

Abstract
The pH in the aqueous pores of poly(lactide-co-glycolide) (PLGA) matrix, also referred to microclimate pH (μpH), is often uncontrolled ranging from highly acidic to neutral pH range. The μpH distribution inside protein-encapsulated PLGA microspheres was quantitatively evaluated using confocal laser scanning microscopy. The fluorescent response of Lysosensor yellow/blue® dextran used to map μpH in PLGA was influenced by the presence of encapsulated protein. The nonprotonated form of pyridyl group on the fluorescence probe at neutral pH was responsible for the interference, which was dependent on the type and concentration of protein. A method for correction of this interference based on estimating protein concentration inside the microspheres was established and validated. After correction of the influence, the μpH distribution kinetics inside microspheres was evaluated for different PLGA 50/50 microsphere formulations under physiological conditions for 4 weeks. Generally, the μpH acidity increased with the progression of incubation time. The co-incorporation of poorly soluble base, magnesium carbonate, in the microspheres prolonged the appearance of detectable acidity for up to 3 weeks. Co-addition of an acetate buffer was able to control the μpH over a slightly acidic range (around pH 4.7) after two weeks incubation. Microspheres prepared from a lower polymer concentration exhibited a higher μpH, likely owing to reduced diffusional resistance to acidic degradation products. The stability of protein was enhanced by addition of MgCO3, acetate buffer, or by reduced polymer concentration in the preparation, as evidenced by more soluble protein recovered after incubation. Hence, the μpH imaging technique developed can be employed in the future for optimization of formulation strategies for controlling μpH and stabilizing encapsulated proteins.
Keywords: microclimate pH, microspheres, confocal laser scanning microscopy, poly(lactide-co-glycolide), pH distribution
INTRODUCTION
Poly(lactide-co-glycolide) (PLGA), as one of the most important class of biodegradable and biocompatible polymers, have long been the research focus of controlled delivery of biomacromolecules, including peptides, proteins, and vaccines.1-6 Despite its excellent safety and versatility, a major drawback associated with this polymer is the common acidification and lack of control of its microenvironment inside polymer matrix during erosion, as a result of acidic polymer impurities and the build-up of acidic monomers and oligomers generated from polymer hydrolysis. Consequently, the integrity of encapsulated acid-labile proteins can be greatly compromised during release. 7-9
Several studies, using indirect methods, have shown evidence of an acidic microclimate within degrading PLGA devices. For example, faster degradation in the center of large PLGA specimens (~1-2 mm dimensions) was observed due to the accelerated hydrolysis of ester bondage catalyzed by the acids accumulated at the matrix core.10 Additionally, Shenderova et al.11 found that camptothecin was stabilized in its acid-stable (and active) lactone form when encapsulated in PLGA microspheres. Furthermore, co-incorporation of antacids, such as Mg(OH)2, MgCO3 and ZnCO3 in PLGAs could strongly inhibit protein structural losses and aggregation for over one month, as demonstrated in studies with model as well as therapeutic proteins.12-15
Moreover, techniques have been developed to directly quantify μpH inside PLGA delivery systems, including 31P nuclear magnetic resonance (NMR),16 electron paramagnetic resonance (EPR),17 potentiometry,18 and confocoal microscopy imaging.19-21 The first three methods are limited to providing an averaged μpH. Confocal microscopy imaging, on the other hand, by encapsulating fluorescent pH-sensitive probes, is capable of delineating a detailed μpH map noninvasively within the polymer matrix.
After early attempts to develop a quantitative ratiometric method of μpH measurement using confocal laser scanning microscopy,19 our group found that SNARF-1® dextran20 and Lysosensor yellow/blue® dextran21, as fluorescent probes encapsulated into PLGA microspheres, could sense pH changes from pH 5.8 to 8.0, and pH 2.8 to 5.8, respectively. Thus, after confocal images processing, an accurate pixel-by-pixel μpH distribution map either in the neutral or acidic range could be created.20,21 This ratiometric method is advantageous in that it eliminates artifacts including photo bleaching, leakage of the dye probe, and non-uniform distribution of dye within microspheres. The dextran-conjugated probes employed are water-soluble macromolecules, thereby localizing themselves in the aqueous pores where protein resides.19
In the present study, μpH inside PLGA microspheres encapsulating both protein and Lysosensor yellow/blue® dextran was accurately quantified using confocal microscopy imaging technique. In order to accomplish this, significant interference of the dye response from the presence of protein was corrected by estimating protein concentration inside the PLGA pores to perform the measurement. The acquired knowledge is beneficial to further our understanding of μpH development and promote formulation designs for optimized delivery of pH-sensitive biomacromolecules.
MATERIALS AND METHODS
Materials
Poly(D,L-lactide-co-glycolide), end capped, 50/50 with inherent viscosity (i.v.) of 0.6 dl/g in hexafluoroisopropanol at 25 °C was purchased from Durect Corporation (Birmingham, AL). The fluorescent pH sensitive probe, Lysosensor yellow/blue® dextran (MW=10 kDa) was purchased from Invitrogen (Eugene, OR). Bovine Serum Albumin (BSA, fraction V), was purchased from Sigma Chemical Co. (St. Louis, MO). Polyvinyl alcohol (PVA, 80% hydrolyzed, MW 9-10 kDa) was supplied by Polysciences Inc. (Warrington, PA). All other chemicals were of analytical grade or higher were obtained from commercial suppliers.
Preparation of Microspheres
Protein-encapsulated PLGA microspheres containing Lysosensor yellow/blue® dextran as an acidic pH sensitive probe were prepared using the w/o/w double emulsion-solvent evaporation method. Briefly, 100 μl of 300 mg/ml BSA with 25 mg/ml dye in double distilled water was added to 1 ml of 400 mg/ml PLGA solution (40% w/v) in methylene chloride. The mixture was then homogenized using a Tempest IQ2 homogenizer (The VirTis Co., Gardiner, NY) at 7,500 rpm for 1 min to generate first w/o emulsion, followed by quickly adding 1 ml of PVA solution (2% w/w). After vortexing for 20 s, the formed w/o/w emulsion was poured slowly into 100 ml of PVA solution (0.5% w/w) and stirred at room temperature for 3 hours to extract and evaporate the organic solvent. Then, the hardened microspheres were harvested and sieved for 45-63 μm size. After washing with double distilled water three times, the microspheres were freeze-dried on a FreeZone 2.5 Liter Benchtop freeze dry system (Labconco, Kansas City, MO).
