1) in the disease trajectory equation is not the ‘true' disease status and is mainly driven by the score used in the modeling, i.e., CDR-SOB. It can also be mentioned that even in the logit domain, the progression of the score could not be modeled by a straight line and required a nonlinear function. This is due to an asymmetry in the score or/and its progression as a function of time. Finally, the use of a differential equation to describe the trajectory was justified by the flexibility it offers in the future to describe different drug actions on disease progression.
With this model, it was identified that the disease progression evolved significantly more slowly in a number of MCI subjects. To capture this difference in disease trajectories, a mixture model was implemented. The time shift approach has already been applied by Yang et al. on ADAS-cog scores.6 However, in our case, the model was not able to align all patients according to one trajectory but rather identified statistically two populations, based on fast or slow progression in the CDR-SOB score. As expected, most of the patients from the AD group (97%) were identified by the model as fast-progressing subjects whereas the slow-progressing population was essentially formed by the patients from the MCI group (44%), indicating that these patients did not transit to AD status during the observation period. This is in agreement with the segregation of MCI patients into two subpopulations described by Samtani.5 Identification of fast-progressing and slow-progressing MCI highlights an important caveat to the analysis of Yang et al.6 in which ADAS-cog 13 is used to synchronize time trajectories across three different populations. The logic of that analysis would be sound if the MCI population was simply a more progressed version of the control population and the AD population were just a more progressed version of the MCI population. That, however, is not the case: the AD population is both a more progressed and a highly selected (by virtue of the study design) version of the MCI population. Because of this selection, it is not the case that, given enough time, the MCI population would eventually be similar to the AD population. This has also an impact on the estimation on the DOT.
The present analysis can be considered as an additional step in the sequential development of AD progression models. In our model, all the trajectories of CDR-SOB scores were defined by individual DOTs and a rate, which could progress slowly in slow-progressing subjects, or faster with more or less acceleration as the disease evolved in fast-progressing subjects. In addition, the disease trajectories were completed by a ‘placebo component' to take into account the transient slight improvement or delay in the score progression observed in numerous patients following study enrolment. Apart from a placebo effect, this drop in CDR-SOB scores may also be interpreted as learning effect but could result from other study-specific effects (inclusion criteria and concomitant drug effect). The decrease in CDR-SOB scores was a challenging aspect of the model development, since it could confound other aspects of disease progression. The way it was modeled allowed taking this effect into account without compromising other aspects of the disease progression model. However, the fact that the placebo effect was introduced after logit back transformation might produce negative scores in a simulation mode. Since the score was digitalized to 0, 0.5, 1, 1.5, etc, no negative scores were produced in the simulations (e.g., visual predictive checks).
CDR-SOB and ADAS-cog scores were identified as covariates for DOT and MMSE as covariate on the disease-increasing rate term. It should be noted that the covariate search only focused on biomarkers available in most of the patients; therefore, the impact of CSF p-tau181p and Aβ1-42 which were only available in half of the subjects in ADNI was not fully explored in this analysis. The potential influence of CSF p-tau181p and Aβ1-42 should be further tested with additional longitudinal CSF and clinical data.
Covariates identified for assignment to the slow- or fast-progressing MCI groups at study entry were CDR-SOB, FAQ, and the hippocampal volume normalized for age and head size. Notably, without normalization, HIPV was only just significant (P = 0.02). Implementation of these biomarkers as covariates of the mixture model significantly improved the model (ΔOF > 200) and patient stratification. On the basis of these prognostic factors, 81% of the subjects could correctly be assigned to the slow or fast-progressing subpopulations and, assuming that fast progressers will convert, 77% of the MCI to AD conversion could be predicted. Notably, the disease progression model described correctly 84% of the conversions (see Supplementary Data online). These biomarkers have also been identified by other authors as important for status definition,8 progression rate,10 and MCI to AD conversion.20 Of note, they combine a cognitive score with a functionality score and brain anatomy. Therefore, the use of these biomarkers to calculate the probability of fast disease progression should improve the clinical diagnosis and prognosis of AD.
