

Abstract
Genes with common profiles of the presence and absence in disparate genomes tend to function in the same pathway. By mapping all human genes into about 1000 clusters of genes with similar patterns of conservation across eukaryotic phylogeny, we determined that sets of genes associated with particular diseases have similar phylogenetic profiles. By focusing on those human phylogenetic gene clusters that significantly overlap some of the thousands of human gene sets defined by their coexpression or annotation to pathways or other molecular attributes, we reveal the evolutionary map that connects molecular pathways and human diseases. The other genes in the phylogenetic clusters enriched for particular known disease genes or molecular pathways identify candidate genes for roles in those same disorders and pathways. Focusing on proteins coevolved with the microphthalmia-associated transcription factor (MITF), we identified the Notch pathway suppressor of hairless (RBP-Jk/SuH) transcription factor, and showed that RBP-Jk functions as an MITF cofactor.
Keywords: HPO, MSigDB, Heme, synaptonemal complex
Introduction
The hundreds of eukaryotic genomes now sequenced allow the tracking of the evolution of human genes, and the analysis of patterns of their conservation across eukaryotic clades. Phylogenetic profiling describes the relative sequence conservation or divergence of orthologous proteins across a set of reference genomes. Proteins that functionally interact in common pathways or in protein complexes can show similar patterns of relaxation of conservation in phylogenetic clades that no longer require that complex, pathway, or function, or conversely show similar levels of relative conservation in organisms that continue to utilize those functions. Phylogenetic profiling has been used to predict gene functions (Eisen and Wu, 2002; Enault et al, 2004; Jiang, 2008), protein–protein interactions (Sun et al, 2005; Kim and Subramaniam, 2006), protein subcellular location (Marcotte et al, 2000; Pagliarini et al, 2008), cellular organelle location (Avidor-Reiss et al, 2004; Hodges et al, 2012), and gene annotation (Merchant et al, 2007).
A number of improvements to phylogenetic profiling have increased its sensitivity and selectivity (Ruano-Rubio et al, 2009; Pellegrini, 2012). A modified phylogenetic profiling method that uses a continuous measure of relative conservation in each species revealed many new components of the Caenorhabditis elegans RNAi machinery (Tabach et al, 2013). Because of the primacy of human health in the biomedical enterprise and in primary research, the sum total of genetic and biochemical annotation for the human genome is much larger than that of C. elegans. Furthermore, because so many genome-scale gene expression experiments are done in human cell lines, many of which are tumor derived, there are thousands of gene sets defined by coexpression under a wide range of conditions available using the human genome as a reference. We therefore used the continuous-scale phylogenetic profiling method to map human genes into coevolved clusters. Our goal was to use this evolutionary mapping of human genes to systematically identify uncharacterized disease and pathway genes while revealing connections between different diseases and biological function groups. To establish the phylogenetic profile of every human protein-coding gene, we surveyed 86 disparate eukaryotic genome sequences, from animals, fungi, plants, and protists, and generated a continuous scale for conservation that is also normalized for evolutionary distance between each species. On the basis of this phylogenetic profile, we sorted 19 017 human proteins into clades of conservation and divergence in the other 85 genome sequences.
The phylogenetic profile revealed many gene clusters already known to function in pathways. To globally study the significance of patterns of coevolution in different classes of functional gene groups, we analyzed >6600 gene sets, ranging in size from 3 to 200 genes, collected in the Molecular Signatures Databases (MSigDB) (Subramanian et al, 2005). These gene sets include genes that are significantly changed in expression in tumors (Brentani et al, 2003; Segal et al, 2004; Subramanian et al, 2005), gene coexpressed under similar conditions, genes that bear similar transcriptional or miRNA regulatory sequences (Xie et al, 2005; Matys et al, 2006), or genes annotated to function in similar processes based on the pathway and ontology databases such as GO (Ashburner et al, 2000), KEGG (Ogata et al, 1999), REACTOME (Matthews et al, 2009), and BIOCARTA. We found that for 20% of these 6600 functional groups, the genes that constitute them have a significant overlap with the gene sets defined only by having similar phylogenetic patterns of conservation and divergence. Importantly, particular clusters of coevolved genes were frequently associated with several functional groups, including groups with no obvious overlap, defined for example by coexpression under some condition and common GO terms. In this way, the phylogenetic profile gene set could be used as a signal-to-noise filter to help discern molecular function from for example coexpression or other molecular signatures.
Because the clustering of human genes based on their phylogenetic profiles overlapped so significantly with the annotation in the MSigDB, we sought to discern similar statistically significant overlap with a Mendelian genetics database, on the assumption that genes that mutate cause a similar phenotype are expected to act in the same pathway and are therefore likely to show the similar patterns of phylogenetic conservation and divergence. We therefore asked whether the genes so far implicated in any particular disease have more similar phylogenetic profiles than would be expected of genes selected at random. We could ask this question for any one disease or we could ask it for all diseases in parallel. Our analysis of the phylogenetic clustering of the current set of thousands of genes so far implicated in a wide range of diseases revealed >100 disease classifications that contain multiple genes that are significantly correlated with each other in the phylogenetic profile clusters.
To generalize the phylogenetic analysis of human genes and intersect this analysis with the similar intersection with molecular signature gene clusters, we developed an alternative clustering protocol that first classified human genes into 1076 coevolved clusters of 3–193 genes bearing similar patterns of conservation or divergence across 86 disparate species. We then detected statistically significant overlap between these coevolved clusters and groups of human disease genes bearing the same Human Phenotype Ontology (HPO) terms. Many of the human phylogenetic profile clusters included an overrepresentation of annotated disease genes for particular diseases. Diseases with associated genes that overlapped phylogenetic gene clusters with the highest statistical significance included mitochondrial diseases, molybdoenzymes, heme biogenesis, or genes associated with cell migration and adhesion. Interestingly, we found that wide range of diseases intersected the phylogenetic profiles. The human genes we delineate here that phylogenetically cluster with the subset of human disease genes identified to date constitute prime candidate genes in which to expect mutations as more humans with various diseases are sequenced.
In parallel, we tested the overlap between each of the 1076 human phylogenetic clusters and the 6600 molecular signatures gene clusters and found that 20% of the molecular signatures clusters showed highly significant overlap with phylogenetic profile clusters. In many cases, the same phylogenetic profile cluster overlapped both human genetic disease gene clusters and molecular signature clusters, strongly informing the molecular pathway that is aberrant in these human genetic diseases.
The human phylogenetic profiles could also be used to query particular genes implicated in one particular disease or pathway for candidate other loci to act in the same pathway. For genes implicated in melanoma, we experimentally tested one of the genes that clusters with a transcription factor implicated in progression of melanoma, microphthalmia-associated transcription factor (MITF). The transcription regulator RBP-Jk (SuH) had a phylogenetic profile very similar to that of MITF. The predicted function of RBP-Jk in the MITF pathways was validated by demonstration of a protein–protein interaction between RBP-Jk and MITF and by experiments showing the requirement for RBP-Jk in MITF transcriptional regulatory activity. Similarly phylogenetic profiling revealed a meiosis-specific chromatin localization for the previously uncharacterized gene ccdc105. Thus, phylogenetic profiling provides a high-resolution view of the evolutionary interplay of human genes and identifies candidate genes involved in signaling pathways and human disease.
Results
Normalized phylogenetic profiling
To systematically identify new human disease genes, we adapted our method of normalized phylogenetic profile (NPP) analysis, previously employed to study the small RNA pathways in C. elegans (Tabach et al, 2013), to study human genes. We used data from 86 high-quality fully sequenced eukaryotic genomes from animal, plant, fungal, and protist phyla. The use of many species increased the statistical power and variance between the conservation of genes in these many genomes; most importantly, this reduced noise due to accidental missed annotation of a gene sequence in one genome sequence (see Materials and methods). To improve the sensitivity for the identification of coevolved proteins, we used a continuous scale of similarity between proteins (Enault et al, 2003; Tabach et al, 2013) that significantly improved the performance in prokaryotes (Enault et al, 2003) compared with a binary method. Normalized phylogenetic profiling uses a continuous scale of conservation that detects even small evolutionary changes by taking into account the evolutionary distances between the organisms and the protein lengths. For example, while sequence similarity of 40% between a human protein and its mouse ortholog may be relatively poor conservation, a very few protist proteins (<600) have 40% similarity to their human orthologs. For each human protein, we calculated the normalized protein BLAST (Blastp) bit score between the human query protein and the most similar protein in each organism (Figure 1A). The result is a human-centered NPP that represents a matrix of the gene products of 19 017 genes and how they are conserved or diverge across the genome sequences of 86 species (Supplementary Table S1). For example, in the heat map of conservation scores (Figure 1A), there are about 2000 H. sapiens proteins that are conserved in all animals but not in fungi, plants, or protists. Other H. sapiens proteins are conserved in some fungi but not in others. Members of the same protein families tend to exhibit similar patterns of phylogenetic conservation and therefore tend to cluster together in the hierarchical clustering, an almost trivial result for members of gene families. In many cases, however, genes in phylogenetic clusters have no homology to other genes in that cluster; only their pattern of conservation in some genomes and divergence in other genomes is correlated, indicating that they are probably comaintained by selection in some genomes and under less stringent selection is some genomes. The change in relative selection on particular proteins or particular domains of proteins is likely to result from the specializations of organisms to particular ecological niches, where certain pathways are no longer required and can now diverge.
Figure 1.

