

Abstract
A new rhodium(I) catalyst allows practical and efficient alkyne carbothiolation reactions to be achieved on synthetically useful ketone-bearing aryl methyl sulfides. The carbothiolation adducts, featuring a ‘recycled methyl sulfide’ activating group, are convenient precursors to highly substituted isoquinolines.
Aromatic heterocycles represent the dominant structural motif in medicinal chemistry. Among the varied range of architectures found within this group, isoquinolines play an important role. In addition to forming the foundation of many medicines (Figure 1), the isoquinoline core, and derivatives, are found in many natural products and in organic materials.1 The majority of “classic” routes to isoquinolines—the Pictet–Spengler, Bischler–Napieralski, and Pomeranz–Fritsch syntheses—are in general centered on an electrophilic substitution process as the key bond-forming event.2 Despite the success of these protocols, reliance on electrophilic aromatic substitution imposes significant limitations on the substitution pattern that can be tolerated on the starting arene, and also on the substitution patterns that can be accessed, due to the inherent electronic bias of this class of transformations. In this Letter we disclose a new Rh-catalyst for alkyne carbothiolation reactions and apply this process as the key step in a practical, one-pot route to isoquinolines. This new synthesis provides a distinct disconnection to these valuable heterocycles, employs simple building blocks, allows access to a variety of substitution patterns, and is performed under mild reaction conditions. It also validates the utility of our recently reported “functional group recycling” approach to arene functionalization.3
Figure 1.

Representative biologically active isoquinolines.
Transition metal catalysis has transformed the synthesis of aromatic heterocycles, providing a set of nontraditional disconnections that complement the many classic approaches to these molecules.4 In particular, cross-coupling processes have opened up new classes of building blocks for heterocycle synthesis.5 The majority of cross-coupling methods rely on an activating group to secure reactivity and to control regioselectivity; an intrinsic feature of these reactions is that at the completion of the transformation the activating group is usually discarded as waste. For example, aryl halides (Ar–X) when combined with an organometallic coupling partner (M–R) deliver (M–X) salts as waste products. Coupling reactions that rely on addition processes, as opposed to substitution reactions, allow the activating group to be reincorporated into the product; effectively, the activating groups are recycled.3,6 The ability to recycle activating groups, and in the process provide a handle for further manipulation, begins to address the demand to produce more sustainable synthetic routes.
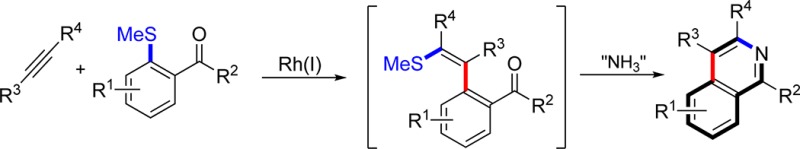
We have recently demonstrated an effective activating group recycling approach to arene functionalization based on Rh-catalyzed alkyne carbothiolation.3 In this system, simple aryl methyl sulfides are combined with alkynes to deliver alkenyl sulfide products resulting from both C–C and C–S bond formation. Crucially, the alkenyl sulfide functionality incorporated in the products represents a masked carbonyl unit, and as such provides a powerful handle for further functionalization.7 Wanting to exploit the utility of this masked carbonyl unit we conceived a new route to substituted isoquinolines.8 The key transformation involves the Rh-catalyzed carbothiolation of alkynes with carbonyl-containing aryl methyl sulfides, leading to the formation of a masked benzo-fused 1,5-dicarbonyl (1 + 2 → 3, Scheme 1); treatment of dicarbonyl 3 with an ammonia source would allow access to isoquinolines (4).
Scheme 1. A Rh-Catalyzed Alkyne Carbothiolation Route to Isoquinolines.

