

Abstract
Background
Insulin receptors are widely distributed in the brain, where they play roles in synaptic function, memory formation, and neuroprotection. Autophosphorylation of the receptor in response to insulin stimulation is a critical step in receptor activation. In neurons, insulin stimulation leads to a rise in mitochondrial H2O2 production, which plays a role in receptor autophosphorylation. However, the kinetic characteristics of the H2O2 signal and its functional relationships with the insulin receptor during the autophosphorylation process in neurons remain unexplored to date.
Results
Experiments were carried out in culture of rat cerebellar granule neurons. Kinetic study showed that the insulin-induced H2O2 signal precedes receptor autophosphorylation and represents a single spike with a peak at 5–10 s and duration of less than 30 s. Mitochondrial complexes II and, to a lesser extent, I are involved in generation of the H2O2 signal. The mechanism by which insulin triggers the H2O2 signal involves modulation of succinate dehydrogenase activity. Insulin dose–response for receptor autophosphorylation is well described by hyperbolic function (Hill coefficient, nH, of 1.1±0.1; R2=0.99). N-acetylcysteine (NAC), a scavenger of H2O2, dose-dependently inhibited receptor autophosphorylation. The observed dose response is highly sigmoidal (Hill coefficient, nH, of 8.0±2.3; R2=0.97), signifying that insulin receptor autophosphorylation is highly ultrasensitive to the H2O2 signal. These results suggest that autophosphorylation occurred as a gradual response to increasing insulin concentrations, only if the H2O2 signal exceeded a certain threshold. Both insulin-stimulated receptor autophosphorylation and H2O2 generation were inhibited by pertussis toxin, suggesting that a pertussis toxin-sensitive G protein may link the insulin receptor to the H2O2-generating system in neurons during the autophosphorylation process.
Conclusions
In this study, we demonstrated for the first time that the receptor autophosphorylation occurs only if mitochondrial H2O2 signal exceeds a certain threshold. This finding provides novel insights into the mechanisms underlying neuronal response to insulin. The neuronal insulin receptor is activated if two conditions are met: 1) insulin binds to the receptor, and 2) the H2O2 signal surpasses a certain threshold, thus, enabling receptor autophosphorylation in all-or-nothing manner. Although the physiological rationale for this control remains to be determined, we propose that malfunction of mitochondrial H2O2 signaling may lead to the development of cerebral insulin resistance.
Background
Insulin receptor is a member of the receptor tyrosine kinase family. Upon insulin binding to the extracellular α-subunits, the receptor undergoes rapid autophosphorylation at three specific tyrosine residues within the activation loop of the cytoplasmic β-subunits [1,2], resulting in more than a 200-fold increase in receptor tyrosine kinase activity [3]. Therefore, the autophosphorylated receptor is regarded as fully activated [4]. Research conducted over 30 years ago revealed that cells generate hydrogen peroxide (H2O2) in response to insulin stimulation [5,6]. Evidence from several studies supports the hypothesis that the main role of insulin-induced H2O2 is inhibition of protein tyrosine phosphatases (PTPs), which otherwise dephosphorylate the autophosphorylated insulin receptor [7-9]. According to this theory, H2O2 prolongs the duration of time for which the insulin receptor remains active, rather than directly influence receptor activation. Additionally, exogenous H2O2 has been shown to facilitate receptor autophosphorylation in immunoprecipitates of the insulin receptor in the presence of phosphate donors [10,11]. The obvious independence of this effect on intracellular PTPs suggests that H2O2 also participates in insulin receptor activation. Insulin receptors are widely distributed in the brain, where they play roles in synaptic function, memory formation, and neuroprotection [12-14]. The neuron-specific isoform A is the predominant insulin receptor type in the brain. Isoform A is generated from alternative splicing and differs from its peripheral counterpart (isoform B) in some notable respects, such as higher affinity for insulin and absence of negative cooperativity in insulin binding [15,16]. Earlier studies by our group demonstrated that neurons generate H2O2 in response to insulin stimulation [17]. This H2O2 is derived from the mitochondrial respiratory chain and plays a role in insulin receptor autophosphorylation. However, the kinetic characteristics of the H2O2 signal and its functional relationships with the insulin receptor during autophosphorylation in neurons remain to be clarified. In the current investigation, these issues have been explored as an extension of our previous study.
Results and discussion
Insulin dose–response for receptor autophosphorylation is well described by hyperbolic function
We characterized insulin-stimulated receptor autophosphorylation in a primary culture of rat cerebellar granule neurons (CGN). The insulin dose–response curve for the autophosphorylation process is depicted in Figure 1A. Fitting the curve to the Hill equation generated ED50 of 16.3±2.2 nM and Hill coefficient, nH, of 1.1±0.1 (R2=0.99), indicating that this process in neurons is described by a classic hyperbolic function. These results suggest that the insulin dose–response for receptor autophosphorylation in neurons is gradual and not switch-like.