The estimation of the DOT allowed a better positioning of each individual in his disease trajectory at study entry. The DOT estimated by the CDR-SOB model could be applied to the time profiles of biomarkers. The application of this concept has already been introduced by Yang.6 It is illustrated for our analysis in Figure 5 in which the hippocampal volume time profiles have been synchronized on the DOT. This approach virtually expands the clinical observation period from 3 to 8 years and provides insights into long-term changes in the biomarkers.
Figure 5.

Hippocampal volume–time profiles. (a) Evolution of the hippocampal volumes during the study for each status. (b) Relationship between hippocampal volumes centered to median of the normal population and model estimated time since disease onset (DOT). (c) Relationship between normalized hippocampal volumes to control values for same age and head size (estimated intracranial volumes) and DOT. Lines are smooth through the data.
In conclusion, the use of the DOT and disease trajectory concepts was powerful for detecting different AD progression rates in the ADNI population and identifying corresponding prognostic factors. Estimation of the DOT by the CDR-SOB model allowed the synchronization of biomarker-time profiles on disease onset rather than on study entry and provided insights in long-term changes in biomarkers. From this analysis, the CDR-SOB score could be considered as complementary to the ADAS-cog score from a modeling and simulation perspective for evaluating disease-modifying drugs in prodromal subjects.
Methods
ADNI data extraction and assembly. Data used in this article were obtained from the ADNI database (adni.loni.ucla.edu). The ADNI was launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the US Food and Drug Administration, private pharmaceutical companies, and nonprofit organizations. The primary goal of ADNI has been to test whether serial magnetic resonance imaging, positron emission tomography, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. The Principal Investigator of this initiative is M.W.W., VA Medical Center and University of California—San Francisco, with efforts of many coinvestigators from a broad range of academic institutions and private corporations. Subjects have been enrolled from over 50 sites across the USA and Canada. For up-to-date information, see www.adni-info.org.
Following approval to access ADNI, original data sets were downloaded on 1 August 2011. At that time, ADNI database consisted of 819 adults, aged 55–91 years, with CDR-SOB records for an observation period up to 4 years: 229 subjects were classified as NL, 397 as MCI, and 193 as AD at enrollment. Biomarker assessments were recorded at 0, 6, 12, 24, 36, and 48 months with an additional evaluation at 18 months for the MCI group. During the observation period, 166 MCI converted to AD, 6 AD converted to MCI, and 19 NL evolved to MCI whom 3 of which subsequently evolved to AD.
Intrinsically, the model considered that control subjects had CDR-SOB scores of zero which was in agreement with the observations. Therefore, control subjects were not included in the final NONMEM dataset for the CDR-SOB analysis since they did not carry information to the model. Patients with a single CDR-SOB record (16 MCI and 13 AD) were also removed. Covariates investigated were age, sex, serum cholesterol, APOE ε4 carrier status, hippocampal, ventricular and estimated intracranial volumes, FAQ, Trail B test, MMSE and ADAS-cog (total 11) scores, CSF Tau, p-tau181P and Aβ1-42, plasma Aβ1-42 and Aβ1-40, MCI to AD conversion, and medication for cognitive disorder (CCMED). The latter was registered as categorical covariates (0, 1) and positive when at least one anticholinesterase drug was recorded. Values at study enrollment were utilized. HIPV was the average of left and right HIPV as used by Samtani.4 For covariate model building, median covariate value of the diagnostic group was imputed for missing covariate when covariate was nearly complete. For GAM analyses, subjects with missing relevant covariates were excluded. The HIPV of the subject at study entry (HIPVbsli) was normalized to the HIPV of a healthy subject with same head size and age (HPNMbsli) to obtain an estimation of the HIPV atrophy of the patient (RHPNMbsli) at study entry as follows:
where HPNMbsli = Agei x (–26.6268 + EICVbsli × 0.0016 + 3340.4395). The calculation was based on regression analysis between HIPV, age, and estimated intracranial volumes performed in healthy subjects; the estimated intracranial volume being the best substitute for head size found in the ADNI database.