Our initial report of a Rh-catalyzed alkyne carbothiolation reaction employed the [Rh(DPEphos)(o-xylene)][BArF4] complex A as the precatalyst, used in o-xylene.3 Although complex A represents an efficient catalyst system, the use of the BArF4 counterion makes this catalyst relatively expensive and also difficult to prepare without the use of a glovebox.9
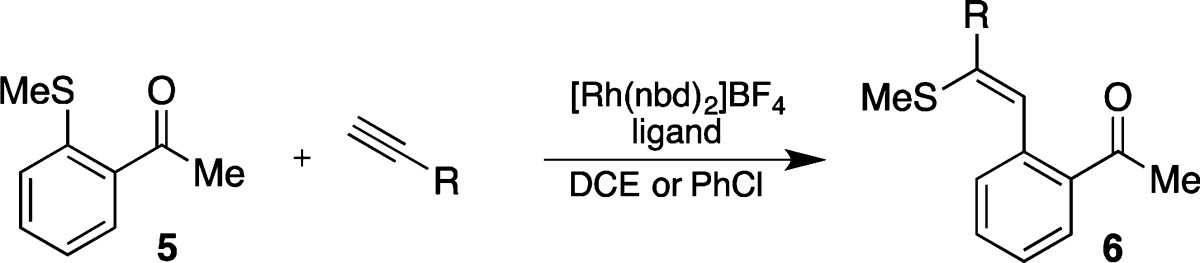
In order to deliver a practical solution, we therefore sought to identify a new catalyst system which retained the activity of complex A, was straightforward to prepare, and avoided use of the BArF4 anion. We began by employing the commercially available precursor [Rh(nbd)2]BF4, which can be employed routinely without recourse to a glovebox and allowed the active catalyst to be generated in situ by addition of the phosphine ligand and subsequent hydrogenation. Due to the significant decrease in catalyst solubility when moving from the BArF4 to BF4 counterions, a new solvent system was established (Table 1). After evaluating a number of solvents, the desired carbothiolation reaction was achieved in good yields with a selection of terminal alkynes and methyl sulfide 5 using 5 mol % [Rh(nbd)2BF4] and 5 mol % DPEphos, in either DCE or chlorobenzene (Table 1, entries 1–4).
Table 1. Use of the BF4 Anion in Rh-Catalyzed Alkyne Carbothiolationa.
| entry | R | ligand | time | yield (DCE, PhCl)b |
|---|---|---|---|---|
| 1 | Ph | DPEphos | 24 h | 80%, 90% |
| 2 | 4-MeO-Ph | DPEphos | 24 h | 73%, 90% |
| 3 | 4-F-Ph | DPEphos | 24 h | 70%, 80% |
| 4 | 3,5-(CF3)2-Ph | DPEphos | 24 h | 24%, 33%c |
| 5 | Ph | Xantphos | 2 h | 99%, 99% |
| 6 | 4-MeO-Ph | Xantphos | 2 h | 96%, 99% |
| 7 | 4-F-Ph | Xantphos | 1.5 h | 99%, 99% |
| 8 | 3,5-(CF3)2-Ph | Xantphos | 24 h | 43%,c 80% |
| 9 | CH2N(Boc)Bn | Xantphos | 1.5 h | 81%, – |
Reaction conditions: 1 (1 equiv), alkyne (1.5 equiv), Rh(nbd)2BF4 (5 mol %), ligand (5 mol %), solvent (0.3 M), 80 °C (DCE), 100 °C (chlorobenzene).
Isolated yields.
Conversion determined by 1H NMR spectroscopy.
The change from a BArF4 anion to BF4 resulted in a lower activity catalyst, and in an attempt to achieve faster catalysis we next focused on the structure of the ligand. The flexibility and hemilabile nature of the DPEphos ligand allows it to adopt a number of coordination modes (Scheme 2). We have previously observed that the reaction of complex A with sulfide 5 results in an equilibrium mixture of A with complexes B and C in a 0.1:1:0.1 ratio (o-xylened10 298 K). We speculated that by locking the ligand into a single, active conformation, the efficiency of the reaction might be improved. To impede this flexibility around the P–O–P bonds present in DPEphos, we explored the use of a rigid DPEphos analogue, XantPhos.10 Pleasingly, when evaluated in the carbothiolation of phenylacetylene, the reaction employing Xantphos achieved completion in only 2 h (cf. 24 h with DPEphos; Table 1, entries 1 and 5). The carbothiolation of both electron-rich and electron-poor alkynes was performed with quantitative conversion to the carbothiolated product (Table 1, entries 6 and 7).
Scheme 2. Comparison of DPEphos and Xantphos Rh-Complexes.