Figure 1.
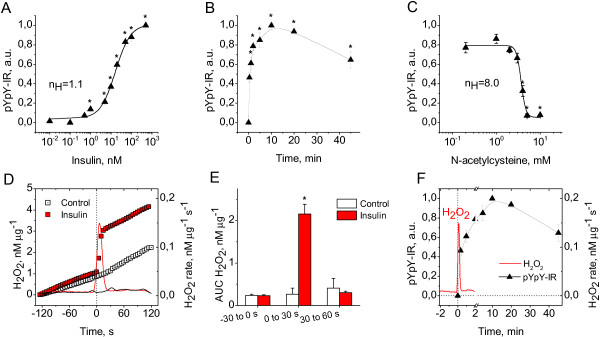
Relationship between H2O2 signal and receptor autophosphorylation in neurons stimulated with insulin. (A) Insulin dose–response for receptor autophosphorylation in CGN exposed to insulin for 10 min (black triangles, mean±SEM of 5 to 9 cultures, *P<0.05 vs. control). (B) Time course of receptor autophosphorylation in CGN exposed to 100 nM insulin (black triangles, mean±SEM of 3 to 4 cultures, *P<0.05 vs. baseline). (C) N-acetylcysteine dose–response for receptor autophosphorylation in CGN exposed to 100 nM insulin for 10 min (black triangles, mean±SEM of 3 to 7 cultures, *P<0.05 vs. 100 nM insulin). (D) Left Y axis: time courses of H2O2 efflux from CGN exposed to vehicle (white squares, mean of 3 culture dishes) or 100 nM insulin (red squares, mean of 10 culture dishes). Right Y axis: first time derivative (rate) of H2O2 efflux from CGN exposed to vehicle (black line) or 100 nM insulin (red line). (E) Areas under curves (AUC) for 30-s periods of H2O2 efflux from CGN exposed to vehicle (white columns, mean±SEM, n=3) or 100 nM insulin (red columns, mean±SEM, n=10, *P<0.05 vs. control). (F) Time courses for insulin-stimulated H2O2 efflux and receptor autophosphorylation. Left Y axis: time course of receptor autophosphorylation in CGN exposed to 100 nM insulin (black triangles, mean±SEM of 3 to 4 cultures). Right axis: first time derivative (rate) of H2O2 efflux from CGN exposed to 100 nM insulin (red line, mean of 10 culture dishes).
The H2O2 signal precedes receptor autophosphorylation during insulin stimulation
To determine the temporal relationship between receptor autophosphorylation and H2O2 generation during insulin stimulation, we compared the kinetics of insulin-induced autophosphorylation and H2O2 production in CGN. The time-course of receptor autophosphorylation in response to insulin stimulation is depicted in Figure 1B. Autophosphorylation peaked at 10 min and dissipated by 30% at 45 min of stimulation. Figure 1D presents the kinetics of the insulin-induced H2O2 signal. H2O2 efflux from neurons into the incubation medium was used as a measure of the signal, given that H2O2 penetrates readily across cellular membranes with an estimated time of gradient formation within 1 s [18]. Our data showed that insulin stimulation evokes a transient single H2O2 spike with a peak at 5–10 s and duration of less than 30 s. We observed a significant difference between the areas under curves (AUCs) calculated for the H2O2 signal in insulin- and vehicle-exposed neurons during the 30 s of insulin stimulation (P<0.001), while no difference was observed at baseline and periods following this time (Figure 1E). A comparison of the kinetic curves for the insulin-induced H2O2 signal and receptor autophosphorylation revealed that the H2O2 signal precedes autophosphorylation (Figure 1F). Notably, at 20 s of stimulation, when the H2O2 signal was 95% complete, receptor autophosphorylation was still in progress.
The H2O2 signal generates strong ultrasensitivity in insulin-induced receptor autophosphorylation
To address the specific function of the H2O2 signal in insulin-stimulated receptor autophosphorylation, we investigated the effects of increasing concentrations of N-acetylcysteine (NAC), a scavenger of hydrogen peroxide, on receptor autophosphorylation in neurons stimulated with insulin. NAC dose-dependently and completely (at concentrations ≥4 mM) inhibited receptor autophosphorylation in CGN cultures exposed to 100 nM insulin (Figure 1C). Approximation of the experimental dose–response data with the Hill function generated IC50 of 3.7±0.2 mM and a Hill coefficient, nH, of 8.0±2.3 (R2=0.97), signifying that the observed dose response is highly sigmoidal and considerably steeper than a hyperbolic curve (nH=1). Therefore, even a small increase in the H2O2 scavenger dose above the threshold results in complete abrogation of receptor autophosphorylation. Given that a steep dose–response with a Hill coefficient nH>1 is defined as ultrasensitive [19,20], the results suggest that the insulin-induced H2O2 signal generates strong ultrasensitivity in receptor autophosphorylation. Autophosphorylation only occurs when the H2O2 signal has surpassed a certain threshold. Conversely, if the H2O2 signal does not reach this threshold, no autophosphorylation occurs, even in response to the highest insulin dose (100 nM). Therefore, H2O2 appears to function as an enabling signal that permits insulin receptor autophosphorylation in an all-or-nothing manner. This mode of switching between two modes of action is often referred to as a decision-making process. In this context, H2O2 signal above the threshold serves as a decision-making step of the neuron to permit activation of the insulin receptor.