For microspheres containing BSA of a specific pH, 100 mg/ml BSA solution was first titrated with HCl to pH 3, 4, and 5, respectively, followed by freeze-drying. The lyophilized powder was then reconstituted with water and encapsulated with dye in PLGA microspheres as described above. Microspheres containing magnesium carbonate were prepared by suspending 3% (w/w) of the base to the polymer solution with all other conditions as described above. To prepare microspheres encapsulating acetate buffer, BSA and dye were dissolved in 100 μl of 0.1 M sodium acetate buffer of pH 4.6 to make the water phase, with other conditions unchanged. Microspheres with a lower polymer concentration (30% w/v) were also prepared following the same procedures.
Confocal Laser Scanning Microscopy for Microspheres Imaging
A ratiometric method based on a confocal microscopy imaging technique was employed similarly as described by Ding et al.21 A Carl Zeiss LSM 510-META laser scanning confocal microscope (LSCM, Carl Zeiss Microimaging, Inc., Thornwood, NY) was equipped with an Enterprise UV laser and a Carl Zeiss Axiovert 100 M inverted microscope. The fluorescent dye that was encapsulated in the microspheres was excited at 364 nm, and the emission at two wavelengths, 450 nm and 520 nm were recorded. All measurements were conducted using a C-Apochromat 63X water immersion objectives lens with numerical aperture of 1.2. The detection gain was set at 650, and the pinhole was 328 μm. The laser power was set at 40% of its full power. The image size was 512×512 pixels and the images were scanned by 8 bit plane mode at a scan speed of 6.40 μs/pixel.
Calibrating Fluorescence Intensity Ratio vs. pH in the Presence of Protein
A set of universal buffers with pH ranging from 2.8 to 5.8 were prepared using combined 0.1 M citric acid and 0.2 M Na2HPO4 solutions. A certain concentration of protein solution (BSA or lysozyme, e.g. 100 mg/ml) were prepared by dissolving protein in the buffers and then titrating the solution to its original pH. Lysosensor yellow/blue® dextran was then dissolved in the protein buffer solutions with a concentration of 1.2 mg/ml.
Images of dye solution were obtained under confocal microscope at 450 nm and 520 nm. The acquired images (n=8) were processed by frame averaging, followed by neighborhood averaging, and applying a median filter as described by Li et al.20 using Image J software (developed by National Institutes of Health and available on the internet at http://rsbweb.nih.gov/ij/) to eliminate the signal noise and obtain accurate pixel value. The standard curves were established by plotting the ratio of mean pixel intensity of the dye solutions at two emission wavelength, 450 nm and 520 nm vs. the pH of that solution.
Microclimate pH Mapping Inside Microspheres
Microspheres (20–25 mg) were incubated in 1 ml phosphate buffer saline (7.74 mM Na2HPO4, 2.26 mM NaH2PO4, 137 mM NaCl and 3 mM KCl) containing 0.02% tween 80 (PBST, 10mM, pH=7.4) at 37°C under mild agitation at 320 rpm by a KS 130 basic shaker (IKA® Works Inc., Wilmington, NC). At pre-determined time points, the release medium was replaced with fresh buffer and a small amount of microspheres were collected and placed under confocal microscope while focusing at the center of microspheres to obtain images (n=5). After image processing20, the ratio of fluorescence intensity I450nm/I520nm at each pixel having intensity above the threshold value (indicating the fluorescence from release media) of the images was calculated and assigned to a pH from the standard curves independent of dye concentration. In the processed images, each pixel was converted to a color corresponding to specific pH. When plotting the μpH distribution curves, the probability of a specific pH inside microspheres was calculated by taking the amount of pixels corresponding to that pH divided by the total pixels of the images. For intensity ratios exceeding the limit of standard curve, the pH was assigned to either below 2.8 or above 5.8. In such cases, their percentage was plotted as the boundaries of the distribution curves accordingly.
The μpH could be accurately mapped within ± 0.2 pH unit over pH from 2.8 to 5.8 (see Supporting Information for statistical analysis).
Determination of Protein Loading and Encapsulation Efficiency
The amount of protein encapsulated in PLGA microspheres was determined by direct recovery from the polymer matrix.13 Eight mg of microspheres were dissolved in 2 ml acetone. The mixture was vortexed and centrifuged at 8,000 rpm for 10 min, followed by removal of the acetone. After repeating the above procedures for three times, the BSA pellet was air-dried and reconstituted in PBST and incubated at 37°C for 1 h. The protein concentration was then determined using Coomassie® Plus protein assay reagent. The working range in this study was from 25 μg/ml to 500 μg/ml (assay sensitivity is from 1 μg/ml to 1500 μg/ml) and not interfered by reagents used in our experiments. Protein loading was calculated from the amount of protein recovered divided by the mass of microspheres. Encapsulation efficiency was obtained from the ratio of actual protein loading to the theoretical protein loading. All measurements were performed in triplicate (n=3).
Release and Stability of Protein from Microspheres
Microspheres (20–25 mg) were incubated in 1 ml PBST (10mM, pH=7.4) at 37 °C under mild agitation at 320 rpm. At pre-determined time points, the release media was removed after centrifugation at 5,000 rpm for 5 min and replaced with fresh buffer. The protein concentration in the release media was determined using Coomassie® Plus protein assay reagent.
At the end of release study, soluble protein was recovered from PLGA microspheres as described in the loading study. Any remaining insoluble aggregates were collected by centrifugation and dissolved in denaturing solvent (PBST/6M urea/1mM ethylenediaminetetraacetic acid (EDTA)) and incubated at 37°C for 30 min to dissolve non-covalent bonded aggregates. Finally, any insoluble aggregates were collected again and dissolved in reducing solvent (denaturing solvent plus 10 mM dithiothreitol (DTT)) to dissolve any disulfide-bonded aggregates. The protein content in each step was all analyzed with Coomassie® Plus protein assay reagent using the appropriate solvent as diluent for protein standards. All measurements were performed in triplicate (n=3).