As Xantphos is analogous to the mer-isomer of a DPEphos complex (B), the improved reactivity with Xantphos might suggest that B is the active species in carbothiolation when complex A is employed.11 The catalytic activities of preformed and in situ prepared Xantphos- and DPEphos-containing catalysts were compared by monitoring the reactions by HPLC (Figure 2). Preformed catalysts containing the BArF4 counterion showed higher rates of conversion relative to the BF4 catalysts. Nevertheless, the [Rh(Xantphos)(nbd)][BF4] complex formed in situ showed exceptional activity with full conversion to the product achieved in less than 2 h.
Figure 2.
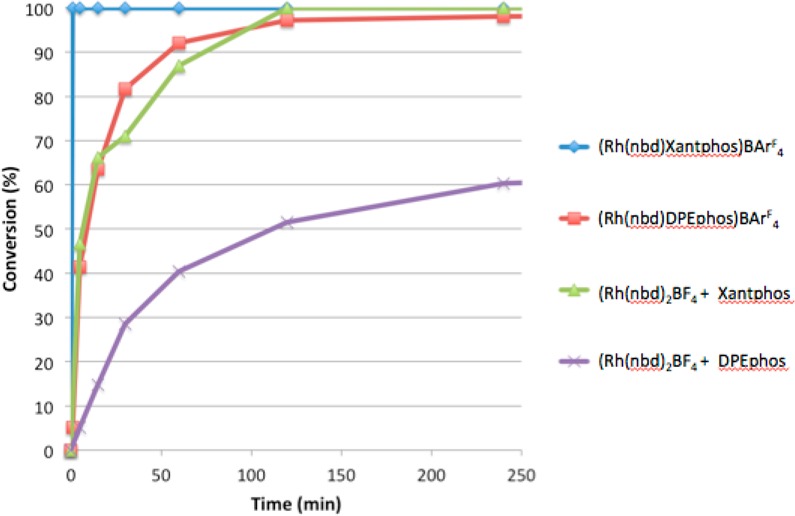
Time course plot for the carbothiolation reaction using DPEphos and Xantphos ligands. Reaction conditions: Rh(nbd)2BF4 (5 mol %), ligand (5 mol %), DCE (0.3 M), H2 (2 min); 5 (1 equiv), phenylacetylene (1.5 equiv), 80 °C.
Having established an efficient and practical catalyst for the carbothiolation step, the next task was to address the subsequent cyclization to form the isoquinoline structure.12 This could be achieved by simply adding NH4OAc and acetic acid directly to the reaction upon completion of the carbothiolation reaction. Heating the reaction mixture at 110 °C led to the isolation of isoquinoline 7a in 90% yield (Figure 3). To investigate the scope of this reaction a range of alkynes were explored. Maintaining sulfide 5 as the standard, a variety of substituents could be employed on the alkyne, including electron-donating (7b) and electron-withdrawing groups (7c). It was also pleasing to note that an aryl bromide substituent remained intact during the reaction, providing a useful handle for further elaboration (7d).
Figure 3.

Rh-catalyzed alkyne carbothiolation-based isoquinoline synthesis: scope of the alkyne component. Conditions: Rh(nbd)2BF4 (5 mol %), Xantphos (5 mol %), PhCl (0.3 M), H2 (2 min); 5 (1 equiv), alkyne (1.5 equiv), 100 °C, then NH4OAc, AcOH, 110 °C.
Heterocyclic substituents, such as thiophene, could be carried through both reactions to generate the biheteroaryl product 7e in good yield. Aliphatic alkynes were also excellent substrates for this reaction, with linear (7f) and cyclic (7g) aliphatic groups efficiently incorporated into the product. Additionally, a ferrocene unit could be installed without incident (7h). Using the newly developed catalyst system, the process was not limited to the use of terminal alkynes; for example, employing 3-hexyne allowed isoquinoline 7i to be isolated in 45% yield, while diphenyl acetylene allowed the formation of 7j. Although the use of an unsymmetrical internal alkyne was successful, a mixture of regioisomers was obtained (7k, 7k′).
The reaction could also be performed at a reduced catalyst loading with no decrease in the yield; employing phenylacetylene as the alkyne component, the reaction was performed on 1.2 mmol scale using 1 mol % Rh, to deliver the isoquinoline product 7a in 93% yield.