Succinate dehydrogenase of mitochondrial complex II regulates the insulin-induced H2O2 signal
To extend our previous finding that the mitochondrial electron transport chain (ETC.) provides the source of insulin-induced H2O2 in neurons [17], we investigated the roles of ETC. complexes I and II in this process. Malonate, a competitive inhibitor of succinate dehydrogenase of complex II, dose-dependently and completely (at concentrations ≥ 4 mM) inhibited receptor autophosphorylation stimulated by 100 nM insulin in neurons, as shown in Figure 2A. Approximation of the malonate dose–response curve for autophosphorylation with the Hill function generated an IC50 of 2.0±0.3 mM and Hill coefficient, nH, of 3.4 (R2=0.92), indicating that the dose response is a highly sigmoidal, and therefore, even a small change in succinate dehydrogenase activity around a certain threshold can have a dramatic effect on insulin receptor autophosphorylation. At concentrations of malonate that completely inhibited receptor autophosphorylation, the insulin-induced H2O2 signal was abolished (Figure 2B,C). Since the inhibitory effects of malonate (on both autophosphorylation and H2O2 generation) were so complete, we conclude that the reducing equivalents for generation of H2O2 do not originate from sources other than the succinate dehydrogenase reaction. The results collectively suggest that activation of succinate dehydrogenase is a possible mechanism underlying insulin-mediated switching of the mitochondrial H2O2 signal critical for receptor autophosphorylation.
Figure 2.
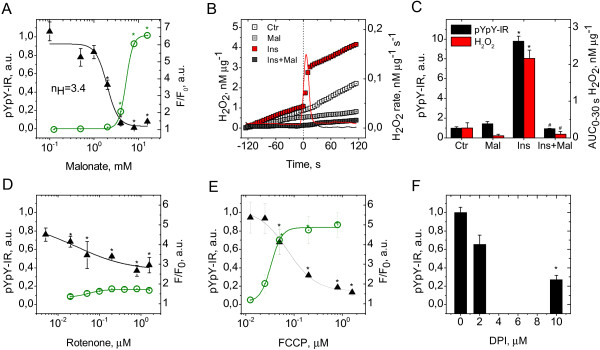
Effects of inhibitors on H2O2 signal and receptor autophosphorylation in neurons stimulated with 100 nM insulin. (A) Left Y axis: malonate dose–response for receptor autophosphorylation in CGN exposed to insulin (triangles, mean±SEM of 3-11 cultures, *P<0.05 vs. insulin). Right Y axis: malonate effect on Rhodamine 123 fluorescence in CGN (circles, mean±SEM of 3-7cultures, *P<0.05 vs. control). (B) Left Y axis: kinetics of H2O2 efflux from CGN exposed to vehicle (mean of 3 cultures), 6 mM malonate (mean of 3 cultures), insulin (mean of 10 cultures), or insulin plus 6 mM malonate (mean of 5 cultures). Right Y axis: rates of H2O2 efflux from CGN exposed to insulin (red line) or insulin plus 6 mM malonate (grey line). (C) Left Y axis: receptor autophosphorylation in CGN exposed to vehicle, 6 mM malonate, insulin, or insulin plus 6 mM malonate (mean±SEM of 5-11 cultures. *P<0.05 vs. control. #P<0.05 vs. insulin). Right Y axis: areas under curves of H2O2 efflux from CGN exposed to vehicle, 6 mM malonate, insulin, or insulin plus 6 mM malonate for 30 s (mean±SEM of 3-10 cultures. *P<0.05 vs. control. #P<0.05 vs. insulin). (D) Left Y axis: rotenone dose–response for receptor autophosphorylation in CGN exposed to insulin (triangles, mean±SEM of 3-4 cultures, *P<0.05 vs. insulin). Right Y axis: rotenone effect on Rhodamine 123 fluorescence in CGN (circles, mean±SEM of 3-4 cultures, *P<0.05 vs. control). (E) Left Y axis: FCCP dose–response for receptor autophosphorylation in CGN exposed to insulin (triangles, mean±SEM of 3-5 cultures, *P<0.05 vs. insulin). Right Y axis: FCCP dose-response for Rhodamine 123 fluorescence in CGN (circles, mean±SEM of 4-8 cultures, *P<0.05 vs. control). (F) DPI effect on receptor autophosphorylation in CGN exposed to insulin (mean±SEM of 3 cultures, *P<0.05 vs. insulin).