Phylogenetic profiles. (A) Phylogenetic profiles of 19 017 human genes across 86 eukaryotic genomes. The matrix was normalized by the evolutionary distances between organisms and the protein length and clustered using average linkage. The entry values are between 0 and 1 where 1 (dark blue) represents 100% identity and 0 (white) corresponds to no detectable homolog. (B) Phylogenetic profile of TCA cycle genes. The specific core profile (green rectangles) is defined mainly by genes lost across three protists: C. parvum, G. intestinalis, and E. histolytica. The detailed analysis of the phylogenetic profile predicts more subtle functions as well. For example, IDH1 and IDH3 function at the same step in the cycle (Supplementary Figure S1), but the gene with the core profile, IDH1, functions more centrally. A less conserved gene, IDH3, has a tissue-specific function. (C) Phylogenetic profile of genes (red box) that include the descriptor respiratory paralysis (HP:0002203). Mutations in these genes cause defects in heme biogenesis and porphyria disorders. Mutations in five other genes with a similar phylogenetic profile (pink box) cause porphyria or coproporphyria. Ten more genes (gray box) with similar phylogenetic profiles are predicted to mediate heme biogenesis and to cause porphyria-like symptoms when defective. (D) Phylogenetic profile of genes associated with urinary xanthine stone and sulfite oxidase deficiency HPO classifications (red and pink boxes) in addition to 18 genes with similar phylogenetic profiles (gray box). Of these 18 genes, MOSC2 and MOSC1 were recently identified as a new family of molybdenum enzymes. AOX1 is associated with xanthine urinary stones although a function was not assigned by the HPO database. Mutations in the HGD gene cause alkaptonuria disease. (E) Conservation pattern of the human genes MITF and RBP-Jk in 86 organisms. The scores are between 0 and 1, with 1 representing 100% identity and 0 corresponding to no detectable homolog.
The ability of phylogenetic profiling to cluster proteins based on the function is demonstrated by the clustering of proteins known from previous biochemical or genetic analysis to function in pathways. For example, the phylogenetic profile matrix clustered many of the TCA cycle genes (Figure 1B), with the loss of the TCA cycle genes in some protists is the hallmark of this phylogenetic cluster (Alberts et al, 2002; Munnich, 2008). About 80% (18/23) of the TCA cycle genes are conserved in most eukaryotes (Supplementary Figure S1, blue circled genes) but are lost in the protists C. parvum, G. intestinalis, and E. histolytica (Figure 1B). These protists lack mitochondria, the cellular organelle that mediates aerobic metabolism, but have mitosomes, organelles that are probably degenerate mitochondria (Tovar et al, 2003). Other genes that share the same profile but are not annotated be TCA cycle genes are known to interact with the TCA cycle (Supplementary Figure S2). As expected, most of these proteins, such as ATP synthase, cytochromes, and pyruvate dehydrogenase, are energy metabolism genes. Other genes that map to this phylogenetic cluster but are not annotated to be metabolic genes may nevertheless directly interact with the TCA cycle and mitochondria. For example, the multivesicular body sorting proteins CHMP4C and CHMP4B have a very similar phylogenetic profile to the TCA cycle genes. These endosomal proteins may mediate mitophagy, which may no longer operate in the protists with vestigial mitosomes. Thus, the TCA cycle phylogenetic profile example shows that phylogenetic profile analysis clusters genes that are already known to function together and identifies new factors that may be associated with this function.
Different classes of functional gene groups have distinct coevolution patterns
The TCA cycle is an extreme example of a well-studied, highly annotated molecular pathway that overlaps significantly with the phylogenetic profile classification of human genes. To systematically query the overlap between our phylogenetic profiling of human genes and many other analyses of human molecular pathways, we tested for significant overlap between the groups of 3–200 genes assigned to 6600 MSigDB (Subramanian et al, 2005) gene sets and the phylogenetic profile clusters defined by our analysis. MSigDB gene sets come from various sources including GO (Ashburner et al, 2000), REACTOME (Matthews et al, 2009), BIOCARTA, and KEGG (Ogata et al, 1999) pathway databases. In addition, they include groups of genes coexpressed under different conditions as well as microarray-derived sets of genes differentially expressed in cancers (Brentani et al, 2003; Segal et al, 2004; Subramanian et al, 2005) and genes sets that share promoter (Matys et al, 2006) or 3′UTR miRNA motifs (Xie et al, 2005). While some of the molecular signature gene sets correspond to GO/KEGG term annotation, with comparable levels of biochemical and genetic research distilled into the gene sets, other gene sets are much more provisional clustering of functions, for example, genes that are coexpressed under the same perturbation or genes bearing the same cis-regulatory sequence. For those gene sets that might include secondary responses or entries such as particular transcription factor or miRNA binding sites that might have true targets present in a haze of false targets, we expected to see less significant overlap with our phylogenetic profile gene compared with the well-curated gene sets.
To determine whether the multiple genes assigned to a specific MSigDB classification are significantly coevolved, we developed metrics for quantifying the statistical significance of the coevolution pattern in a set of genes. We calculated coevolution scores for the genes assigned to each MSigDB group, termed Co10 (see Materials and methods). Briefly, we defined for each gene the 10 non-homologous genes that are most phylogenetically correlated with it (the 10 nearest neighbors when correlating phylogenetic conservation across 86 genomes compared). Then for a group of genes in an MSigDB classification, we counted the number of times these genes were found in each other’s 10 nearest neighbors in the phylogenetic profile (Figure 2A, blue x’s). The P-value was estimated by performing the same operation on 1 000 000 random groups (Figure 2A, red and brown dots). The Co10 scoring system scores significantly any MSigDB gene group with a substantial fraction of its member genes having similar phylogenetic profiles. For the NPP with 86 organisms, 340 out of 6600 MSigDB gene groups had Co10 scores higher than those of the 1 000 000 random sets of the same size (P-values<10−6), and 1277 sets had q-values of 0.05 (see Materials and methods), implying an upper boundary of 5% false positives (q-values are the name given to the adjusted P-values found using an optimized false discovery rate (FDR) approach). As expected, different MSigDB categories contained different percentages of significantly coevolved groups (Table I; Supplementary Table S2). For example from the KEGG pathway subcategory, 20% of the KEGG groups have multiple members that are significantly coevolved. It is noteworthy for example that among the top 25 KEGG subgroups that cluster phylogenetically are intensively studied signaling pathways such as the MAP kinase Wnt, VEGF, and cytokine as well as biochemical pathways such as porphyrin biosynthesis (Supplementary Table S2). Interestingly, we also found many coregulated gene sets to be significantly coevolved. For example, a significant fraction of the 251 genes with FOXC1 (forkhead box C1) binding sites in their promoter regions are coevolved. Mutations in FOXC1 cause various glaucoma phenotypes including primary congenital glaucoma, autosomal dominant iridogoniodysgenesis anomaly, and Axenfeld-Rieger anomaly. In addition, coevolved gene sets contain binding sites for AP4 or the uncharacterized promoter sequences SMTTTTGT, TTTNNANAGCYR, or WTTGKCTG (Supplementary Table S2). Finally, the target genes of several microRNAs, for example, mir-218, mir-524, and mir-30 family are also significantly coevolved. Thus even though coexpressed genes or genes that are regulated by the same microRNA are less likely to have similar phylogenetic profiles since expression patterns differ substantially among even closely related primate species (Khaitovich et al, 2006), we can easily see that the genes bearing this cis-regulatory sites show a significant phylogenetic clustering. These results suggest that coevolved genes tend to gain additional and new regulation by gaining promoter and microRNA sites.
Figure 2.

Coevolution scores (Co10) and q-values for MSigDB gene sets. (A–E) Scatter plots of the Co10 scores (y axis) and the number of genes (x axis) in each classification of the MSigDB sets (blue x’s) were calculated using normalized phylogenetic profiling (NPP) with 86, 64, 43, and 22 organisms. Binary phylogenetic profiles were calculated with 86 organisms. The blue x’s represent the Co10 scores of human gene sets from MSigDB, which includes KEGG-annotated sets such as MAP kinase signaling-annotated genes or ribosomal-annotated genes. The dots represent the distribution of Co10 scores associated with 100 000 randomly generated gene sets derived from randomized MsigDB gene lists. The color scale of the dots represents the number of random groups found at that position (red—one random group to purple when >10 random groups with the same size have the same Co10 score). The white lines represent the average of the random data (bold line) and 1–4 standard divisions from the average. (F) The q-value distribution of the MSigDB sets obtained using NPP with 86, 64, 43, and 22 organisms and BPP with 86 organisms.
Table 1. Number and percentage of significant coevolved MSigDB groups in different methods.