Model building and evaluation. Data were analyzed by nonlinear mixed effects modeling in NONMEM, version 7.2.0.21 The first-order conditional estimation method was used. Technical details about the NONMEM implementation of the model are given in the Supplementary Data online.
Influences of covariates were evaluated as possible explanatory variables of IIV in model parameters and for subpopulations stratification in $MIX and included in the model as shown in Table 2. The covariates were selected based on the change in objective function.
Models of interest were implemented in the trial simulator TS2 version 2.1.2 (Pharsight, Mountain View, CA). Simulations were performed based on the population NONMEM parameter estimates and bootstrap of covariates from the original dataset. Visual predictive checks to evaluate the predictive ability of the models were based on 100 simulated datasets. S-PLUS 6.2 (Insightful Corporation, Seattle, WA) was used for data management, statistical analysis, and generation of graphic outputs.
Uni- and multivariate GAM analyses on slow and fast MCI. Slow and fast MCI were analyzed as binary variables using the GAM in which the effect of available biomarkers was examined as prognostic factors, separately or in combination (uni- or multivariate GAM analysis).22 Multivariate GAMs were built using the automated step-wise search developed in S-PLUS (version 6.2; Insightful Corporation, Seattle, WA). The automated step-wise search selects the best GAM using alternating forward selection and backward deletion given the range of models. A series of candidate relationships (e.g., linear, log transformation, spline, and Loess smooth) that describe how each particular prognostic factor might enter the model is defined for every prognostic factor. The final models were built by evaluating all candidate forms for each prognostic factor in a step-wise manner.
Author contributions
P.J., I.D., R.G., S.R., and J-E.C. designed the analysis. P.J. and I.D. from SGS Exprimo NV were contracted to assemble the data and perform the modeling work and analysis on behalf of F. Hoffman-La Roche. All were involved in writing and reviewing the plan and manuscript.
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.ucla.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.ucla.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Conflict of interest
P.J. and I.D. from SGS Exprimo NV were paid consultants to F. Hoffmann-La Roche. J-E.C., R.G., and S.R. are employees of F. Hoffmann-La Roche. The authors declare that they have no other competing interests.
Study Highlights

Acknowledgments
Data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), and through generous contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; BioClinica; Biogen Idec; Bristol-Myers Squibb Company; Eisai; Elan Pharmaceuticals; Eli Lilly and Company; F Hoffmann-La Roche and its affiliated company Genentech; GE Healthcare; Innogenetics NV; IXICO; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace; Merck & Co.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer; Piramal Imaging; Servier; Synarc; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30AG010129 and K01 AG030514. The authors also thank Dr. Janet Wade (SGS Exprimo NV) for her comments on this manuscript.