As shown in Figure 2B, the rate of H2O2 generation exhibits biphasic behavior, showing a fast initial increase followed by a rapid decrease to the baseline steady-state level while insulin stimulation is sustained. In keeping with common theory, such a response can be generated by an incoherent feed-forward loop (IFFL) in which insulin modulates the H2O2 rate via at least two intermediate pathways with opposite functions of “fast activation” and “delayed inhibition”. We speculate that fast activation and delayed inhibition of succinate dehydrogenase generates the biphasic response. In this scheme, delayed inhibition of succinate dehydrogenase is attributed to the downstream metabolites of succinate oxidation, oxaloacetate [21] and H2O2[22-24]. These metabolites act in orchestrated manner to yield optimal inhibition, since oxaloacetate binds the oxidized form of succinate dehydrogenase more effectively than the reduced form [25] and oxidative conditions induce more significant inhibition of succinate dehydrogenase by oxaloacetate [26-29]. Given that the transition times between the active and inactive states of succinate dehydrogenase [26,30] correspond well to the timing of the insulin-induced biphasic response, we propose that succinate dehydrogenase has sufficient regulatory capacity to generate the transient H2O2 signal in response to insulin stimulation.
To clarify the factors influencing generation of the insulin-induced H2O2 signal, we briefly reviewed current knowledge on succinate-supported H2O2 generation in isolated mitochondria. Succinate promotes the highest rates of H2O2 production among all respiratory substrates [31-35]. Most mitochondrial H2O2 is produced from superoxide generated by reduction of molecular oxygen in the electron transport chain (ETC.) [36,37]. Although several ETC. sites may generate superoxide in mitochondria respiring on succinate, the majority is produced at complex II [38] and at complex I during reverse electron transport (RET) from complex II [32-36], where complex I-associated superoxide production is sensitive to modulation of the mitochondrial membrane potential, ΔΨm[32,39]. Notably, RET-associated superoxide generation is observed at the highest non-physiological succinate concentrations at which superoxide production at complex II is suppressed, whereas generation at complex II occurs only within the range of lower physiologically relevant succinate levels [38]. At succinate concentrations favoring RET-associated H2O2 production, generation of H2O2 depends on the metabolic state of mitochondria and changes in parallel with succinate dehydrogenase activity. The highest H2O2 rates [37] and succinate dehydrogenase activity [27,28] are observed at metabolic state 4. On transition to state 3, H2O2 production rates drop rapidly [34,39] and succinate dehydrogenase activity decreases [27,28]. Protonophores completely inhibit H2O2 production [33,39] and deactivate succinate dehydrogenase to the lowest observable activity [27]. In summary, the documented literature suggests that ΔΨm and activity of mitochondrial complexes I/II are the candidate factors that influence generation of the insulin-induced H2O2 signal and thus play a role in the control of insulin-stimulated receptor autophosphorylation in neurons.
Mitochondrial complex I is involved in control of insulin-induced receptor autophosphorylation, but to a lower extent than complex II
To investigate whether ETC. complex I functions in the control of insulin-stimulated receptor autophosphorylation, we examined the effects of rotenone on receptor autophosphorylation in neurons stimulated with 100 nM insulin. Rotenone is a selective inhibitor of the ubiquinone reduction site at mitochondrial complex I. Rotenone inhibited insulin-stimulated receptor autophosphorylation, as shown in Figure 2D. The effect of rotenone was significant, but incomplete, and even at the highest concentration, autophosphorylation was reduced by less than 50%. Fitting the dose–response curve generated a Hill coefficient, nH, of 0.7 (R2=0.83), indicating that the observed dose response is gradual and not switch-like. Our results suggest that ETC. complex I is involved in control of insulin receptor autophosphorylation, presumably as part of the mitochondrial machinery of RET-associated H2O2 generation. However, its role does not appear to be as important as that of ETC. complex II.