| Collections | Subcollections | No. of groups |
q-values<0.05 |
P-value<10−6 |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NPP (86 organisms) | NPP (64 organisms) | NPP (43 organisms) | NPP (22 organisms) | BPP (86 organisms) | NPP (86 organisms) | NPP (64 organisms) | NPP (43 organisms) | NPP (22 organisms) | BPP (86 organisms) | |||||||||||||
| C1: positional gene sets | Positional gene sets | 320 | 10 | 3% | 9 | 3% | 6 | 2% | 4 | 1% | 4 | 1% | 2 | 1% | 3 | 1% | 1 | 0% | 1 | 0% | 0 | 0% |
| C2: curated gene sets | Chemical and genetic perturbations | 2365 | 250 | 11% | 208 | 9% | 193 | 8% | 159 | 7% | 33 | 1% | 47 | 2% | 40 | 2% | 43 | 2% | 34 | 1% | 9 | 0% |
| BioCarta | 217 | 41 | 19% | 34 | 16% | 27 | 12% | 7 | 3% | 8 | 4% | 2 | 1% | 3 | 1% | 3 | 1% | 1 | 0% | 3 | 1% | |
| KEGG | 186 | 98 | 53% | 93 | 50% | 85 | 46% | 61 | 33% | 22 | 12% | 38 | 20% | 34 | 18% | 31 | 17% | 24 | 13% | 6 | 3% | |
| REACTOME | 430 | 163 | 38% | 159 | 37% | 154 | 36% | 110 | 26% | 58 | 13% | 67 | 16% | 71 | 17% | 60 | 14% | 63 | 15% | 23 | 5% | |
| C3: motif gene sets | MicroRNA targets | 219 | 54 | 25% | 41 | 19% | 43 | 20% | 20 | 9% | 0 | 0% | 4 | 2% | 5 | 2% | 1 | 0% | 1 | 0% | 0 | 0% |
| Transcription factor targets | 584 | 143 | 24% | 135 | 23% | 89 | 15% | 54 | 9% | 5 | 1% | 6 | 1% | 6 | 1% | 2 | 0% | 4 | 1% | 0 | 0% | |
| C4: computational gene sets | Cancer modules | 443 | 81 | 18% | 77 | 17% | 71 | 16% | 76 | 17% | 33 | 7% | 25 | 6% | 23 | 5% | 23 | 5% | 27 | 6% | 8 | 2% |
| Cancer gene neighborhoods | 427 | 149 | 35% | 132 | 31% | 141 | 33% | 108 | 25% | 22 | 5% | 48 | 11% | 46 | 11% | 51 | 12% | 48 | 11% | 9 | 2% | |
| C5: GO gene sets | GO biological process | 793 | 136 | 17% | 121 | 15% | 100 | 13% | 64 | 8% | 42 | 5% | 38 | 5% | 28 | 4% | 30 | 4% | 16 | 2% | 7 | 1% |
| GO cellular component | 215 | 50 | 23% | 41 | 19% | 43 | 20% | 40 | 19% | 9 | 4% | 18 | 8% | 18 | 8% | 19 | 9% | 8 | 4% | 3 | 1% | |
| GO molecular function | 395 | 102 | 26% | 99 | 25% | 93 | 24% | 70 | 18% | 38 | 10% | 45 | 11% | 37 | 9% | 40 | 10% | 21 | 5% | 8 | 2% | |
| Total | 6594 | 1277 | 19% | 1149 | 17% | 1045 | 16% | 773 | 12% | 274 | 4% | 340 | 5% | 314 | 5% | 304 | 5% | 248 | 4% | 76 | 1% | |
Many other gene sets (>80%) did not significantly overlap with the phylogenetic profiles of human genes. These gene sets may include many genes that act in multiple gene pathways, essentially obscuring the phylogenetic profile signatures, or may correspond to gene groupings that do not correspond to actual molecular pathways. For example, among gene sets that barely registered in this analysis, in the sub-category ‘positional gene sets’, which maps genes based on the chromosomal location, there were only two significant (P-value<10−6) phylogenetically clustered gene sets, <1% of the total number of groups in this sub-category. The most two significant sets contain >50 keratin proteins, several chemokine, and Schlafen family members on chr17q12. And chromosomal band, chr6p22, is enriched with histones and olfactory receptors.
Thus, phylogenetic profiling can be used to shift through thousands of molecular signature gene groupings to discern those gene sets that may correspond to pathways and to highlight the particular genes in any molecular signature group that have the strongest phylogenetic profile signatures. To examine the impact of the number of species included in the phylogenetic profile on the power to detect overlap with MSigDB, we repeated the analysis with genome sequences of 86 disparate species (the full set, Figure 2A), and groups of 64 species (Figure 2B), 43 species (Figure 2C), and 22 species (Figure 2D). A small but consistent difference of at least 10% more significant molecular signature groups with overlap of phylogenetic profiles detected favored the use of data from all 86 organisms (Table I). In addition, inclusion of more organisms in phylogenetic clustering improved the ability to separate functional gene groups from random sets of genes as reflected by the improved P-values when more organisms are used for the phylogenetic profiling (Figure 2F). Although we did not systematically compare the NPPs with other methods, Enault et al (2003) showed that normalizing the phylogenetic profiles in prokaryotes increases the number of Ecocyc enzymes identified as being evolutionarily related by about 25% compared with the binary (absent/present) form of phylogenetic profiling. Even more importantly, while our analysis identified that 20% of the GO groups are significantly coevolved, several studies have claimed that mouse or human phylogenetic profiling does not create functionally cohesive clusters and was informative for only a small fraction of GO terms (Loganantharaj and Atwi, 2007; Ko and Lee, 2009; Ruano-Rubio et al, 2009). To demonstrate the difference between binary and normalized phylogenetic profiling approaches, (see Materials and methods) we repeated the analysis using the binary method of phylogenetic profiling with the same 86 organisms (Figure 2E). Using the normalized phylogenetic profiling method, we found 4.5 times more functional groups that are significantly coevolved than using the binary method (Table I). The use of NPPs reduced the variance dramatically in the random grouped Co10 scores compared with the binary method (Figure 2A–E, red dots). In fact, the difference between the methods is so significant that normalized phylogenetic profiling performs significantly better with only 25% of the organisms than binary profiling using the full set of organism comparisons (Figure 2F).
Phylogenetic profile analysis of genes sets with similar disease phenotypes
Phylogenetic profile analysis has previously been a powerful tool for the study of human Bardet-Biedl syndrome (Mykytyn et al, 2004) and mitochondrial diseases (Pagliarini et al, 2008). Just as phylogenetic profiling could detect significant overlap with about 20% of the molecular signatures gene groups, we sought to detect a similar fraction of the smaller set of genes annotated at present to be variant in human genetic diseases. Even though only a subset of human disease loci have been identified at this intermediate stage in human genetic analysis, we expected to detect similar phylogenetic profiles for the known genes for any disorder for which multiple genes have been implicated. For many human gene variants, including many that have been assigned to specific genetic loci, a suite of symptoms are associated with descriptors such as ‘ataxia’ or ‘alveolar cell carcinoma-associated’ in various data sources. Such descriptors have been systematized in the HPO database (Robinson and Mundlos, 2010). The different HPO groups include genes that cause similar symptoms or phenotypic manifestations as characterized by the curated data from the OMIM (On Mendelian Inheritance in Man) database, or OMIM. For example, 215 human genes have the descriptor ‘ataxia’ associated with them in the HPO database. In a specific example, missense and deletion mutations in the inositol 1,4,5-triphosphate receptor (ITPR1) are the cause of spinocerebellar ataxia 15, and the OMIN/HPO file for ITPR1 uses the word ataxia 12 times. Thus, ITPR1 is one of the 215 genes associated with the HPO ataxia term.
To identify the HPO groups that contain a significant fraction of human genes with similar phylogenetic profiles, we calculated the Co10 scores, P-values, and q-values for each HPO group that contains between 3 and 500 genes, similarly to as described for the MSigDB analysis. As in the analysis of the MSigDB data, use of all 86 organisms and the normalized phylogenetic profiling method detected the most significant overlap between HPO groups and phylogenetic profiles (data not shown).
Our analysis revealed 156 out of 3413 HPO classifications (Figure 3, see Supplementary Table S3 for the entire list of HPO and their P-values) that contain multiple genes that are significantly correlated with each other in the phylogenetic profile clusters (q-values<0.05). Among HPO classifications with the most significant associated gene clustering (P-values≤10−6), we found hypoglycemia (HP:0001943, with 80 genes), abnormality of mitochondrial metabolism (HP:0003287, with 39 genes), cerebral edema (HP:0002181, with 24 genes), respiratory paralysis (HP:0002203, with 3 genes), and abnormality of blood glucose concentration (HP:0011015, with 88 genes). The significant HPOs (q-values<0.05) map in several clusters that share groups of coevolved genes (Figure 4; Supplementary Table S4). Two of these clusters that include many of the most significant HPOs (Figure 4) share large sets of mitochondrial genes. These data suggest that the phylogenetic signature of mitochondrial genes and mitochondrial-associated symptoms define the strongest signal in this phylogenetic profile analysis. There are many disease loci in these phylogenetic clusters that are not understood to be mitochondrial diseases but in fact may have a mitochondrial disease basis. Other clusters are defined either molecularly, for example genes coding for molybdoenzymes or heme biogenesis genes, or phenotypically by associations with pathologies, such as aberrant bone epiphyses (Figure 4).
Figure 3.

Genes associated with particular diseases have significantly higher coevolution scores. The dots denote the random distribution of Co10 scores that emerge from a randomized set of 100 000 HPO gene sets. The color scale of the dots represents the number of random groups found at that position (red—one random group to purple when >10 random groups with the same size have the same Co10 score). The white lines represent the average of the random data (bold line) and 1–4 standard divisions from the average. Notice that there are a significant number of bona fide HPO groups with numbers of genes in those groups ranging from a few to hundreds that are far from the random expectation cloud of dots.
Figure 4.

Many of the diseases associated with high coevolution scores share genetic components. Significant HPOs (q-value<0.05) with <100 genes are present as nodes. The color code scale represents the Co10 significance score from gray (q-value=0.05) to cherry (P-value <10−6). The size of the HPO reflects the fraction of coevolved genes out of the all the genes in the HPO (i.e., the genes that contribute to the Co10 score). Two HPOs are connected by edge if they share two or more coevolved genes (see Supplementary Table S4), such that the number of the shared genes reflected in the edge width and color, with more genes are represented by a thicker and darker line.
One HPO classification that has the most significant overlap with the phylogenetic profile is respiratory paralysis (HP:0002203). Respiratory paralysis is the main cause of death in patients with genetic deficiencies in the enzymes of the heme biosynthesis pathway such as the porphyria disorders. We examined the other proteins with similar profiles to the known heme biosynthetic genes. These proteins include all five known genes in which mutations cause porphyria disorders. All these genes share very similar phylogenetic profiles, amusingly, given our laboratory expertise in C. elegans, the hallmark of which is loss of these genes in the Nematoda among the animals and in some protists (Figure 1C, pink square). In addition to these known heme biosynthetic genes, there are 10 other genes (Figure 1C, white square) with similar phylogenetic profiles. Three of these genes are ORM1-like genes, previously implicated in sphingolipid biosynthesis and asthma (Breslow et al, 2010). Our data suggest that the sphingolipid defects of ORM1 mutations in yeast may have a connection to heme biogenesis as their molecular basis. Interestingly, the organisms that have lost these heme pathway genes, for example, L. scapularis (deer tick), nematodes, C. parvum and G. intestinalis (intestinal parasites), Leishmania (a blood cell parasite), and T. brucei (the cause of African trypanosomiasis) obtain heme from their hosts or from bacterial food sources.