Water Uptake of Microspheres
Microspheres (20–25 mg) were incubated in 1 ml PBST (10mM, pH=7.4) at 37 °C under mild agitation at 320 rpm. At pre-determined time points, the microspheres were collected and the surface water was removed by filtration and the wet weight (W1) of the microspheres was recorded. The samples then were dried under vacuum to a constant weight and the dry weight (W2) was recorded.
To correct for the interparticle water, dry microspheres were suspended in PBST at room temperature and rapidly filtered and dried as described above. Assuming little water uptake by the microparticles between suspension and filtration, the weight differences between wet and dry particles accounted for the portion of interparticle water (Wi), as defined by
| (1) |
Where W1’ and W2’ are the weights of wet microspheres and dry microspheres after immediate collection (t=0), respectively. The water uptake of microspheres at time t (WP(t)) was estimated by:
| (2) |
Where W1 and W2 are the wet and dry microsphere weights at time t. Note that in control experiments the interparticle water estimation did not significantly depend on the temperature of water used, e.g. 4°C, 25°C and 37°C (data not shown). All measurements were performed in triplicate (n=3).
Correction of Protein Interference on μpH Mapping
In order to account for the influence of protein on dye emission, corrections were necessary to acquire an accurate estimation of the μpH in the presence of significant BSA. The influence of lysozyme was significantly less, and therefore, its correction was not considered further. Since protein concentration inside microspheres changes during incubation due to the protein release and water uptake by the polymer matrix, corrections were done for each time point of pH mapping. The average protein concentration (Cp(t)) inside the microsphere aqueous pores at time t can be estimated by the following equation:
| (3) |
where Mp is the mass of protein in microspheres. Vpores is the volume of aqueous pores. MP,0, MP(t) are the initial and time dependent mass of microspheres respectively. l is the fraction of protein loaded. f(t) is the fraction of protein release from microspheres. And ρw is the density of water.
When the estimated protein concentration was not the same as those in known standard curves (Figure 1B), the corresponding fluorescence ratio vs. pH curve was interpolated. (see Supporting Information)
Figure 1.
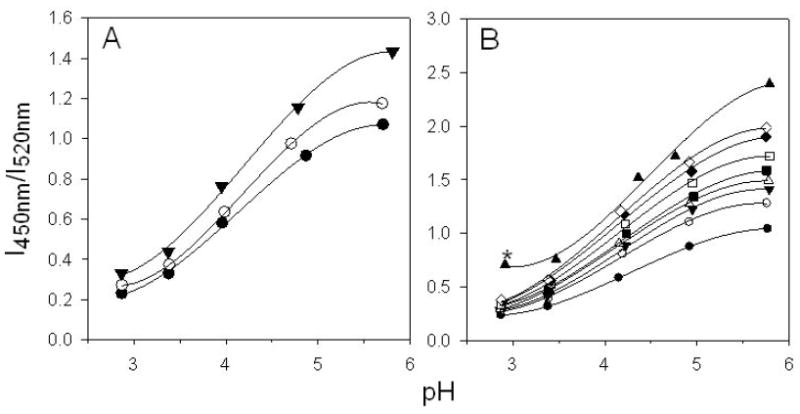
Interference of confocal pH measurement of Lysosensor yellow/blue® dextran as a function of pH (A) by the presence of 100 mg/ml of BSA (▼), 100 mg/ml of lysozyme (○), or absence of protein (●); (B) by the presence of BSA at the concentration of 0 mg/ml (●), 25 mg/ml (○), 50 mg/ml (▼), 75 mg/ml (△), 100 mg/ml (■), 150 mg/ml (□), 200 mg/ml (◆), 250 mg/ml (◇), and 500 mg/ml (▲). The concentration of fluorescence dye was 1.2 mg/ml. Lines represent best fits to a third order polynormial function of the experimental data. SD for all data points were less than 2% of mean (n=8). * BSA formed a gel-like phase at this protein concentration and pH.
RESULTS AND DISCUSSION
Interference of Protein on Fluorescent Response of the Dye
Lysosensor yellow/blue® dextran, which is sensitive to changes in acidity from roughly pH 2.8 to 5.8, was selected as a fluorescence probe to investigate the μpH inside PLGA microspheres, as previously reported.21 Adding protein to the dye solutions also provided a fluorescent intensity emission ratio (I450 nm/I520 nm) vs. pH standard curve well fitted to a third-order polynomial function (r2=0.999) from pH 2.8 to 5.8 (Figure S3). The pH sensitivity of the dye is concentration-independent as well, which ensures the standard curve is not affected when the dye concentration changes in microspheres during incubation. However, as protein concentration was raised to elevated levels (e.g. >25 mg/ml of BSA), the fluorescent response of the dye was significantly influenced by the presence of protein, and this interference was dependent on the specific protein. For example, as shown in Figure 1A, the pH sensitivity differed for dye solutions with or without presence of protein, with the presence of BSA giving more pronounced changes in emission intensity ratio compared to that of lysozyme. As expected from Figure 1A, the fluorescence ratio was significantly affected by the protein concentration over wide range. As shown in Figure 1B, the intensity ratio at a certain pH for BSA concentration of 0 to 500 mg/ml rose as protein concentration was increased, with little or no influence at pH 2.8 to an extensive effect at pH 5.8. The sensitivity of dye at high pH corresponding to the presence of the non-protonated form of the dye’s pyridyl group implicates this dye species as responsible for the protein interference. Note that by 500 mg/ml BSA at highly acidic pH resulted in a gel formation, consistent with the low pH unfolding of the protein22 and noncovalent aggregation of BSA in PLGA,12,13 which was associated with an unexpected increase in the intensity ratio at the pH of 2.8 (Figure 1B).
The mechanism of protein interference on the fluorescent response of the non-protonated form of dye is not well understood, although it was demonstrated that the presence of protein would quench the emission of dye at 520 nm and shift the emission peak at 450 nm slightly to a shorter wavelength in the fluorescence spectrum (Figure S4). Ground state interactions between dextran-dye and protein, e.g., binding, was not likely to cause the interference, considering the emission ratio did not depend on dye concentration in presence of either lysozyme or BSA. (Figure S3). Processes involved excited state of fluorophores, such as energy transfer or collisional quenching induced by the protein, were more likely responsible for the interference.