Insulin-induced receptor autophosphorylation is sensitive to mitochondrial depolarization
To address whether receptor autophosphorylation is sensitive to ΔΨm, we compared the effects of insulin and other additives on autophosphorylation and ΔΨm in neurons. Rhodamine 123 was used as a measure of mitochondrial ΔΨm[40]. Insulin did not have a significant effect on Rhodamine 123 fluorescence in neurons. Malonate induced a dose-dependent increase in Rhodamine 123 fluorescence, signifying that inhibition of succinate dehydrogenase results in mitochondrial depolarization (Figure 2A). Fitting the fluorescence curve to the Hill equation led to ED50 of 5.2±0.1 mM and Hill coefficient, nH, of 5.4 (R2=0.99). Comparison of the malonate dose–response curves for autophosphorylation and fluorescence revealed that at a malonate concentration of 2 mM (inducing 50% inhibition of autophosphorylation), mitochondria were depolarized by less than 1%. This finding suggests that ΔΨm in neurons is ultrasensitive to succinate dehydrogenase activity and mitochondrial depolarization is not a causative factor for the inhibitory effect of malonate on receptor autophosphorylation. Figure 2D shows that the inhibitory effect of rotenone on autophosphorylation is not accompanied by mitochondrial depolarization. The protonophore, FCCP, known to dissipate the transmembrane proton gradient, evoked an increase in Rhodamine 123 fluorescence and inhibited insulin-stimulated receptor autophosphorylation in a dose-dependent manner (Figure 2E). Fitting the FCCP dose–response curves for fluorescence and autophosphorylation gave ED50 of 0.030±0.003 (R2=0.99) and IC50 of 0.07±0.02 (R2=0.98), respectively. At a FCCP concentration of 0.07 μM (inducing 50% inhibition of autophosphorylation), >95% mitochondrial depolarization was observed. These data suggest that FCCP-induced mitochondrial depolarization leads to inhibition of insulin-stimulated receptor autophosphorylation. Given that protonophores deactivate succinate dehydrogenase to the lowest observable activity [27], we propose that the inhibitory effect of depolarization on autophosphorylation is mediated via deactivation of succinate dehydrogenase. Taken together, these results suggest that insulin receptor autophosphorylation is sensitive to factors inducing mitochondrial depolarization, while modulation of ΔΨm is not implicated in the mechanism of insulin-triggered receptor autophosphorylation.
Nox is not involved in control of insulin-induced receptor autophosphorylation in neurons
A number of previous studies using non-neuronal cells have assigned the insulin-induced H2O2 signal to activation of NADPH oxidase (Nox) that is sensitive to inhibition by diphenyleneiodonium (DPI) [41-43]. Accordingly, we investigated the effect of DPI on receptor autophosphorylation in neurons exposed to 100 nM insulin. DPI treatment at a concentration of 10 μM significantly inhibited insulin-stimulated receptor autophosphorylation. Moreover, the inhibitory action of DPI was dose-dependent (Figure 2F). Since selective mitochondrial inhibitors that do not inhibit Nox completely abrogate both insulin-induced H2O2 generation and receptor autophosphorylation, the results of the DPI experiment cannot be interpreted to conclude that Nox is a source of the insulin-induced H2O2 signal in neurons. Although the nonspecific flavoprotein inhibitor, DPI, is often claimed to be a specific inhibitor of Nox, compelling evidence shows that DPI inhibits many targets [44], including ROS generation at the mitochondrial complex I [45,46] and succinate-supported H2O2 generation in rat brain mitochondria [33]. In this context, data from the DPI experiment do not contradict our main conclusion that the mitochondrial ETC. is the only source of insulin-induced H2O2 in neurons. Another argument against a role of Nox in neuronal insulin receptor autophosphorylation is that the kinetics of the insulin-induced H2O2 signal is unusually fast, compared to that previously observed for Nox in non-neuronal cells [8,43]. The neuronal H2O2 signal is a single spike with a sharp peak at 5–10 s and duration of less than 30 s. In contrast, the insulin-induced oxidant signal in adipocytes reaches a peak at 5 min and begins to dissipate by 10 min [8]. In HepG2 cells, ROS generation is optimal at about 10 min and dissipates after 30 min of insulin stimulation [43]. This significant difference in the timing of H2O2 signals indicates that the source of insulin-induced H2O2 in neurons differs from that in adipocytes and HepG2 cells, which is Nox [8,43]. Our results collectively suggest that Nox is not involved in H2O2 signaling that controls insulin-stimulated receptor autophosphorylation in neurons.
Data from the present study raise the issue of whether the mitochondrial origin of the insulin-induced H2O2 signal is a neuron-specific phenomenon. Although abundant literature is available on insulin-induced H2O2 generation in cells, limited studies have focused on the origin of insulin-induced H2O2 implicated in insulin receptor autophosphorylation. In experiments with 3T3-L1 adipocytes [41,42] and HepG2 cells [43], the source of H2O2 was assigned to activated Nox on the basis of data obtained with DPI only. Strong evidence from experiments with dominant-negative Nox4 constructs and Nox4 siRNA further suggested that Nox4 is the source of the insulin-induced H2O2 signal in adipocytes [47]. The results from the present study, together with previous data [17], indicate that mitochondrial ETC. is the only source of insulin-induced H2O2 in neurons. In summary, owing to limited data, it is not currently possible to address whether mitochondria represent the only neuronal source of insulin-induced H2O2 implicated in receptor activation.