Systematic identification of genes that coevolve with known pathways and diseases
In the mapping of genes classified by HPO groups or by MSigDB groups to phylogenetic clusters, we noted that some of the same genes were correlating with distinct diseases and distinct molecular signature gene groups. For example, a set of 4–6 nuclearly encoded mitochondrial proteins constitute the overlap with MSigDB groups such as KEGG oxidative phosphorylation and HPO terms such as abnormal cerebrospinal fluid, not an obvious mitochondrial disorder to the untrained eye. The correlation of MSigDB pathways with human disease classifications promised to illuminate both the possible biochemical and molecular pathways that might be aberrant in diseases not as well understood as mitochondrial disease and to highlight particular genes from within a phylogenetic profile that might be tested for mutations in various human genetic diseases. To systematically identify other gene pathways that have coevolved with known genes associated with diseases, or related pathways, we phylogenetically clustered (see Materials and methods) all the human genes, into 1076 coevolved clusters (Supplementary Table S5). Thus, genes with similar patterns of conservation in the 86 other genomes analyzed were grouped into clusters of phylogenetic similarity, each containing between 3 and 193 coevolved genes. We then tested whether the lists of genes in each of the coevolved cluster significantly overlapped the lists of genes assigned by HPO human genetic or MSigDB molecular pathways analysis.
Compared with random sets of genes, these phylogenetic clusters have an astonishing overrepresentation of genes associated with similar HPO groups (Figure 5A and B). We termed these phylogenetic clusters that are enriched with disease genes in PhyloDisease clusters. Homologous proteins that have high sequence similarity, by definition, have similar phylogenetic profiles. Those profiles with multiple gene family members can inflate the overlaps we measured. To eliminate the effect of homologous genes, we calculated the overrepresentation of P-values conflating members of gene family into one instead of multiple proteins of the same family (see Materials and methods; Supplementary Table S6, columns L and M) and used this P-value as our main filter of significance. After correcting for multiple testing and removing the statistical complication of paralogous genes, we identified 54 PhyloDisease clusters (Supplementary Table S6). Each of these clusters contains coevolved genes that associate significantly with one and frequently several HPO disease classifications. The genes in the same PhyloDisease clusters are strong candidates for roles in those same disorders.
Figure 5.

P-value distribution of the overlap between coevolved clusters and random or functional gene groups. (A) Distribution of the hypergeometric P-values of the significant overlap between every pair of coevolved cluster with HPO group (white bars), or coevolved cluster with randomly permutated HPO in the same size (black bars). (B) The P-value distribution using only the best P-value per HPO group (white bars) or randomly permutated HPO groups (black bars). (C) Distribution of the hypergeometric P-values using 6600 MSigDB functional groups (white bars) or randomly permuted MSigDB groups (black bars). (D) The P-value distribution using the best P-value per MSigDB.
The connection between the malfunction of biological networks and disease symptoms is not fully understood in many cases. To associate a possible biological function to the PhyloDisease clusters, we tested whether the genes in these clusters are enriched for known biological function. We therefore determined which particular molecular signatures gene clusters overlap the same human phylogenetic profile clusters as those overlapped by human disease HPO gene clusters. The interleaved highly significant human phylogenetic clusters that overlap either MSigDB or HPO clusters are shown in Figure 6 (and in the Supplementary Information, we show a figure of how a particular intersection of MsigDB or HPO clusters with phylogenetic clusters is generated). Of the 54 PhyloDisease clusters, 48 clusters contained an overrepresentation of genes that also significantly overlapped genes grouped in MSigDB to have a known biological function or coregulation under particular conditions. In general, coevolved clusters have a significantly overrepresentation of genes that share biological function (Figure 5C and D).
Figure 6.

(A) Heat map of the overlap between coevolved clusters and functional and disease gene sets (rows). Each dot in row i and column j represented the P-value of the overlap between functional/disease group (in row i) and coevolved cluster (in column j). The color code represents the P-values indicating the significance of the overlap. The white boxes delineate the indicated insets representing coevolved clusters that are associated with either (B) mitochondria, (C) ribosome (D) RAS signaling, and E2F (E) immune response, or (F) muscle stiffness. The data can be found in Supplementary Table S5.
Mapping human genes into coevolved clusters uncovered many known and many unexpected connections between different diseases and functional gene groups (Supplementary Table S6). The strongest signal (Figure 6B), as expected from our previous analysis, was generated by the 6 mitochondria-related clusters: cluster 474 (complex I), cluster 305 (complex I), cluster 21 (TCA cycle), and clusters 8, 220, and 19 (mitochondria membrane and other mitochondrial subassemblies). The genes in these clusters are also associated with a wide range of disease symptoms (HPOs), such as cerebral edema, coma, acidosis, Babinski sign, vomiting, and abnormal pyramidal tracts in addition to defects in metabolic processes like insulin secretion, valine, leucine, and isoleucine degradation, or genes for which expression is downregulated during kidney aging. Some of these disease symptoms are associated with specific mitochondrial subpathways (i.e., hypertrophic cardiomyopathy coevolves with complex I genes), while others such as abnormal acid-base homeostasis are correlated with many mitochondrial functions (Figure 6B). Many of the HPO descriptors are known to be associated with mitochondrial defects; for example, the association between neurodegenerative diseases such as Huntington’s, Alzheimer’s and Parkinson’s and mitochondrial defects (Lin and Beal, 2006). Our results show clear overlap (P-value<10−6) between neurodegenerative disease proteins that show patterns of coevolution with mitochondrial proteins. (Figure 6B; Supplementary Table S6).
Cell-cycle and cancer genes also emerged from this analysis. One of the most significant overlaps between MSigDB groups and phylogenetic clusters was cluster 102 that overlaps for example multiple MSigDB groups including for example coexpression on microarrays with the APEX1 DNA repair nuclease or coexpression with the PRKDC DNA directed kinase and includes the cell-cycle regulatory genes CCT4, CCT5, CDK2, IARS, MCM6, PPP1CC, PSMA1, and YWHAQ. Another phylogenetic cluster strongly associated with both disease genes and molecular attributes contains many ‘RAS GTPASE’ genes. RAS is one of the most common oncogenes in human cancer—mutations that activate RAS are found in 20–25% of human tumors and up to 90% in certain types of cancer (Goodsell, 1999). RAS family genes are found in cluster 368; the genes in this coevolved cluster are enriched with E2F and ELK1 binding motifs in their promoters and mir-194 binding motifs in their 3′UTRs (Figure 6D). The E2F family has a crucial role in the control of cell-cycle and cancer progression in p53 and RAS pathways (Milyavsky et al, 2005; Tabach et al, 2005). The microRNA mir-194 has a role in the p53 pathway (Takwi and Li, 2009) and suppresses metastasis of mouse liver cancer cells (Meng et al, 2010).
Another phylogenetic cluster 867, with 69 genes with similar patterns of evolutionary conservation, overlaps both HPO terms and MSigDB terms with high significance (Supplementary Table S6). The genes that overlap ADAMTS2, FERMT1, and ITGA6 are annotated by MSig to be involved in focal adhesion, consistent with the protease and integrin annotation, and the HPO terms implicate this cluster in the related assembly of skin and gingiva and blood vessels. The genes of this cluster are also upregulated in neural crest cells and in lung metastasis and overrepresented in the KEGG small cell lung cancer module.
Ribosomal proteins are clustered into six distinct coevolved clusters, the hallmark of which is extremely high conservation in nearly every organism (Figure 6C). Cluster 30 groups ribosomal proteins (RPL4, RPS12, RPS15A, RPS4X, RPS4Y1, and RSL24D1) with aminoacyl-tRNA synthetase proteins (CARS, CARS2, LARS, SARS, and SARS2) and RNA degradation proteins (ENO1, ENO2, ENO3, EXOSC2, PAPOLA, PAPOLB, and PAPOLG). These three coevolved clusters have a probable function in modulating protein expression and mRNA levels.
The binding motifs of two transcription factors LHX3 (LIM Homeobox) and TITF1/NKX2-1 (thyroid transcription factor 1) are significantly overrepresented in the promoters of the genes in the phylogenetic cluster 469. Disruption of Nkx2-1 in mice results in ablation of the pituitary gland (Takuma et al, 1998), and LHX3 is required for pituitary development. Mutations in the LHX3 gene cause combined pituitary hormone deficiency 3. The protein expression levels of both TFs are almost identical and both highly expressed in blood plasma and the kidney (Supplementary Figure S3). Several pathologies are associated with cluster 469, for example ‘xanthine urinary stone’, molybdenum cofactor deficiency, myoclonic spasms, lens dislocation, and kidney stones; these disease symptoms are caused by mutations in the genes (MOCS1, MOCS2, GPHN, XDH, and GPHN) that are coevolved (Figure 1D) and have roles in molybdenum cofactor biosynthesis and xanthine biogenesis. Cluster 469 predicts other genes to act in molybdenum cofactor synthesis or xanthine biosynthetic pathways (Supplementary Table S5; Figure 1D, gray box) (Wahl et al, 2010). Aldehyde oxidase 1 (AOX1) is associated with xanthine urinary stones (Gok et al, 2003) but was not assigned to the ‘xanthine urinary stone’ phenotype by the HPO database. AOX1 is involved in the metabolism of the drug thiopurine as are two other proteins encoded by genes in this group, XDH and MOCOS. Another gene, HGD, is active chiefly in the liver and kidneys and can, when mutated, cause alkaptonuria, a disease associated with prostatic and renal stones (Introne et al, 1993).
Two other clusters 576 and 675 (Figure 6E) have an overrepresentation of chemokine and other immune-related genes; cluster 442 groups together coevolved genes that have roles in coma, hypotonia, and muscle stiffness are regulated by several transcription factors, for example, TEF1, MEF2, and NKX25 (Figure 6F).