We next examined the scope of the substitution possible on the sulfide precursors and began by exploring variation on the carbocyclic ring (Figure 4). Both electron-donating (7l) and electron-withdrawing groups (7m) were tolerated. Isoquinoline 7n containing an −SMe group positioned para to the acyl tether remained untouched during the one-pot synthesis, demonstrating the high levels of regiocontrol possible. Again, an aryl bromide, this time positioned para to the ketone (7o), remained intact throughout the reaction. Variation of the substitution adjacent to the carbonyl group was also possible, including longer aliphatic ketones (7l–n), cyclopropyl (7p) and cyclohexyl ketones (7q), and an α,β-unsaturated ketone (7r). Good functional group tolerance was also displayed by the incorporation of a pendant oxindole (7s).
Figure 4.

Rh-catalyzed carbothiolation based isoquinoline synthesis: scope of the sulfide component. Conditions: Rh(nbd)2BF4 (5 mol %), Xantphos (5 mol %), PhCl (0.3 M), H2 (2 min); sulfide (1 equiv), phenylacetylene (1.5 equiv), 100 °C, then NH4OAc, AcOH, 110 °C.
To demonstrate the utility of the developed method we undertook the preparation of a simple derivative of the phosphodiester inhibitor moxaverine (Scheme 3). Three simple steps from commercially available o-F-benzaldehyde 8 provided the key methyl sulfide 9. Combination of sulfide 9 with 1-pentyne, employing the optimized carbothiolation/cyclization conditions, delivered 3-propyl moxaverine in good yield. The intermediacy of sulfide 9 serves as a useful diversity generating branch point, as simply exchanging 1-pentyne for alternative alkynes would allow the rapid preparation of further derivatives.
Scheme 3. Rh-Catalyzed Alkyne Carbothiolation Route to 3-Propyl Moxaverine.

To provide additional utility we explored the use of alternative N-nucleophiles in the cyclization process. For example, by substituting ammonium acetate with hydroxylamine hydrochloride, we were able to access the isoquinoline N-oxide 11 (Scheme 4). Using primary anilines in the one-pot carbothiolation/cyclization sequence, followed by a NaBH4 mediated reductive step, allowed access to N-arylated dihydroisoquinolines (12). Finally, by exploring bulky primary amines as the nitrogen source, we were able to prevent formation of the isoquinolinium salts and gain access to amino-naphthalene products. Both isopropyl- and cyclohexylamine delivered the amino-naphthalene products in good yield (13 and 14).
Scheme 4. Access to Alternative Heterocycles/Arenes by Variation of the N-Nucleophile.

In conclusion, we have developed a second-generation catalyst system for alkyne carbothiolation using simple methyl sulfides. This new catalyst offers reduced reaction times and low catalyst loadings and allows an ‘activating group recycling’ approach to arene functionalization to be achieved in a practical manner. Importantly, the catalyst is assembled in situ from commercial components. We have utilized this approach to arene functionalization in a one-pot, three-component synthesis of isoquinolines and have delivered a convergent and efficient synthetic sequence that allows for the preparation of a diverse family of isoquinolines.
Acknowledgments
EPSRC for support of this study.
Supporting Information Available
Experimental procedures and full characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Pike V. W.; Halldin C.; Crouzel C.; Barre L.; Nutt D. J.; Osman S.; Shah F.; Turton D. R.; Waters S. L. Nucl. Med. Biol. 1993, 20, 503. [DOI] [PubMed] [Google Scholar]; b Takeuchi K.; Sakamoto S.; Nagayoshi Y.; Nishizawa H.; Matsubara J. Eur. J. Cardio-Thoracic Surgery 2004, 26, 956. [DOI] [PubMed] [Google Scholar]; c Weissmam B. A.; Raveh L. J. Neurochem. 2003, 84, 432. [DOI] [PubMed] [Google Scholar]; d Rinehart K. L. Med. Res. Rev. 2000, 20, 1. [DOI] [PubMed] [Google Scholar]