The insulin-induced H2O2 signal and receptor autophosphorylation in neurons are pertussis toxin-sensitive
A pertussis toxin-sensitive G protein links the insulin receptor to the insulin-induced H2O2 signal in non-neuronal cells [48]. To determine whether G protein signaling is involved in generation of insulin-induced H2O2 in neurons, we examined the effects of pertussis toxin (PTX), an inhibitor of Gi/0 protein-receptor coupling, on H2O2 generation and receptor autophosphorylation in CGN exposed to 100 nM insulin. PTX treatment completely inhibited insulin-stimulated receptor autophosphorylation at the highest dose of 2 mg/L, and this suppression was dose-dependent (Figure 3A). Approximation of the curve to the Hill equation gave IC50 of 0.16±0.06 mg/L and Hill coefficient, nH, of 1.7±2.0 (R2=0.96), implying that insulin-induced receptor autophosphorylation is ultrasensitive to PTX. PTX (2 mg/L) completely eliminated the insulin-induced H2O2 signal (Figure 3B). Thus, at the dose at which PTX completely abrogated the H2O2 signal (2 mg/L), receptor autophosphorylation was abolished, as shown in Figure 3C. Based on these results, we suggest that a PTX-sensitive G protein links the insulin receptor to the H2O2-generating system in neurons during generation of the insulin-induced H2O2 signal critical for insulin receptor autophosphorylation.
Figure 3.
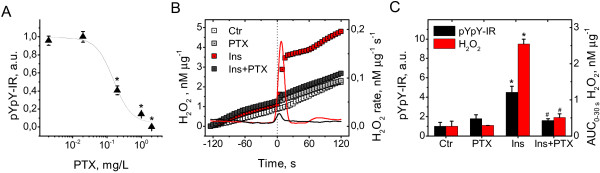
Effects of pertussis toxin on H2O2 generation and receptor autophosphorylation neurons stimulated with insulin. (A) PTX dose–response curve for receptor autophosphorylation in CGN exposed to 100 nM insulin for 10 min (black triangles, mean±SEM of 3 to 8 cultures, *P<0.05 vs. 100 nM insulin). (B) Left Y axis: time courses of H2O2 efflux from CGN exposed to control buffer (white squares, mean of 3 culture dishes), 2 mg/L PTX (light grey squares, mean of 3 culture dishes), 100 nM insulin (red squares, mean of 3 culture dishes), or 100 nM insulin plus 2 mg/L PTX (grey squares, mean of 3 culture dishes). Right Y axis: first time derivatives (rates) of H2O2 efflux from CGN exposed to 100 nM insulin (red line) or 100 nM insulin plus 2 mg/L PTX (black line). (C) Left Y axis: receptor autophosphorylation in CGN exposed to control buffer, 2 mg/L PTX, 100 nM insulin, or 100 nM insulin plus 2 mg/L PTX. Black columns represent the mean±SEM of values obtained from 3 to 8 cultures. *P<0.05 vs. control. #P<0.05 vs. insulin. Right Y axis: Areas under curves (AUC) of H2O2 efflux for 30 s from CGN exposed to control buffer, 2 mg/L PTX, 100 nM insulin, or 100 nM insulin plus 2 mg/L PTX. Red columns represent the mean±SEM of values obtained from 3 culture dishes. *P<0.05 vs. control. #P<0.05 vs. insulin.
Although our data show for the first time that a PTX-sensitive G protein is possibly involved in activation of the insulin receptor in CNS, the involvement of a PTX-sensitive G protein in insulin receptor activation in peripheral tissues is widely documented. In mice, Gαi2 deficiency results in impaired tyrosine phosphorylation of the insulin receptor substrate, IRS1, and frank insulin resistance in peripheral tissues [49], whereas targeted expression of a constitutively active form of Gαi2 enhances insulin action through amplifying tyrosine phosphorylation of the insulin receptor and IRS1 [50-52]. In human adipocytes, insulin recruits Gαi2 to activate Nox-dependent H2O2 generation [48] and regulate insulin receptor autophosphorylation [53]. Although no data supporting the involvement of Gαi2 in brain insulin signaling are available, Gαi2, and especially its short-lived splice variant, sGi2, are widely distributed throughout rat and monkey brain, where sGi2 is detected in both axons and dendrites at presynaptic and postsynaptic sites [54]. Gαi2 operates largely in plasma membranes, while sGi2 is localized in a variety of subcellular locations, including mitochondria [54-56]. Our present results are generally in keeping with current knowledge on the amplification role of pertussis toxin-sensitive Gi protein in insulin receptor activation. However, the specific G protein isoform implicated in neuronal insulin receptor activation remains to be determined.