Phylogenetic profiling identifies a new MITF-associated factor
While phylogenetic profiling could be used to seek the particular diseases with the strongest phylogenetic profile overlap, we could also query for particular known components of diseases whether they have similar phylogenetic profiles to any other genes. The proteins with the same profile are much more likely to act in the same pathway. As an example, we used phylogenetic profiling to investigate the role of MITF, the master regulator of the melanocyte lineage that also serves as a driver for melanoma (Garraway et al, 2005). MITF both directs melanocytes toward terminal differentiation and paradoxically promotes their malignancy. Although the role of MITF in the melanocyte lineage is established, the mechanism by which MITF promotes these two seemingly contradictory roles is mostly unknown, and the identification of MITF coregulators is probably the key to solving this puzzle. We ranked ∼20 000 human proteins based on their phylogenetic correlation with MITF (Supplementary Table S7). Several of the highly correlated genes encode known MITF paralogs for example, the bHLH-family proteins TFEB, TFEC, and TFE3 (Supplementary Table S7). MITF forms either a homodimer or a heterodimer with TFEC and TFEB, when binding to DNA (Levy et al, 2006). From the phylogenetically correlated genes with no homology to MITF and no previous indication of a function related to that of MITF, we looked for genes that have higher probability to interact as transcription cofactor with MITF. For that we filtered for nuclear localized proteins that are known to act as transcription regulators. We found that the transcription factor Suppressor of Hairless (also known as RBP-Jk and SuH) and Forkhead box protein R2 (FOXR2) met these criteria. To increase our confidence that either RBP-Jk or FOXR2 could be a cofactor for MITF, we looked for co-occurrence of the promoter binding sites of MITF and either RBP-JK or FOXR2. We found that RBP-Jk DNA binding sites are significantly enriched (P-value<1.3 × 10−7, see Materials and methods) in known MITF target gene promoters (Levy et al, 2006) (Supplementary Table S8). Transcriptional coexpression studies also support a role for RBP-Jk in MITF function. Among the 50 most coevolved genes to MITF in the phylogenetic profile, RBP-Jk showed the highest correlation to MITF across 100 different expression data sets (P-value=1 × 10−19), calculated using Multi-Experiment Matrix (MEM) for gene expression similarity searches across many data sets (Adler et al, 2009).
RBP-Jk functions downstream of Notch signaling in Drosophila, C. elegans, and mammals (Tanigaki and Honjo, 2010) and although RBP-Jk is more conserved than MITF across the 86 organisms analyzed, their phylogenetic profiles are similar (Figure 1E). We experimentally validated RBP-Jk function in the regulation of the known MITF target gene, TRPM1 (Miller et al, 2004). We found that RBP-Jk occupies the TRPM1 promoter but its interaction with this promoter is MITF dependent (Figure 7A; Supplementary Figure S4 and Supplementary Table S9 for list of primers and siRNA sequences). Although RBP-Jk occupancy is MITF dependent, MITF occupancy was unchanged after RBP-Jk depletion (Supplementary Figure S4b). Moreover, RBP-Jk occupied a region without any known RBP-Jk DNA binding sites, suggesting DNA interaction via protein complex with MITF. Indeed we found, using coimmunoprecipitation, that MITF and RBP-Jk proteins interact directly in the cell (Figure 7B). RBP-Jk depletion caused a decrease in Pol-II occupancy on the gene locus (Figure 7C) and a decrease in TRPM1 mRNA levels (Figure 7D). To further examine whether MITF and RBP-Jk activities are mutually dependent, we tested their cooperation in activation of gene expression. Mutations in MITF binding sites in TRPM1 caused a loss of RBP-Jk-mediated promoter activation (Figure 7E), and RBP-Jk depletion caused a decrease in MITF-mediated transcriptional activity. Together, these observations suggest that MITF recruits RBP-Jk to the promoter site to enhance transcriptional activity. RBP-Jk is a central mediator of Notch signaling (Liang et al, 2002) that interacts directly with other transcription factors (Obata et al, 2001; Beres et al, 2006) including Myc (Agrawal et al, 2010). MITF is a member of the same basic helix-loop-helix zipper (bHLH-Zip) superfamily (Steingrimsson et al, 2004) as Myc. Also in support of these data, Notch signaling, and RBP-Jk activity in particular (Aubin-Houzelstein et al, 2008), has been implicated in melanocyte differentiation (Schouwey et al, 2011). Our finding indicates that RBP-Jk directly affects MITF transcriptional activity and demonstrates the ability of normalized phylogenetic profiling to discover novel cofactors.
Figure 7.

RBP-Jk is an MITF gene coregulator. (A) Binding of RBP-Jk to an endogenous TRPM1 promoter in an MITF-dependent manner. ChIP analyses were performed using log-phase primary human melanomas on TRPM1 promoter upon depletion of MITF (by treatment with siMITF) or RBP-Jk (by treatment with siRBP-Jk). Protein:chromatin-crosslinked complexes were immunoprecipitated with RBP-Jk antibody. RBP-Jk occupancy was calculated relative to cells transfected with control siRNA (siCont), normalized to input, and represent mean±s.d. of three independent experiments. Controls are shown in Supplementary Figure S3. (B) MITF and RBP-Jk directly interact. CoIP was performed using MITF and RBP-Jk antibodies. Reactions were treated with DNase to exclude the possibility that the MITF:RBPJk interaction is DNA dependent. (C) Pol-II occupancy on the TRPM1 locus was performed upon MITF or RBP-Jk depletion with indicated siRNA. (D) Endogenous TRPM1 levels were affected by RBP-Jk perturbation. Primary human melanomas were transfected with siMITF, siRBP-Jk, or scrambled control siRNA (siCont). TRPM1 mRNA levels were measured by qRT–PCR. HES1 mRNA levels were measured as a control for a gene known to be controlled by and an RBP-Jk target gene and PZL2 was used as an irrelevant control gene (upper panel). TRPM1 levels were also increased upon RBP-Jk overexpression (lower panel). Melanoma cells were transfected with MITF or RBP-Jk expression vectors or empty vector. Results are normalized to actin and are relative to siCont or empty vector, respectively, and represent mean±s.d. of five replicates. (E) MITF and RBP-Jk activity is mutually dependent. Upper panel shows that RBP-Jk activity is MITF dependent. Human primary melanoma cells were transiently transfected with empty vector, MITF, or RBP-Jk (0.5 μg each), TRPM1 promoter reporters (0.3 μg), and Renilla luciferase construct (0.1 μg). At 48 h after transfection, cell lysates firefly and Renilla luciferase activities were measured. Lower panel shows that when RBP-Jk is absent MITF-dependent transcriptional activity is compromised. Human primary melanoma cells were transiently transfected with siControl or siRBP-Jk. The next day, cells were transfected as indicated. Data are presented as mean values and s.d. for at least three independent experiments compared with the level of luciferase activity obtained in the presence of empty vector.
Phylogenetic profiling identifies ccdc105 as a meiosis-specific chromatin localization gene
Proteins that constitute components of specialized multiprotein complexes are also expected to have similar phylogenetic profiles. As a test for the use of phylogenetic profiles to generate candidate components of such protein complexes, we analyzed proteins of the synaptonemal complex. The synaptonemal complex is specialized for meiotic cell divisions and is essential for proper meiotic recombination and segregation (Fraune et al, 2012). The known protein components of the synaptonemal complex clustered strongly with other, with known meiotic proteins as well as with a number of previously uncharacterized proteins (Supplementary Figure S5a). Several of these newly identified proteins share a predicted coiled-coil secondary structure that is also shared by many known proteins of the synaptonemal complex (Costa and Cooke, 2007; Fraune et al, 2012). Since meiosis is limited to germ cells that produce spermatozoa and oocytes, meiotic genes should be expressed in the germ cell lineage (Schramm et al, 2011). We analyzed the mRNA expression pattern of these uncharacterized genes and found that several are highly expressed in the testes and ovaries (Supplementary Figure S5b). ccdc105 had a phylogenetic profile most similar to the well-annotated synaptonemal complex components and showed tissue-specific expression in testes and ovary (Figure 8A and B; Supplementary Figure S5a). CCDC105 protein colocalized with the heterochromatin surrounding the centromeres in one of the extremes of the synaptonemal complex at diplotene (Figure 8C–F, arrowheads). Diplotene is the stage of meiotic prophase I that directly precedes the first chromosomal segregation. CCDC105 also colocalized with the heterochromatin of the clustered centromeres in post-meiotic round spermatids (Figure 8C–F, arrows). Together, these results suggest a role for CCDC105 in late stages of meiotic prophase I and in spermatogenesis. CCDC105 may be a variant in humans with infertility (Handel and Schimenti, 2010).
Figure 8.

Ccdc105 characterization. (A) RT–PCR analysis of Ccdc105 mRNA expression in mouse tissues. Actin was used as a positive control for mRNA amplification. (B) Western blot of CCDC105 in protein extracts from testes. Purified antibodies against peptides 1 and 3 recognized a band of around 57 kilodaltons (kD), the predicted size for this protein, in total protein extracts from testes (Total). CCDC105 protein is mostly detected in nuclear extracts (Nuc), whereas in cytoplasmic extracts (Cyto) little or no signal was detected. (C–F) Immunolabeling of CCDC105 on spread cells from testes. Anti-SYCP3 antibody labels the synaptonemal complex throughout the meiotic prophase I, and its labeling pattern was used to identify cells in meiosis. Anti-CREST antibody labels centromeres and anti-CCDC105 antibody (anti-peptide 1) is localized in a region where SYCP3 and CREST colocalize at diplotene stage (meiotic cells) and in clustered centromeres in round spermatids (post-meiotic cells) (arrowheads in D and E). z, zygotene cells; p, pachytene cells; d, diplotene cells; rs, round spermatids. Bars represent 10 micrometers (μm).