Correction of BSA Effect on Dye Interference and BSA Buffering Capacity
As described in the Materials and Methods, we estimated the BSA concentration in the microspheres to correct for the BSA interference on the μpH reporting of the dye. Key assumptions involved estimates of interparticle water and assuming uniform BSA concentration in the pores. In addition, for this polymer molecular weight, very little water partitions in the polymer phase until late stages of polymer erosion.23 To validate our approach of correction, we compared the μpH measured inside microspheres encapsulating BSA after 1 day incubation in PBST at 37°C as a function of various pH of BSA solutions used to form the primary emulsion during microsphere preparation. We hypothesized that after 1-day incubation, a concentrated protein solution would be formed due to the water penetration into the polymer matrix. Therefore, the μpH would be dictated by the pH of the encapsulated protein solution in aqueous pores as significant degradation of polymer is not expected at such an early time of incubation.23,24 Moreover, from μpH measurement of microspheres without encapsulating protein, little acidity was observed (See Figure 3B), indicating the lack of significant acid impurities. Encapsulated BSA of different pH was prepared by titrating 100 mg/ml BSA solution to a specific pH and then freeze-drying. The estimated protein concentration after 1-day incubation inside polymer pores was roughly 500 mg/ml (within ± 10%) for each formulation, as calculated from (3). Thus, μpH values were estimated from fluorescence ratio vs. pH standard curve with 500 mg/ml BSA. The processed confocal images and μpH distribution curves are shown in Figure 2 & Figure 3, respectively, and corresponding results are summarized in Table 1. The estimated μpH after correction was very close (difference within 0.1 pH unit) to the pH of the concentrated protein solution, strongly supporting the approach of correction.
Figure 3.
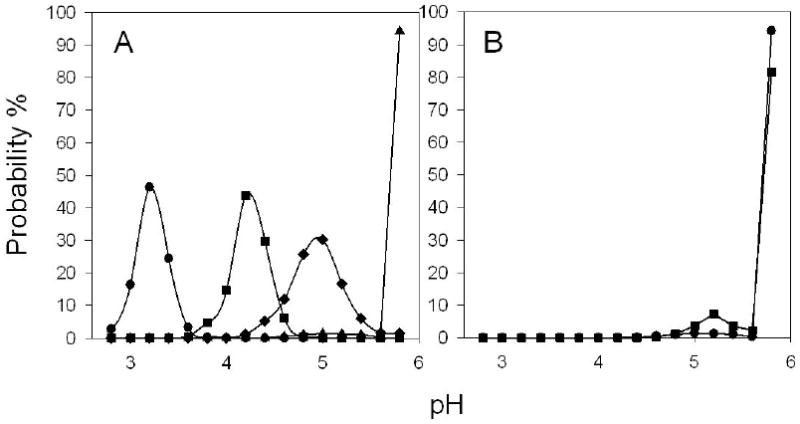
μpH distribution kinetics of microspheres encapsulating (A) dye and BSA of pH of 3 (●); dye and BSA of pH of 4 (■); dye and BSA of pH of 5 (◆); and dye and BSA of pH of 7 (▲) (B) dye only (●) and dye with BSA (pH of 7)(■) after incubation at 37°C in PBST buffer for 1 day. The μpH was controlled by the inner water phase pH, as described in Materials and Methods.
Figure 2.
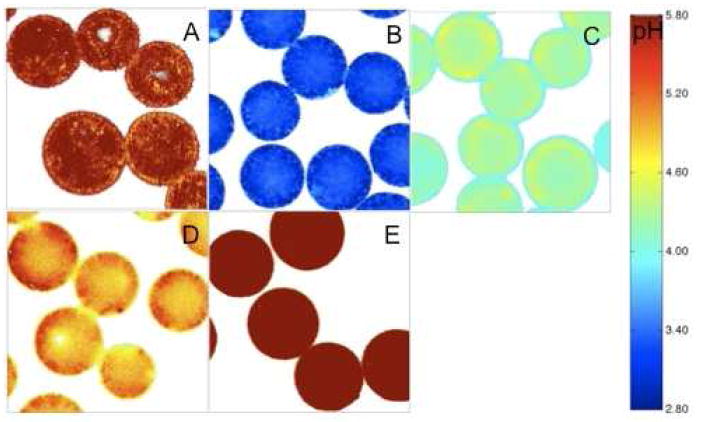
Processed confocal images of microspheres encapsulating dye only (A); dye and BSA of pH of 3 (B); dye and BSA of pH of 4 (C); dye and BSA of pH of 5 (D); and dye and BSA of pH of 7 (E) after incubation at 37°C in PBST buffer for 1 day. The μpH was controlled by the inner water phase pH, as described in Materials and Methods.
Table 1.
pH comparison of concentrated BSA solution and average μpH after 1 day incubation from confocal microscopy after correction of protein interference.
| pH of 100 mg/ml BSA solution | pH of 500 mg/ml BSA solution a | Average μpH from confocal imaging b |
|---|---|---|
| 3.0 | 3.2 | 3.2 |
| 4.0 | 4.1 | 4.2 |
| 5.0 | 5.1 | 5.0 |
| 7.0 | 7.0 | >5.8 |
The solution was made by reconstitution of lyophilized BSA power from 100 mg/ml solution of specific pH, as described in Materials and Methods.
The μpH was controlled by the inner water phase pH, as described in Materials and Methods.
A slight acidity was observed in PLGA microspheres encapsulating only dye after the 1-day incubation (Figure 2A), which could be ascribed the existence of a very low level of acidic impurities in the polymer. However, upon incorporation of BSA, pH in most aqueous pores were raised above 5.8, with more than 95% of pixels out of detection limit compared to 80% in microspheres without protein (Figure 3B), consistent with significant buffering capacity of the encapsulated BSA. Moreover, the μpH was more homogenously distributed inside microspheres encapsulating protein (Figure 2E).