Functional association of the insulin receptor and mitochondria during receptor activation in neurons
It is possible to draw some tentative conclusions regarding the functional relationship between the insulin receptor and mitochondria during receptor activation in neurons (Figure 4). Insulin stimulation induces receptor autophosphorylation, which reaches a peak at 10 min. Upon autophosphorylation, the receptor becomes fully activated and initiates signaling to the inside of the neuron. At times preceding autophosphorylation, insulin induces a transient H2O2 signal, which plays a permissive role in activation of the insulin receptor. Autophosphorylation only occurs once the H2O2 signal has surpassed a certain threshold. Under conditions where the H2O2 signal does not reach this threshold, no autophosphorylation occurs, even in response to the highest insulin dose. In this context, H2O2 signal above the threshold serves as the neuron’s decision to activate the insulin receptor. The insulin-induced H2O2 signal is derived from mitochondria. Succinate dehydrogenase in complex II plays a key role in control of H2O2 generation. An unknown pertussis toxin-sensitive G protein links the insulin receptor to the mitochondrial H2O2-generating system during H2O2 signal activation.
Figure 4.
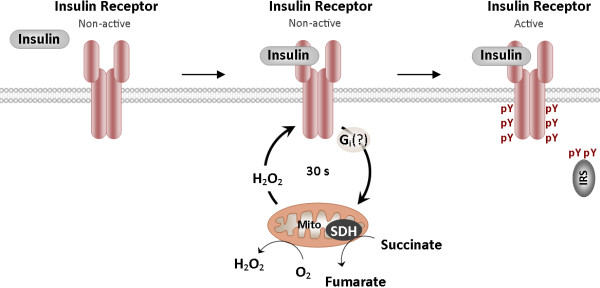
Scheme of functional relationship between insulin receptor and mitochondria during receptor activation in neurons. Insulin stimulation evokes a transient single H2O2 spike with a peak at 5–10 s and duration of less than 30 s. Mitochondrial complex II and, to a lesser extent, I are involved in H2O2 generation. Autophosphorylation only occurs when the H2O2 signal has surpassed a certain threshold. Conversely, if the H2O2 signal does not reach this threshold, no autophosphorylation occurs, even in response to the highest insulin dose. Upon the autophosphorylation, receptor tyrosine kinase becomes fully activated and initiates signaling to the inside of the neuron. Therefore, the receptor is activated if two conditions are met: 1) insulin binds to the receptor, and 2) the H2O2 signal exceeds a certain threshold, enabling receptor autophosphorylation.
The current study provides novel insights into the mechanisms underlying neuronal response to insulin. The insulin receptor is activated if two conditions are met: 1) insulin binds to the receptor, and 2) the H2O2 signal exceeds a certain threshold, enabling receptor autophosphorylation. Generally, this mode of control means that activation of the neuronal insulin receptor is conditional on mitochondrial functioning. Moreover, receptor activation may be conditional on neural activity. Neural activity evokes insulin release from synaptosomes within nerve endings into the synaptic cleft [57,58] and induces mitochondrial migration to dendritic spines [59], which are commonly poor in mitochondria. Therefore, periods of synaptic activity favor insulin receptor activation in the postsynaptic density of dendritic spines where the two above conditions are met.
Possible links between impairment of H2O2 signaling and insulin resistance
Given the importance of the insulin-induced H2O2 signal in autophosphorylation of the neuronal insulin receptor, factors that disrupt H2O2 signaling may generate unresponsiveness or resistance to insulin. In line with the above findings, mitochondrial dysfunction that affects generation of the H2O2 signal may induce central insulin resistance. Moreover, insulin resistance may be induced by pathologically elevated activity of antioxidant enzymes that metabolize H2O2, e.g., arising as a compensatory response to oxidative stress. Accumulating evidence from animal and human studies supports this hypothesis. Glutathione peroxidase (Gpx1) and peroxiredoxins (Prx) are primary H2O2 scavengers capable of metabolizing the insulin-induced H2O2 signal, according to kinetic estimations based on published second-order rate constants of ~107 M-1 s-1 and abundance data [60-62]. Mice overexpressing Gpx1 are insulin-resistant, obese, and along with hyperinsulinemia, display a 70% reduction in insulin-stimulated phosphorylation of insulin receptors, compared to wild-type control mice [63]. In contrast, mice lacking Gpx1 are protected from high fat diet-induced insulin resistance, while administration of the H2O2 scavenger, NAC, renders them more insulin-resistant [64]. Brain insulin resistance is an early and common feature of Alzheimer’s disease (AD). Compared to age-matched normal brains, AD brains are characterized by diminished tyrosine phosphorylation of the insulin receptor and its substrate, IRS1 [65], and significant overexpression of glutathione peroxidase [66,67] and peroxiredoxins (Prx1 and Prx2) [68-71]. Elevated antioxidant activity in early AD is considered the result of compensatory response to oxidative stress [72,73]. From this viewpoint, oxidative stress seems to be another factor that interferes with the insulin-induced H2O2 signal and consequently induces central insulin resistance.