- a Whaley W. M.; Govindachari T. R. Org. React. 1951, 6, 74. [Google Scholar]; b Bischler B. N. A.; Napieralski B. Chem. Ber. 1893, 26, 1903. [Google Scholar]; c Pictet A.; Spengler T. Chem. Ber. 1911, 44, 2030. [Google Scholar]; d Whaley W. M.; Govindachari T. R. Org. React. 1951, 6, 151. [Google Scholar]; e Pomeranz C. Monatsh. Chem. 1893, 14, 116. [Google Scholar]; f Fritsch P. Chem. Ber. 1893, 26, 419. [Google Scholar]
- Hooper J. F.; Chaplin A. B.; Gonzalez-Rodriguez C.; Thompson A. L.; Weller A. S.; Willis M. C. J. Am. Chem. Soc. 2012, 134, 2906. [DOI] [PubMed] [Google Scholar]
- a Nakamura I.; Yamamoto Y. Chem. Rev. 2004, 104, 2127. [DOI] [PubMed] [Google Scholar]; b Zeni G.; Larock R. C. Chem. Rev. 2006, 106, 4644. [DOI] [PubMed] [Google Scholar]; c Patil N. T.; Yamamoto Y. Chem. Rev. 2008, 108, 3395. [DOI] [PubMed] [Google Scholar]; d Donohoe T. J.; Bower J. F.; Chan L. K. M. Org. Biomol. Chem. 2012, 10, 1322. [DOI] [PubMed] [Google Scholar]
- a Palladium in Heterocyclic Chemistry, 2nd ed.; Li J. J., Gribble G. W., Eds.; Elsevier: Oxford, U.K., 2007. [Google Scholar]; b Sadig J. E. R.; Willis M. C. Synthesis 2011, 1. [Google Scholar]; c Ball C.; Willis M. C. Eur. J. Org. Chem. 2013, 425. [Google Scholar]
- For related examples of “activating group recycling”, see:; a Schomaker J. M.; Grigg R. D. Synlett 2013, 401. [Google Scholar]; b Alcaide B.; Almendros P.; Alonso J. M.; Cembellín S.; Fernández I.; del Campo T. M.; Torres R. M. Chem. Commun. 2013, 49, 7779. [DOI] [PubMed] [Google Scholar]; c Grigg R. D.; Van Hoveln R.; Schomaker J. M. J. Am. Chem. Soc. 2012, 134, 16131. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Newman S. G.; Howell J. K.; Nicolaus N.; Lautens M. J. Am. Chem. Soc. 2011, 133, 14916. [DOI] [PubMed] [Google Scholar]; e Newman S. G.; Lautens M. J. Am. Chem. Soc. 2011, 133, 1778. [DOI] [PubMed] [Google Scholar]; f Liu H.; Li C.; Qiu D.; Tong X. J. Am. Chem. Soc. 2011, 133, 6187. [DOI] [PubMed] [Google Scholar]
- Schaumann E. Top. Curr. Chem. 2007, 274, 1. [Google Scholar]
- For selected recent reports of transition metal catalyzed approaches to isoquinolines, see:; a Guimond N.; Fagnou K. J. Am. Chem. Soc. 2009, 131, 12050. [DOI] [PubMed] [Google Scholar]; b Chiba S.; Xu Y.; Wang Y. J. Am. Chem. Soc. 2009, 131, 12886. [DOI] [PubMed] [Google Scholar]; c Jayakumar J.; Parthasarathy K.; Cheng C. Angew. Chem., Int. Ed. 2012, 51, 197. [DOI] [PubMed] [Google Scholar]; d Donohoe T. J.; Pilgrim B. S.; Jones G. R.; Bassuto J. A. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 11605. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kornhaass C.; Li J.; Ackermann L. J. Org. Chem. 2012, 77, 9190. [DOI] [PubMed] [Google Scholar]; f Wang H.; Grohmann C.; Nimphius C.; Glorius F. J. Am. Chem. Soc. 2012, 134, 19592. [DOI] [PubMed] [Google Scholar]; g Meng L.; Ju J.; Bin Y.; Hua R. J. Org. Chem. 2012, 77, 5794. [DOI] [PubMed] [Google Scholar]
- Yakelis N. A.; Bergman R. G. Organometallics 2005, 24, 3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg M.; Vanderburgt Y. E. M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Goubitz K.; Fraanje J. Organometallics 1995, 14, 3081. [Google Scholar]
- Xanthphos can adopt a number of coordination modes:; a Williams G. L.; Parks C. M.; Smith C. R.; Adams H.; Haynes A.; Meijer A. J. H. M.; Sunley G. J.; Gaemers S. Organometallics 2011, 30, 6166. [Google Scholar]; b Dallanegra R.; Chaplin A. B.; Weller A. S. Organometallics 2012, 31, 2720. [Google Scholar]
- For a pyridine synthesis which employs alkenyl sulfide substrates, see:Okada E.; Masuda R.; Hojo M. Heterocycles 1992, 34, 1927. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.