Mapping μpH Distribution and Kinetics in Degrading PLGA Microspheres
After the successful test of the correction for BSA interference on μpH measurement, μpH distribution and kinetics were examined and compared in degrading PLGA microspheres prepared from different formulations during a one-month incubation. The processed confocal images visualized the μpH distribution by color, and the distribution curves provided a quantitative illustration.
Little acidity was observed in microspheres made from polymer concentration of 40% (w/v) at the beginning of incubation, as evidenced by more than 95% of pixels in the images out of detectable limit of the dye (pH 5.8). As the incubation progressed, the μpH decreased steadily until day 21 in accordance with the accumulation of water-soluble acids generated by the degradation of the polymer. After 21 days, the μpH maintained mostly in the range of 4 to 5.8 (Figure 5A), probably due to the onset of polymer erosion and liberation of water-soluble acids out of the polymer to balance acid production rate. From the processed confocal images (Figure 4A), the acidity was observed to be higher in the center of microspheres than in the peripheral regions, consistent with development of an expected diffusion/reaction mechanism governing polymer distribution of acidic degradation products. The blank regions inside the microspheres indicate no detectable fluorescence, corresponding to regions of extensive dye release from the polymer.
Figure 5.
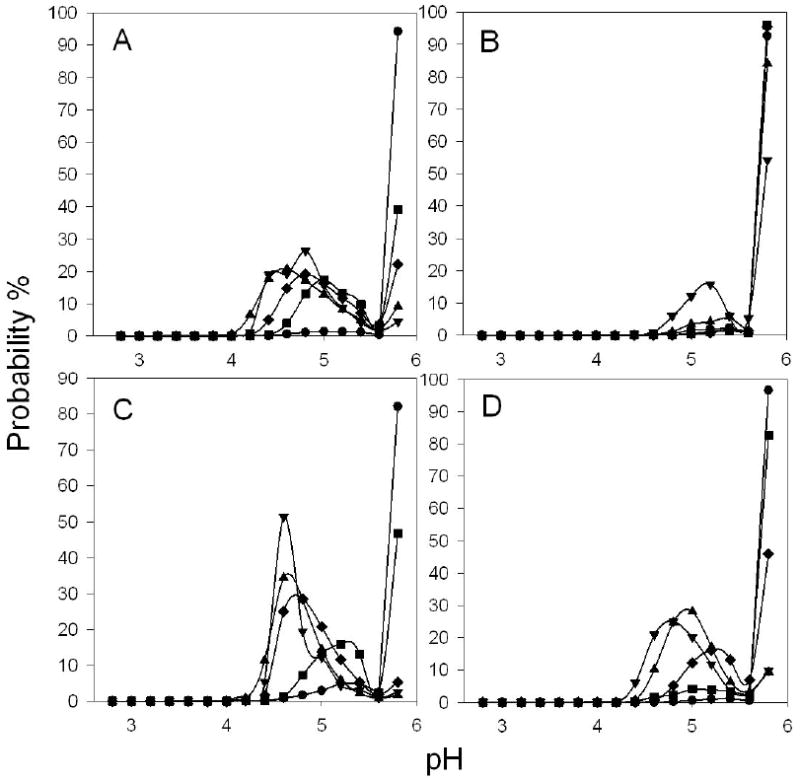
μpH distribution kinetics of microsphere formulations during incubation in PBST at 37°C for 1 day (●), 7 days (■), 14 days (◆), 21 days (▲), and 28 days (▼). Microspheres were prepared from 40% (w/v) PLGA (A), 40% (w/v) PLGA + MgCO3 (B), 40% (w/v) PLGA + acetate buffer (C) and 30% (w/v) PLGA (D).
Figure 4.
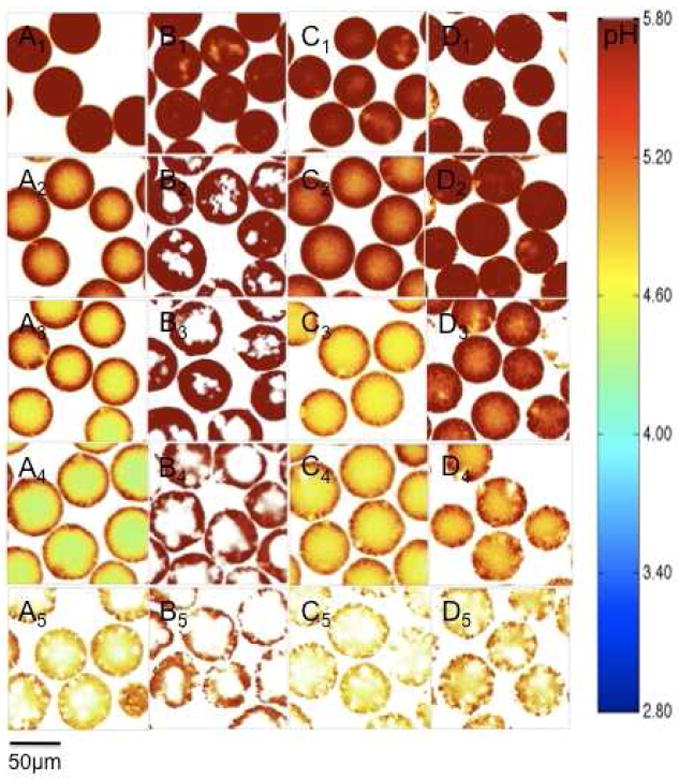
Processed confocal images of microsphere formulations during incubation in PBST at 37°C for 4 weeks. Microspheres were prepared from 40% (w/v) PLGA (A), 40% (w/v) PLGA + MgCO3 (B), 40% (w/v) PLGA + acetate buffer (C) and 30% (w/v) PLGA (D). Images were taken at 1 (A1-D1), 7 (A2-D2), 14 (A3-D3), 21 (A4-D4) and 28 (A5-D5) days.