Discussion
Phylogenetic profiling of human genes has been used most successfully to date to discover genetic causes of mitochondrial disease and disorders of ciliated neurons (Mykytyn et al, 2004; Pagliarini et al, 2008). The discovery that Bardet-Biedl syndrome is a disease of the cilia basal body and that associated genes can be identified using phylogenetic profiling accelerated the discovery of more cilia disease loci. Our analysis of the overlap between human genetic or molecular signatures gene clusters and the human phylogenetic profile clusters we assembled identified almost 1400 disease phenotypes, pathways, coexpressed genes and other biological groups that are significantly coevolved. By mapping a large fraction of human genes into phylogenetic clusters, we found that many clusters are associated with one or more diseases or functional gene groups. In many cases, the coevolved clusters are associated with multiple annotated biological functions and diseases, which immediately suggest functional connections between those diseases and pathways.
From the pattern of organisms that retain those genes and those organisms that have lost those genes, the biological context of the pathway can be inferred. For example, the molybdenum cluster 469 that is associated with kidney stones and involuntary muscle contractions has a phylogenetic profile that shows a loss of these genes in those organisms with few introns and no RNAi pathway, as if purine biosynthetic demands are decreased in those organisms, allowing the molybdenum-requiring purine biosynthesis enzymes to be lost. The genes are needed in humans and deficits in those pathways that cause the disorders above, associated with purine biosynthetic defects. In our analysis of 86 eukaryotes, we were able to link multiple genes with particular disease phenotype descriptions. We validated experimentally our identified links between transcription factor RBP-Jk and a master regulator of melanocyte differentiation MITF and the suggested role of ccdc105 as a novel meiosis-specific chromatin localization gene.
The genome sequence of an organism reveals important characters of its habitat, organelles, and biochemical pathways that are maintained or lost compared with other genomes. For example, parasites that have lost their heme biosynthesis genes live in a heme-rich environment (for example, the bloodstream). The loss of pathways or genes can highlight the vulnerabilities of these organisms and offer potential therapeutic strategies (Rao et al, 2005). Many primates have lost their ability to synthesize vitamins now found in their diet. Other pathways identified by phylogenetic profile analysis can identify analogous vitamins and nutrients that might be important to human health.
This analysis also highlights hundreds of genes (based on their phylogenetic profile) as priority candidates to test for association with particular pathways or diseases (Supplementary Tables S5 and S6). Phylogenetic profile analysis provides a dictionary of biological gene sets and is more cost-effective and robust than other genome-scale analysis methods such as mRNA expression, proteomics, and RNAi screens, which are labor intensive, costly, and dependent on experimental conditions. Nevertheless, the integration of the coevolved clusters with ∼10 000 functional and disease groups offers us a powerful way to find evolutionary and functional interactions between different diseases and pathways (Figure 6) (Lee et al, 2011).
Phylogenetic profiling has the potential to reveal the placements of hundreds of genes in protein complexes, metabolic networks, and cellular processes. From a clinical perspective, these opportunities to connect different diseases or symptoms based on similar phylogenetic profiles have a great potential to provide better understanding of human diseases by highlighting those genes that cluster with known causes of diseases. As we enter the stage of human genetics when thousands of patients with a wide range of diseases will be sequenced and compared with non-disease expressing patients, there will be millions of gene variants revealed. If some of the mutations detected in patients with particular diseases map to loci with similar phylogenetic profiles as the small subset of loci known to cause this disease, then the problem of assigning genetic causes to such diseases may be simplified.
Materials and methods
Species database generation
Protein-coding sequences for human genes were downloaded using BioMart version 0.7 from the Ensembl project (release 60). Ensembl includes both automatic annotation, in which transcripts are determined and annotated genome-wide by automated bioinformatic methods, and manual curation. When different splice variants existed for a gene, the longest variant was used. The resulting 19 017 protein-coding genes of human were compared using Blastp to all open reading frames (ORFs) of 85 organisms (Supplementary Table S1). From the existing genomes available in the Ensembl database (release 60), we obtained a set of 53 fully sequenced eukaryotic genomes that represent most eukaryotic clades with sequenced genomes. Since Ensembl has a limited number of fungi and protists, 33 additional high quality genomes form the NCBI genome database were added to supplement the analysis. To get the most analytical power while at the same time avoiding degradation in data quality due to incorrect or incomplete genome annotation, we tried to use all available high-quality genomic data across the eukaryotic tree of life, while applying certain filters to remove poorly annotated genomes. The uses of nearly 90 high quality genomes reduced the effect of occasional errors on the correlation between the gene. As an additional quality control, we calculated the correlation of the protein conservation among species. We removed several species that showed low correlation with their closely related species, like Nasonia vitripennis or Equus caballus, since low correlation might reflect problems in the genome assembly or annotation. Finally, the expected effect of random noise in our data is to reduce rather than increase clustering, and as such, we believe that random noise in general does not increase the likelihood chance of having false positives. Hence, although noise in genomic annotation data may weaken the significance of some coevolved clusters, genomes with moderate noise in their annotations can nevertheless contribute overall to the significance of our phylogenetic clusters while at the same time introducing little risk of predicting false associations.
Preprocessing and clustering the phylogenetic profile
The Blastp comparison generates a Protein-Protein Best Hit Matrix of size 19 017 × 86 (85 organisms plus human) where each entry Pab is the best Blastp bit score between a human gene ‘a’ and the top hit in organism ‘b’. Preprocessing and normalization were applied to the profile matrix before clustering or test for correlation between proteins profile as we previously described (Tabach et al, 2013). The result is a NPP matrix NPP that gives different organisms of similar weight, independently of their global evolutionary relation to humans. The normalized matrix NPP was used for the correlation tests and the clustering process.
Calculation of the list of most correlated genes (List10)
A Pearson correlation coefficient (R) was calculated using the NPP matrix to generate a correlation matrix. High correlation can be the result of coevolution or a by-product of homology between gene sequences and in the later only corresponds to paralogous genes. To remove phylogenetic profile correlation scores that resulted from homology between the sequences of two human genes Gi to Gj, we assigned R=0 if the Blastp score was >100 or if the sequence identity between Gi and Gj genes was higher than 10%. List10 for Gi includes the 10 genes without a significant sequence homology that were most correlated with the particular gene.
Calculation of Co10 scores
To test whether sets of functional annotated genes are significantly coevolved, we calculated a Coevolution (Co10) score. We determined for each gene the 10 non-homologous genes (the 10 nearest neighbors) that are most phylogenetically correlated with it (List10—see Materials and methods). We also tested 20, 50, and 100 nearest neighbors and this analysis yielded similar results (data not shown). Then for a group of genes in, we counted the number of times these genes were found in each other’s 10 nearest neighbors in the phylogenetic profile. High Co10 scores indicate genes in a set that share similar phylogenetic profiles across the species that were examined. To evaluate the significance of the Co10 score for each functional set, we generated 1 000 000 random sets of the same size as the curated set and calculated Co10 scores for all the random sets. A P-value of <1 × 10−6 was obtained if the score of the curated sets was higher than any of the scores of the 1 000 000 random sets with similar size. To avoid biases inherited from the databases that were used (HPO and MSigDB), the random sets were generated only from the genes found in the database that was tested (i.e., MSigDB or HPO database). To control for the number of false discoveries found in the MSigDB and the HPO database, we applied FDR procedure and calculated q-values. The q-value is similar to the well-known P value, except it is a measure of significance in terms of the FDR rather than the false positive rate. For example, an FDR adjusted P-value (or q-value) of 0.05 implies up to a limit of 5% false positive results.
Generation of binary phylogenetic profile and NPP with different organism sets
To test for the effect of different numbers of species on the performance of phylogenetic profiling, we resampled our data using 75, 50, or 25% of our original species list. To keep similar phylogenetic representation of the organisms that were used, we chose organisms from the entire eukaryotic tree. From the entire list of organisms found in Supplementary Table S1, we removed every fourth organism to generate the 75% list (indices 1, 2, 3, 5, 6, 7, 9…). The 50% list was generated by removing every second organism (indices 1, 3, 5…). The 25% list was generated by inclusion of every fourth organism (indices 1, 5, 9, 13…).
Generation of coevolved gene clusters
For each protein A, we ranked the top 50 most correlated genes to it, using Pearson’s correlation coefficient (R) on the NPP matrix. The most correlated protein to A received a rank score of 50 and the others the score of 49, 48, …, 1. The 50th protein got the rank score of one. The other genes got the rank score of zero. Since the rankings are asymmetric (i.e., Rank A to B is not necessary identical to the rank B to A), a ranking score between two genes (ranksocreAB) was calculated.
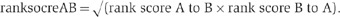 |
We generated a ‘distance’ matrix M to define the correlation rand between two genes A and B, such that each entry Mab=ranksocreAB. Using the MATLAB environment, we clustered the data using the following code:
Z=linkage(DistanceMatrix, 'weighted', 'Euclidean');
C=cluster(Z, 'cutoff', 2);
this yield 1076 phylogenetic clusters with 3–193 genes.
Calculation of functional group enrichment in coevolved gene clusters.
The significance of the overlap between the coevolved gene clusters and the groups obtained from MSigDB and HPO database was calculated using a hypergeometric test. To correct for multiple comparisons, we applied an FDR procedure and calculated q-values. We reported (Supplementary Table S6) only the significant overlap with q-value <0.05. A similar phylogeny can be resulted from sequence homology; to remove this effect, we calculated the hypergeometric considering only one protein per family of protein homologs (e.g., a group of 10 proteins, 5 of which have a sequence similarity, would be considered to have a size of 6). Similarly, if the overlap between the functional gene group (HPO or MSigDB) and coevolved protein cluster contains homologous proteins, then we considered only one protein per homolog family. Proteins are considered to be in the same family if they have a blast score of >100 or sequence identity larger than 10%. In Supplementary Table S6, we reported only the functional groups that have overlap with coevolved clusters of at least three protein families.