References
- Dubois B., et al. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol. 2010;9:1118–1127. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- Weiner M.W., et al. Alzheimer's Disease Neuroimaging Initiative The Alzheimer's disease neuroimaging initiative: progress report and future plans. Alzheimers. Dement. 2010;6:202–211. doi: 10.1016/j.jalz.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner M.W., et al. Alzheimer's Disease Neuroimaging Initiative The Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers. Dement. 2012;8:S1–68. doi: 10.1016/j.jalz.2011.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., et al. Alzheimer's Disease Neuroimaging Initiative Disease progression model for cognitive deterioration from Alzheimer's Disease Neuroimaging Initiative database. Alzheimers. Dement. 2011;7:151–160. doi: 10.1016/j.jalz.2010.03.018. [DOI] [PubMed] [Google Scholar]
- Samtani M.N., et al. Alzheimer's Disease Neuroimaging Initiative An improved model for disease progression in patients from the Alzheimer's disease neuroimaging initiative. J. Clin. Pharmacol. 2012;52:629–644. doi: 10.1177/0091270011405497. [DOI] [PubMed] [Google Scholar]
- Samtani M.N., et al. Disease progression model in MCI subjects from Alzheimer's disease neuroimaging initiative: CSF biomarkers predict population subtypes. Br. J. Clin. Pharmacol. 2013;75:146–161. doi: 10.1111/j.1365-2125.2012.04308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E., et al. Alzheimer's Disease Neuroimaging Initiative Quantifying the pathophysiological timeline of Alzheimer's disease. J. Alzheimers Dis. 2011;26:745–753. doi: 10.3233/JAD-2011-110551. [DOI] [PubMed] [Google Scholar]
- De Meyer G., et al. Alzheimer's Disease Neuroimaging Initiative Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch. Neurol. 2010;67:949–956. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley N., Andrieu S., Jaros M., Weiner M., Cedarbaum J., Vellas B. Suitability of the Clinical Dementia Rating-Sum of Boxes as a single primary endpoint for Alzheimer's disease trials. Alzheimers. Dement. 2011;7:602–610. doi: 10.1016/j.jalz.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Petersen R.C., et al. Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–209. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneman W.J., et al. Hippocampal atrophy rates in Alzheimer disease: added value over whole brain volume measures. Neurology. 2009;72:999–1007. doi: 10.1212/01.wnl.0000344568.09360.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes J., et al. A meta-analysis of hippocampal atrophy rates in Alzheimer's disease. Neurobiol. Aging. 2009;30:1711–1723. doi: 10.1016/j.neurobiolaging.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes J., et al. Head size, age and gender adjustment in MRI studies: a necessary nuisance. Neuroimage. 2010;53:1244–1255. doi: 10.1016/j.neuroimage.2010.06.025. [DOI] [PubMed] [Google Scholar]
- Jernigan T.L., Gamst A.C. Changes in volume with age–consistency and interpretation of observed effects. Neurobiol. Aging. 2005;26:1271–1274. doi: 10.1016/j.neurobiolaging.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Jack C.R., Jr, et al. Steps to standardization and validation of hippocampal volumetry as a biomarker in clinical trials and diagnostic criterion for Alzheimer's disease. Alzheimers. Dement. 2011;7:474–485. doi: 10.1016/j.jalz.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keihaninejad S., Heckemann R.A., Fagiolo G., Symms M.R., Hajnal J.V., Hammers A. Alzheimer's Disease Neuroimaging Initiative A robust method to estimate the intracranial volume across MRI field strengths (1.5T and 3T). Neuroimage. 2010;50:1427–1437. doi: 10.1016/j.neuroimage.2010.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Ahadieh S., Corrigan B., French J., Fullerton T., Tensfeldt T. Alzheimer's Disease Working Group Disease progression meta-analysis model in Alzheimer's disease. Alzheimers. Dement. 2010;6:39–53. doi: 10.1016/j.jalz.2009.05.665. [DOI] [PubMed] [Google Scholar]
- Carlsson K.C., Savic R.M., Hooker A.C., Karlsson M.O. Modeling subpopulations with the $MIXTURE subroutine in NONMEM: finding the individual probability of belonging to a subpopulation for the use in model analysis and improved decision making. AAPS J. 2009;11:148–154. doi: 10.1208/s12248-009-9093-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell A.M, et al. CSF biomarkers in prediction of cerebral and clinical change in mild cognitive impairment and Azheimer's disease. J. Neurosc. 2010;30:2088–2101. doi: 10.1523/JNEUROSCI.3785-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., et al. Alzheimer's Disease Neuroimaging Initiative Sparse learning and stability selection for predicting MCI to AD conversion using baseline ADNI data. BMC Neurol. 2012;12:46. doi: 10.1186/1471-2377-12-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal S.L., Sheiner L.B., Boeckmann A.J., Bauer R.J. Icon Development Solutions; Ellicott City, MA; 1989. 2011. NONMEM user's guides. [Google Scholar]
- Hastie T.J., Tibshirani R.J. Monographs on Statistics and Applied Probability. Chapman & Hall; London; 1990. Generalized additive models. [Google Scholar]