Addition of magnesium carbonate postponed the appearance of detectable acidity inside microspheres up to 3 weeks (Figure 4B & 5B) by multiple mechanisms, including: i) dissolution and direct neutralization of PLGA-produced acids and ii) water uptake and pore formation imparted by the osmotic Mg-carboxylate salts resulting from acid-base titration, which increases liberation of sequestered acids. The minimization of the pH drop conferred by incorporation of base in PLGAs was supported indirectly in a previous study from decreasing the degradation rate of the polymer.12 Moreover, this effect was further confirmed in a quantitative way using a neutral pH sensitive dye.20 In this study, the effect of MgCO3 incorporation was examined by the changes in acidic pH in BSA-containing PLGA 50/50 microspheres. Consistent with previous studies with larger millicylidrical implants,13 elevated water uptake was observed in microspheres upon incorporation of MgCO3 (Figure 6B), as a result of the osmotic pressure generated by the Mg-carboxylate salts described above. Since more water channels were created, encapsulated protein was released slightly faster than that of without base (Figure 6A). The same trend was also expected for dye release in microspheres with MgCO3. The rapid release of dye, therefore, was associated with a higher fraction of blank regions in confocal images than in base-free formulations. In addition, the decrease of the μpH by 28 days of incubation may have been caused by the depletion of base from the polymer. Note that water uptake (Figure 6B) and protein release kinetics (Figure 6A) were used to estimate protein concentration kinetics inside polymer pores (Figure 6C) for correction of protein interference on μpH mapping, as described in the Materials and Methods.
Figure 6.
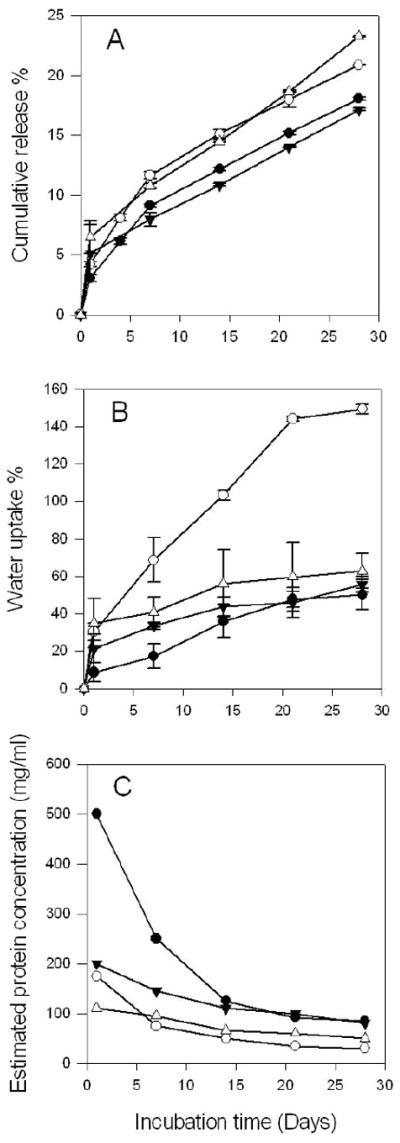
Kinetics of protein release (A), water uptake of microspheres (B), and estimated protein concentration in polymer pores (C) from PLGA microsphere formulations during incubation in PBST at 37°C for 4 weeks. Microspheres were prepared from 40% (w/v) PLGA (●), 40% (w/v) PLGA + MgCO3 (○), 40% (w/v) PLGA + acetate buffer (▼) and 30% (w/v) PLGA (△). Symbols represent mean ± SD, n=3 for A and B, SD is not applicable for C because the value is calculated from independent parameters from equation 3.
In order to control μpH over a moderate acidic range, water-soluble buffering species were co-encapsulated with protein into PLGA microspheres. This was achieved by adding to the protein inner water phase a buffer solution (0.1 M acetic acid and sodium acetate, pH=4.6). During incubation, the aqueous pores inside microspheres would be filled with buffering species with protein so long as these species are retained in the polymer. From confocal images (Figure 4C) and μpH distribution curves (Figure 5C), the μpH gradually dropped from neutral pH as incubation progressed. After 14 days incubation, the average μpH maintained around from 4.6 to 4.8. The relatively high pH during the initial stage of incubation was due to the very high protein concentration in aqueous pores, which acted also as a buffer, undermining the buffering capacity of acetate salts. As water imbibed into and protein released out of polymer matrix, protein concentration decreased. Meanwhile, despite the possible removal of acetic acid, water-soluble acids were generated from polymer degradation, leading to μpH approach to the pH of acetate’s maximal buffer capacity (pKa=4.7).
The effect of polymer concentration on μpH distribution kinetics was also examined in our study. Microspheres were prepared from a lower polymer concentration of 30% (w/v). Compared to that made of 40% (w/v), the μpH was much less acidic upon 14 days incubation (Figure 4D & 5D). This could be rationalized by the fact that microspheres made from solution of lower polymer concentration usually possess more porous internal structure,25 which likely caused a higher effective diffusivity of acidic degradation products through the polymer matrix26 and facilitated their liberation as a result. After 21 days incubation, the effect of polymer concentration on μpH was not apparent, which was presumably due to the changes in polymer properties, e.g. degradation rate, so that the difference in diffusion rate of acids was not significant.
Assumptions for Correction and Anticipated Error
The correction for BSA’s effect on the dye’s fluorescent response was based on multiple assumptions. One important assumption for correction is that protein is evenly distributed inside aqueous pores in microspheres. This was supported by lysozyme’s homogeneous distribution inside PLGA microspheres prepared by w/o/w double emulsion method, as observed by confocal laser scanning microscopy and infrared microscopy.27 In the confocal images recorded in this study (Figure 4), the fairly homogenous distribution of dye (being a water-soluble macromolecule as protein) was observed with the exception of formulations with base added and at the very last time point of 28 days, indicating the similar behavior of protein inside microspheres. Another assumption is that the protein concentration in the cavities of microspheres was relatively uniform. In the case that local protein concentration in some pores were higher than the estimated average protein concentration, the μpH distribution curves would generally shift to a lower pH.