MSigDB and HPO database
The Molecular Signature Database (MSigDB v3.0) contains 6800 gene sets collected from various sources such as online pathway databases (KEGG, BIOcharta), Gene Ontology (GO groups), publications in PubMed and genes that share cis-regulatory motifs or are coexpressed. We used the 6594 sets with fewer than 500 genes. From each MSigDB set, we removed the genes that were not annotated. The remaining genes in each set were used to calculate a coevolution score and the related P-value. The number of genes in each set after removing the annotated genes was considered as the effective group size. The HPO database was downloaded from http://www.human-phenotype-ontology.org/contao/index.php/downloads.html in August 2012. To calculate the probability that genes in a HPO group have similar phylogenetic profiles to each other, we used a similar procedure as described for the MSigDB.
Plasmids
pcDNA3-MITF and PGL4.11-TRPM1 promoter luciferase were described in previous publications. pSG5-RBP-Jk was kindly provided by Dr E Manet (INSERM U758, Unité de Virologie humaine, Lyon, France).
Cell cultures, transfections, and luciferase reporter assays
Human WM3526, WM3682, and WM3314 melanoma cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. Cells were transfected with jetPEI™ for plasmids or Hiperfect (QIAGEN) for the siRNAs targeting MITF (40 nm) or RBP-Jk (10 nm) according to the manufacturer’s instructions. For luciferase assays, cells were grown in 12-well dishes until 60–70% confluence and then were transfected with 0.3 μg pGL4.11-TRPM1 promoter Luciferase, 0.01 μg Renilla luciferase reporter (Promega) and RBP-Jk or MITF expression vectors as indicated in figure legends. The total amount of DNA was adjusted to equal amounts with empty vector. At 48 h after transfection, luciferase levels were measured using the dual luciferase assay kit (Promega), and the firefly luciferase activity was normalized to Renilla luciferase activity. Data are presented as mean values±standard deviation (s.d.) for at least three independent experiments done in duplicate relative to the level of luciferase activity obtained in the presence of empty vector.
Coimmunoprecipitation and immunoblotting analysis
For coimmunoprecipitation analyses, cells were solubilized in lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 0.2% Nonidet P-40), and extracts were clarified by centrifugation at 12 000 g for 30 min at 4°C. Total cell lysate (2 mg) was incubated with the specific antibody for 18 h at 4°C and then incubated, with rotation, with protein-A beads (Santa Cruz Biotechnology) for 2 h at 4°C. Beads were collected by centrifugation, washed three times in lysis buffer, and dissolved in Laemmli buffer. Following SDS–PAGE, proteins were transferred onto nitrocellulose membranes, and after blocking with 5% low-fat milk, filters were incubated with the specific primary antibody. Anti-RBP-Jk polyclonal antibodies were purchased from Cell Signaling and anti-MITF monoclonal antibody was kindly provided by Dr David Fisher (CBRC, MGH, Harvard University, Boston, MA, USA). Membranes were washed in 0.001% Tween-20 in phosphate-buffered saline (PBS) and incubated for 45 min with a secondary antibody. After washing in Tween/PBS, membranes were subjected to enhanced chemiluminescence (ECL) detection analysis (Amersham Biosciences) using horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology).
RNA purification and RT–PCR
Total RNA was purified using Trizol (Invitrogen) according to the manufacturer’s instructions followed by treatment with RNase-free DNase (Qiagen). For mRNA analysis, 100 ng RNA was subjected to one-step RT–PCR using a QuantiTect RT-PCR kit (Qiagen) and SYBRgreen Supermix (Roche). Primer sequences are listed in Supplementary Table S6.
Chromatin immunoprecipitation
ChIP assays were performed with melanoma cells grown to logarithmic phase. Cells were subjected to 1% formaldehyde in PBS for 20 min at room temperature with gentle shaking. Cells were then harvested by scraping and homogenized in hypotonic buffer on ice using a Dounce homogenizer. The nuclei were isolated by centrifugation over a 10% sucrose pad. Nuclei were then spun down, resuspended in ChIPs buffer and sonicated. Antibodies to RNA polymerase II CTD4H8 (Covance), MITF, RBP-Jk (AbCam), or Placental Protein 4 (Assay Designs), as a non-specific control antibody, were added to a 10-fold ChIPs buffer diluted sample and incubated on a rotator for 10 h at 4°C. Ultralink protein-A/G-beads (Pierce) were added to the sample and a control sample and incubated for an additional hour at room temperature. Immunoprecipitates were then washed twice with ChIPs buffer, twice with 500 mM NaCl ChIPs buffer, and once with TE (pH 8). The immunoprecipitates were released from the beads by incubating at 65°C for 20 min in 1% SDS/TE and treated with proteinase K side by side with an unprecipitated sample as an input control. Crosslinks were released by heating at 70°C for 10 h, and DNA was recovered by extraction with phenol and chloroform in high salt buffer (0.6 M sodium acetate, pH 8) and then ethanol precipitated. qRT–PCR was performed to amplify fragments occupied by RBP-Jk, Pol-II, or MITF. Buffers compositions were described previously. PCR primers spanning the TRPM1 promoter were employed as indicated: MITF binding sites (Eboxes 1, 2, 3), intron 1 promoter region (int1 A, B) and RBP-Jk binding sites (Rbox) region. RBP-Jk occupancy was calculated relative to cells transfected with control siRNA (siCont), normalized to input and represent mean±s.d. of three independent experiments. HES5 promoter primers were used as a positive control for RBP-Jk. For negative controls, we used TRPM1 downstream primers and performed ChIP with control antibody (Supplementary Figure S3b).
Promoter binding site analysis
A list of MITF target genes and promoter sequences of 2000 nucleotides were downloaded from ensembl.org. Each promoter contained one or more of the following MITF E-box sites: CATGTG, CACATG, CACGCG, and CACGTC. Each promoter was searched for one of the following RBP-Jk binding sites: TTCCCAC, ATGGGAG, TTCCCAG, and TGGGAAT. Each MITF target gene listed in Supplementary Table S8 was labeled with a ‘yes’ if an RBP-Jk site was found, and ‘no’ if not. The significance was calculated under the parameters that there exists an MITF e-box of length 6 in each promoter of length 2000, each nucleotide has an equal chance to be a, t, g, or c and that every possible position for an RBP-Jk binding site of length 7 are statistically independent of each other. The formula q calculates the probability that one site of length R (RBP-JK binding site nucleotide length) exists in a sequence of 2000 nucleotides with one MITF site of length M (MITF binding site E-Box length). The formula s calculates the probability that q occurs in 10 out of 13 MITF target genes.
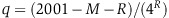 |
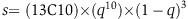 |
Phylogenetic profiling of meiotic genes
Proteins that phylogenetic clustered together with one or more of the meiosis-specific genes (synaptonemal complex genes), Sycp3, Sycp2, Rec8, Sycp1, Syce1, Syce2, Syce3, and Tex12 were predicted to function in meiosis. We tested these genes in mouse. For more accurate predictions (although results were similar), we generated a phylogenetic profile of mouse proteins relative to the other 85 eukaryotes (Supplementary Figure S5a). Candidate meiotic-specific genes were selected if: (1) the candidate gene was not annotated to function in meiosis or any other cellular process and (2) tissue expression profiles were testes and/or ovary specific. We selected five genes that satisfied these criteria. Using databases that report the mRNA expression levels in testicle and/or oocytes, we found that genes 1700040L02Rik, 4930503L19Rik, 2010007H12Rik, Ccdc105, and Cenpw are expressed in testicle and no function has been annotated for these genes (except for Cenpw). To corroborate testes- and or ovary-specific expression, RT–PCR assays were performed using a set of cDNAs from several mouse tissues using specific primers to amplify the cDNA of each of the five selected genes (Supplementary Figure S5c). Specific antibodies were raised against two short peptides of CCDC105 (peptide 1: amino acids 7–30 and peptide 3: amino acids 473–499). Spreading of meiotic cells and immunoblotting, and immunolabeling of CCDC105 were done as described before.
Supplementary Material
Acknowledgments
We thank Jonathan Urbach, Sunil Wadhwa, and the Ruvkun laboratory members for the comments and suggestions that have been helpful to improve the manuscript. YT and GR supported by NIH R01GM44619. CL thanks Meenhard Herlyn and Levi Garraway for supplying melanoma short-term cultures. TG and CL supported by grants from the Israeli Centers for Research Excellence (I-CORE), Israel Cancer Association (ICA), Israel Cancer Research Fund (ICRF), US-Israel Binational Science Fund (BSF), Fritz Thyssen Stiftung, Marie Curie Career Integration Grants (CIG), German Cancer Research Center (DKFZ).
Author contributions: YT, CL, and GR designed experiments and supervised the project; YT, AH-H, CL, and GR wrote the paper. YT wrote the code. TG, ARM, and CL carried out the RBP-JK experiments. AH-H, TF, AK, J-GL, and IL carried out the ccdc105 experiments.