Since the average protein concentration in PLGA pores at each point during controlled release was based on estimation from equation (3), errors may be associated with the deviation of estimation from the actual protein concentration, affecting the μpH measurement. The kinetics of estimated protein concentration inside microspheres during one month incubation are displayed in Figure 6C. Among all the parameters used to calculate the averaged protein concentration, the time dependent water uptake is most variable because the fraction of interparticle water may be changing during incubation depending on the property of polymer. Assuming there was 20% of error involved in experiments of estimating water of microspheres, the μpH kinetics corrected from protein concentration accounting for this ± 20% of error (-17% to +25% of protein concentration) for microspheres with or without encapsulating MgCO3 are shown in Figure S5. As demonstrated, the resulted μpH was not significantly affected (within 0.2 pH unit). Hence, the correction was only modestly influenced by small deviations based on the interparticle water assumption.
Formulation Effects on Protein Stability
Insoluble protein aggregation has been observed when encapsulated in PLGAs, which has been linked to the acidic environment in PLGAs. For example, BSA was found to become hydrolyzed and form noncovalent aggregates by hydrophobic interactions when encapsulated in PLGA 50/50 millicylindrical implants.12,13,15 Therefore, we analyzed the composition of residual protein inside PLGA microspheres after 4 weeks incubation in terms of soluble and insoluble protein12 and the results were summarized in Table 2.
Table 2.
Release and stability of various microsphere formulations after 28 days incubation.
| Formulation Polymer conc. (w/v)/excipient | Loading (%) | Encapsulation efficiency (%) | Released (%)b | Soluble residue (%)b | Insoluble residue (non- covalent) (%)b | Insoluble residue (covalent) (%)b | Recovery (%) b, c |
|---|---|---|---|---|---|---|---|
| 40 % (w/v) | 4.7 ± 0.2a | 68 ± 3 | 18.1 ± 0.2 | 9 ± 2 | 34 ± 5 | 8 ± 1 | 71 |
| 40 % (w/v) w/MgCO3 | 5.2 ± 0.1 | 75 ± 2 | 20.9 ± 0.3 | 44 ± 5 | 21 ± 2 | 4 ± 1 | 90 |
| 40 % (w/v) w/acetate buffer | 4.9 ± 0.1 | 69 ± 1 | 17.1 ± 0.7 | 41 ± 2 | 16 ± 3 | 5 ± 1 | 79 |
| 30 % (w/v) | 3.9 ± 0.1 | 62 ± 1 | 23.3 ± 0.4 | 46 ± 1 | 12 ± 2 | 2 ± 1 | 83 |
All data are reported as mean ± SD, n=3.
Determined after 28-day release.
Recovery (%) = released (%) + soluble residue (%) + insoluble residue (%).
Incorporation of poorly soluble base (MgCO3) significantly improved protein stability in terms of aggregation, as 44 ± 5 % of soluble residue remained after 28 days release relative to 9 ± 2 % in microspheres without any excipients. The mechanism of stabilization is believed to occur primarily via raising the acidic μpH in degrading PLGA matrix, as displayed in our confocal images and μpH distribution curves. This stabilization effect conferred by antacids was also shown in other therapeutic proteins.12,15 Some degree of aggregation persisting in these formulations is consistent with our previous data with microspheres prepared with the ester-end-capped PLGA 50/50.12 The 90% recovery in this formulation also suggested reduced protein hydrolysis than the other samples (71-83% recovery), as low recovery likely results from a lack of recognition of hydrolyzed protein by the Coomassie® Plus protein assay reagent.28 Note that higher water content and slightly faster release (and thus, less remaining encapsulated protein to become damaged) are also potential effects to decrease the 28-day aggregation values.13,15
Addition of acetate buffer also reduced protein aggregation inside PLGA microspheres during one-month incubation. In this case, the pH profile was very similar to that recorded in the most unstable preparation (prepared with 40% w/v polymer concentration), albeit just slightly higher in the vicinity of the high buffering capacity of the acetate buffer. This data suggests perhaps other factors involved, e.g., the elevated water content and strongly reduced protein concentration (see Figure 6) or the different ionic strength anticipated in the microclimate of this formulation affecting protein’s stability. We also note that the small changes in pH in the vicinity of the first unfolding transition of BSA22 may have been important.
Preparing microspheres from a lower polymer concentration also resulted in enhanced protein stability. This can be attributed to a less acidic microclimate developed in degrading PLGA specimen during the course of incubation. A more porous internal structure can take more water, thereby increasing the effective diffusion coefficient of the detrimental water-soluble acids in the polymer matrix26 and accelerating their release from the microspheres.
CONCLUSIONS
An uncontrolled and often acidic μpH is regarded as one of the most deleterious factors responsible for the instability of encapsulated protein in PLGA delivery systems. Therefore, it is important to develop methods of quantitative description of the microenvironment in PLGA. In our study, we demonstrated that μpH mapping in the polymer was affected by the presence of encapsulated protein, whose interference on fluorescent response of dye depends on the type and concentration of protein. μpH distribution in microspheres with protein and/or excipients could be quantitatively evaluated using confocal laser scanning microscopy after correction of the interference of protein. This μpH mapping technique presented as a valuable tool for study of μpH development mechanisms and design of formulation methodologies that control μpH with stabilized biomacromolecules.
Supplementary Material
Acknowledgments
We thank Mr. Bruce Donohoe at Microscopy and Image Analysis Laboratory at the University of Michigan for training and help with confocal microscope. This work was supported by NIH R01 HL 68345 and the University of Michigan Warner Lambert/Parke Davis Fellowship and Gordon Amidon Fellowship to Yajun Liu.
Footnotes
Supporting Information Available:
Statistical analysis of deviation in pH mapping from confocal images, the interpolation of standard curves from estimated protein concentrations, the concentration independency of pH sensitivity of dye, the fluorescence spectrum of dye in presence and absence of BSA, and the comparison of μpH distribution kinetics estimated from protein concentrations considering possible errors from water uptake measurement. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Okada H. One-and three-month release injectable microspheres of the LH-RH superagonist leuprorelin acetate. Adv Drug Deliv Rev. 1997;28:43–70. doi: 10.1016/s0169-409x(97)00050-1. [DOI] [PubMed] [Google Scholar]
- 2.Hutchinson F, Furr B. Biodegradable polymer systems for the sustained release of polypeptides. J Controlled Release. 1990;13:279–294. [Google Scholar]
- 3.Putney SD, Burke PA. Improving protein therapeutics with sustained-release formulations. Nat Biotechnol. 1998;16:153–157. doi: 10.1038/nbt0298-153. [DOI] [PubMed] [Google Scholar]
- 4.Cohen S, Yoshioka T, Lucarelli M, Hwang LH, Langer R. Controlled delivery systems for proteins based on poly (lactic/glycolic acid) microspheres. Pharm Res. 1991;8:713–720. doi: 10.1023/a:1015841715384. [DOI] [PubMed] [Google Scholar]
- 5.Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly (lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev. 2005;57:391–410. doi: 10.1016/j.addr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Schwendeman SP, Costantino HR, Gupta RK, Langer R. Peptide, protein, and vaccine delivery from implantable polymeric systems: progress and challenges. Controlled drug delivery: challenges and strategies. 1997:229–267. [Google Scholar]
- 7.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit Rev Ther Drug Carrier Syst. 2002;19:73–98. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 8.van de Weert M, Hennink WE, Jiskoot W. Protein instability in poly (lactic-co-glycolic acid) microparticles. Pharm Res. 2000;17:1159–1167. doi: 10.1023/a:1026498209874. [DOI] [PubMed] [Google Scholar]
- 9.Fu K, Klibanov A, Langer R. Protein stability in controlled-release systems. Nat Biotechnol. 2000;18:24–25. doi: 10.1038/71875. [DOI] [PubMed] [Google Scholar]
- 10.Li SM, Garreau H, Vert M. Structure-property relationships in the case of the degradation of massive aliphatic poly-(α-hydroxy acids) in aqueous media. J Mater Sci Mater Med. 1990;1:123–130. [Google Scholar]
- 11.Shenderova A, Burke TG, Schwendeman SP. The acidic microclimate in poly (lactide-co-glycolide) microspheres stabilizes camptothecins. Pharm Res. 1999;16:241–248. doi: 10.1023/a:1018876308346. [DOI] [PubMed] [Google Scholar]
- 12.Zhu G, Mallery SR, Schwendeman SP. Stabilization of proteins encapsulated in injectable poly (lactide-co-glycolide) Nat Biotechnol. 2000;18:52–57. doi: 10.1038/71916. [DOI] [PubMed] [Google Scholar]
- 13.Zhu G, Schwendeman SP. Stabilization of proteins encapsulated in cylindrical poly (lactide-co-glycolide) implants: mechanism of stabilization by basic additives. Pharm Res. 2000;17:351–357. doi: 10.1023/a:1007513425337. [DOI] [PubMed] [Google Scholar]
- 14.Jiang W, Schwendeman SP. Stabilization of tetanus toxoid encapsulated in PLGA microspheres. Mol Pharmaceutics. 2008;5:808–817. doi: 10.1021/mp800027f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang J, Schwendeman SP. Comparison of the effects of Mg (OH) 2 and sucrose on the stability of bovine serum albumin encapsulated in injectable poly (,- lactide-co-glycolide) implants. Biomaterials. 2002;23:239–245. doi: 10.1016/s0142-9612(01)00101-6. [DOI] [PubMed] [Google Scholar]
- 16.Burke P. Determination of internal pH in PLGA microspheres using 31P NMR spectroscopy. Proc Int Symp Control Release Bioact Mater. 1996:133–134. [Google Scholar]
- 17.Brunner A, Mäer K, Göferich A. pH and Osmotic Pressure Inside Biodegradable Microspheres During Erosion1. Pharm Res. 1999;16:847–853. doi: 10.1023/a:1018822002353. [DOI] [PubMed] [Google Scholar]
- 18.Shenderova A, Ding AG, Schwendeman SP. Potentiometric method for determination of microclimate pH in poly (lactic-co-glycolic acid) films. Macromolecules. 2004;37:10052–10058. [Google Scholar]
- 19.Fu K, Pack DW, Klibanov AM, Langer R. Visual evidence of acidic environment within degrading poly (lactic-co-glycolic acid)(PLGA) microspheres. Pharm Res. 2000;17:100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Schwendeman SP. Mapping neutral microclimate pH in PLGA microspheres. J Controlled Release. 2005;101:163–173. doi: 10.1016/j.jconrel.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 21.Ding AG, Schwendeman SP. Acidic microclimate pH distribution in PLGA microspheres monitored by confocal laser scanning microscopy. Pharm Res. 2008;25:2041–2052. doi: 10.1007/s11095-008-9594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters T., Jr Serum albumin. Adv Protein Chem. 1985;37:161–245. doi: 10.1016/s0065-3233(08)60065-0. [DOI] [PubMed] [Google Scholar]
- 23.Ding AG, Shenderova A, Schwendeman SP. Prediction of microclimate pH in poly (lactic-co-glycolic acid) films. J Am Chem Soc. 2006;128:5384–5390. doi: 10.1021/ja055287k. [DOI] [PubMed] [Google Scholar]
- 24.Ding AG, Schwendeman SP. Determination of water-soluble acid distribution in poly (lactide-co-glycolide) J Pharm Sci. 2004;93:322–331. doi: 10.1002/jps.10524. [DOI] [PubMed] [Google Scholar]
- 25.Yang YY, Chung TS, Ping Ng N. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials. 2001;22:231–241. doi: 10.1016/s0142-9612(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 26.Kang J, Schwendeman SP. Determination of diffusion coefficient of a small hydrophobic probe in poly (lactide-co-glycolide) microparticles by laser scanning confocal microscopy. Macromolecules. 2003;36:1324–1330. [Google Scholar]
- 27.van de Weert M, van’t Hof R, van der Weerd J, Heeren R, Posthuma G, Hennink WE, Crommelin DJA. Lysozyme distribution and conformation in a biodegradable polymer matrix as determined by FTIR techniques. J Controlled Release. 2000;68:31–40. doi: 10.1016/s0168-3659(00)00227-3. [DOI] [PubMed] [Google Scholar]
- 28.Kang J, Lambert O, Ausborn M, Schwendeman SP. Stability of proteins encapsulated in injectable and biodegradable poly (lactide-co-glycolide)-glucose millicylinders. Int J Pharm. 2008;357:235–243. doi: 10.1016/j.ijpharm.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.