References
- Adler P, Kolde R, Kull M, Tkachenko A, Peterson H, Reimand J, Vilo J (2009) Mining for coexpression across hundreds of datasets using novel rank aggregation and visualization methods. Genome Biol 10: R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal P, Yu K, Salomon AR, Sedivy JM (2010) Proteomic profiling of Myc-associated proteins. Cell Cycle 9: 4908–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002) Molecular Biology of the Cell, 4th edn. Garland Science, New York, USA
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin-Houzelstein G, Djian-Zaouche J, Bernex F, Gadin S, Delmas V, Larue L, Panthier JJ (2008) Melanoblasts' proper location and timed differentiation depend on Notch/RBP-J signaling in postnatal hair follicles. J Invest Dermatol 128: 2686–2695 [DOI] [PubMed] [Google Scholar]
- Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS (2004) Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 117: 527–539 [DOI] [PubMed] [Google Scholar]
- Beres TM, Masui T, Swift GH, Shi L, Henke RM, MacDonald RJ (2006) PTF1 is an organ-specific and Notch-independent basic helix-loop-helix complex containing the mammalian Suppressor of Hairless (RBP-J) or its paralogue, RBP-L. Mol Cell Biol 26: 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentani H, Caballero OL, Camargo AA, da Silva AM, da Silva WA Jr, Dias Neto E, Grivet M, Gruber A, Guimaraes PE, Hide W, Iseli C, Jongeneel CV, Kelso J, Nagai MA, Ojopi EP, Osorio EC, Reis EM, Riggins GJ, Simpson AJ, de Souza S et al. (2003) The generation and utilization of a cancer-oriented representation of the human transcriptome by using expressed sequence tags. Proc Natl Acad Sci USA 100: 13418–13423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Weissman JS (2010) Orm family proteins mediate sphingolipid homeostasis. Nature 463: 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Y, Cooke HJ (2007) Dissecting the mammalian synaptonemal complex using targeted mutations. Chromosome Res 15: 579–589 [DOI] [PubMed] [Google Scholar]
- Eisen JA, Wu M (2002) Phylogenetic analysis and gene functional predictions: phylogenomics in action. Theor Popul Biol 61: 481–487 [DOI] [PubMed] [Google Scholar]
- Enault F, Suhre K, Abergel C, Poirot O, Claverie JM (2003) Annotation of bacterial genomes using improved phylogenomic profiles. Bioinformatics 19(Suppl 1): i105–i107 [DOI] [PubMed] [Google Scholar]
- Enault F, Suhre K, Poirot O, Abergel C, Claverie JM (2004) Phydbac2: improved inference of gene function using interactive phylogenomic profiling and chromosomal location analysis. Nucleic Acids Res 32: W336–W339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraune J, Schramm S, Alsheimer M, Benavente R (2012) The mammalian synaptonemal complex: protein components, assembly and role in meiotic recombination. Exp Cell Res 318: 1340–1346 [DOI] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, Golub TR, Rimm DL, Meyerson ML, Fisher DE, Sellers WR (2005) Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436: 117–122 [DOI] [PubMed] [Google Scholar]
- Gok F, Ichida K, Topaloglu R (2003) Mutational analysis of the xanthine dehydrogenase gene in a Turkish family with autosomal recessive classical xanthinuria. Nephrol Dial Transplant 18: 2278–2283 [DOI] [PubMed] [Google Scholar]
- Goodsell DS (1999) The molecular perspective: the ras oncogene. Stem Cells 17: 235–236 [DOI] [PubMed] [Google Scholar]
- Handel MA, Schimenti JC (2010) Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nature reviews Genetics 11: 124–136 [DOI] [PubMed] [Google Scholar]
- Hodges ME, Wickstead B, Gull K, Langdale JA (2012) The evolution of land plant cilia. New Phytol 195: 526–540 [DOI] [PubMed] [Google Scholar]
- Introne WJ, Gahl WA (1993–2013) Alkaptonuria. In GeneReviews™ [Internet], Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K (eds). Seattle, WA: University of Washington
- Jiang Z (2008) Protein function predictions based on the phylogenetic profile method. Crit Rev Biotechnol 28: 233–238 [DOI] [PubMed] [Google Scholar]
- Khaitovich P, Enard W, Lachmann M, Paabo S (2006) Evolution of primate gene expression. Nat Rev Genet 7: 693–702 [DOI] [PubMed] [Google Scholar]
- Kim Y, Subramaniam S (2006) Locally defined protein phylogenetic profiles reveal previously missed protein interactions and functional relationships. Proteins 62: 1115–1124 [DOI] [PubMed] [Google Scholar]
- Ko S, Lee H (2009) Integrative approaches to the prediction of protein functions based on the feature selection. BMC Bioinformatics 10: 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Blom UM, Wang PI, Shim JE, Marcotte EM (2011) Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res 21: 1109–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy C, Khaled M, Fisher DE (2006) MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med 12: 406–414 [DOI] [PubMed] [Google Scholar]
- Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D (2002) The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev 16: 1977–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443: 787–795 [DOI] [PubMed] [Google Scholar]
- Loganantharaj R, Atwi M (2007) Towards validating the hypothesis of phylogenetic profiling. BMC Bioinformatics 8(Suppl 7): S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EM, Xenarios I, van Der Bliek AM, Eisenberg D (2000) Localizing proteins in the cell from their phylogenetic profiles. Proc Natl Acad Sci USA 97: 12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, Kanapin A, Lewis S, Mahajan S, May B, Schmidt E, Vastrik I, Wu G, Birney E, Stein L, D'Eustachio P (2009) Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res 37: D619–D622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Fu X, Chen X, Zeng S, Tian Y, Jove R, Xu R, Huang W (2010) miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology 52: 2148–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Marechal-Drouard L, Marshall WF, Qu LH, Nelson DR, Sanderfoot AA, Spalding MH, Kapitonov VV, Ren Q, Ferris P, Lindquist E, Shapiro H et al. (2007) The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AJ, Du J, Rowan S, Hershey CL, Widlund HR, Fisher DE (2004) Transcriptional regulation of the melanoma prognostic marker melastatin (TRPM1) by MITF in melanocytes and melanoma. Cancer Res 64: 509–516 [DOI] [PubMed] [Google Scholar]
- Milyavsky M, Tabach Y, Shats I, Erez N, Cohen Y, Tang X, Kalis M, Kogan I, Buganim Y, Goldfinger N, Ginsberg D, Harris CC, Domany E, Rotter V (2005) Transcriptional programs following genetic alterations in p53, INK4A, and H-Ras genes along defined stages of malignant transformation. Cancer Res 65: 4530–4543 [DOI] [PubMed] [Google Scholar]
- Munnich A (2008) Casting an eye on the Krebs cycle. Nat Genet 40: 1148–1149 [DOI] [PubMed] [Google Scholar]
- Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B, Braun T, Casavant T, Stone EM, Sheffield VC (2004) Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci USA 101: 8664–8669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata J, Yano M, Mimura H, Goto T, Nakayama R, Mibu Y, Oka C, Kawaichi M (2001) p48 subunit of mouse PTF1 binds to RBP-Jkappa/CBF-1, the intracellular mediator of Notch signalling, and is expressed in the neural tube of early stage embryos. Genes Cells 6: 345–360 [DOI] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27: 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M (2012) Using phylogenetic profiles to predict functional relationships. Methods Mol Biol 804: 167–177 [DOI] [PubMed] [Google Scholar]
- Rao AU, Carta LK, Lesuisse E, Hamza I (2005) Lack of heme synthesis in a free-living eukaryote. Proc Natl Acad Sci USA 102: 4270–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Mundlos S (2010) The human phenotype ontology. Clin Genet 77: 525–534 [DOI] [PubMed] [Google Scholar]
- Ruano-Rubio V, Poch O, Thompson JD (2009) Comparison of eukaryotic phylogenetic profiling approaches using species tree aware methods. BMC Bioinformatics 10: 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouwey K, Aydin IT, Radtke F, Beermann F (2011) RBP-Jkappa-dependent Notch signaling enhances retinal pigment epithelial cell proliferation in transgenic mice. Oncogene 30: 313–322 [DOI] [PubMed] [Google Scholar]
- Schramm S, Fraune J, Naumann R, Hernandez-Hernandez A, Hoog C, Cooke HJ, Alsheimer M, Benavente R (2011) A novel mouse synaptonemal complex protein is essential for loading of central element proteins, recombination, and fertility. PLoS Genet 7: e1002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Friedman N, Koller D, Regev A (2004) A module map showing conditional activity of expression modules in cancer. Nat Genet 36: 1090–1098 [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Copeland NG, Jenkins NA (2004) Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet 38: 365–411 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci US A 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Xu J, Liu Z, Liu Q, Zhao A, Shi T, Li Y (2005) Refined phylogenetic profiles method for predicting protein-protein interactions. Bioinformatics 21: 3409–3415 [DOI] [PubMed] [Google Scholar]
- Tabach Y, Billi AC, Hayes GD, Newman MA, Zuk O, Gabel H, Kamath R, Yacoby K, Chapman B, Garcia SM, Borowsky M, Kim JK, Ruvkun G (2013) Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature 493: 694–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabach Y, Milyavsky M, Shats I, Brosh R, Zuk O, Yitzhaky A, Mantovani R, Domany E, Rotter V, Pilpel Y (2005) The promoters of human cell cycle genes integrate signals from two tumor suppressive pathways during cellular transformation. Mol Syst Biol 1: 2005.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma N, Sheng HZ, Furuta Y, Ward JM, Sharma K, Hogan BL, Pfaff SL, Westphal H, Kimura S, Mahon KA (1998) Formation of Rathke's pouch requires dual induction from the diencephalon. Development 125: 4835–4840 [DOI] [PubMed] [Google Scholar]
- Takwi A, Li Y (2009) The p53 Pathway Encounters the MicroRNA World. Curr Genomics 10: 194–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigaki K, Honjo T (2010) Two opposing roles of RBP-J in Notch signaling. Curr Top Dev Biol 92: 231–252 [DOI] [PubMed] [Google Scholar]
- Tovar J, Leon-Avila G, Sanchez LB, Sutak R, Tachezy J, van der Giezen M, Hernandez M, Muller M, Lucocq JM (2003) Mitochondrial remnant organelles of Giardia function in iron-sulphur protein maturation. Nature 426: 172–176 [DOI] [PubMed] [Google Scholar]
- Wahl B, Reichmann D, Niks D, Krompholz N, Havemeyer A, Clement B, Messerschmidt T, Rothkegel M, Biester H, Hille R, Mendel RR, Bittner F (2010) Biochemical and spectroscopic characterization of the human mitochondrial amidoxime reducing components hmARC-1 and hmARC-2 suggests the existence of a new molybdenum enzyme family in eukaryotes. J Biol Chem 285: 37847–37859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M (2005) Systematic discovery of regulatory motifs in human promoters and 3' UTRs by comparison of several mammals. Nature 434: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.