Conclusions
In summary, we have demonstrated for the first time that the receptor autophosphorylation occurs only if mitochondrial H2O2 signal exceeds a certain threshold. Our findings provide a novel insight into the mechanisms underlying neuronal response to insulin. The neuronal insulin receptor is activated if two conditions are met: 1) insulin binds to the receptor, and 2) the H2O2 signal surpasses a certain threshold, thus, enabling receptor autophosphorylation in all-or-nothing manner. Although the physiological rationale for this control remains to be determined, we propose that malfunction of insulin-induced H2O2 signaling may lead to cerebral insulin resistance, in view of the critical significance of the H2O2 signal for insulin receptor activation in neurons.
Methods
Materials
PhosphoDetect™ Insulin Receptor (pTyr1162/1163) ELISA kit and Insulin Receptor (β-Subunit) ELISA Kit were from Calbiochem. Other materials were purchased from Sigma, ICN, Gibco, Biosource, Invitrogen and Acros.
Neuronal culture
Cerebellar granule neurons were prepared from 7- to 8-day-old Wistar rats, as described earlier [74]. Cerebellum was dissected and placed in ice-cold Ca2+/Mg2+-free Hanks’ buffered salt solution (HBSS) without Phenol Red (Gibco). After mincing with fine scissors, tissue was placed in Ca2+/Mg2+-free HBSS with Phenol Red and 0.1% trypsin for 15 min at 37°C. Trypsin was inactivated by washing with normal HBSS. Cells were dissociated via trituration and pelleted in HBSS. Next, cells were resuspended in Neurobasal Medium (Gibco) supplemented with serum-free B-27 Supplement (100X, Gibco), 20 mmol/L KCl, GlutaMax (100X, Gibco) and antibiotic-antimycotic (100X, Gibco), and plated at a density of 5×106 cells/ml onto 35X10 mm sterile cell culture dishes or 6×24 plates (Corning) previously coated with polyethyleneimine (10 mkg/ml). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air, and fed with supplemented Neurobasal Medium. Cultures were treated on day 3 with 5 μM cytosine arabinoside (Sigma) for 24 h to prevent glial proliferation, and the medium subsequently changed. Neurons on days 7 to 9 were used for experiments.
Measurement of hydrogen peroxide
H2O2 efflux from CGN cultures was measured using fluorimetry employing the cell-impermeable Amplex Red dye in the presence of horseradish peroxidase (Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit, Cat. No. A22188, Invitrogen). CGN cultures were pre-incubated for 120 min in Hepes-buffered salt solution composed of 156 mM NaCl, 3 mM KCl, 2 mM MgSO4, 1.25 mM KH2PO4, 2 mM CaCl2, 0.5 mM glucose, 20 mM HEPES, and the pH adjusted to 7.4 with NaOH. Cultures were subsequently exposed to Hepes-buffered salt solution supplemented with 50 μM Amplex Red and 0.1 U/ml horseradish peroxidase (incubation medium), and H2O2 efflux recorded every 5 s for 2 min. The incubation medium was rapidly exchanged with fresh medium containing no insulin (control) or 100 nM insulin, and H2O2 efflux recorded every 5 s for 2 min. Where indicated, PTX (2 μg/ml) was added 120 min before and during insulin stimulation. Malonate (6 mM) was added 5 min before and during insulin stimulation. Fluorescence was measured with an epifluorescent inverted microscope Axiovert 200 (Carl Zeiss, Germany) equipped with a 20X fluorite objective, using excitation at 550±10 nm and fluorescence detection at 610±30 nm. All imaging data were collected and analyzed using Metafluor 6.1 software (Universal Imaging Corp., USA). Standard curves of H2O2 concentrations were linear up to 1500 nM and used to convert fluorescence values to H2O2 values. The calculated detection limit of the assay was 7 nM. Data were normalized to total amounts of cell protein, and expressed as H2O2 nM μg-1.
Insulin receptor autophosphorylation assay
Amounts of the autophosphorylated β-subunit of the insulin receptor were measured with the PhosphoDetect™ insulin receptor (pTyr1162/1163) ELISA kit (Calbiochem) suitable for studies with the rat insulin receptor. CGN cultures were incubated in Hepes-buffered salt solution (145 mM NaCl, 5.6 mM KCl, 1.8 mM СаCl2, 1 mM MgCl2, 20 mM HEPES, and 0.5 mM glucose) at pH 7.4 for 30 min, followed by exposure to insulin (10 min, 37°C) or no insulin (control) in the presence or absence of other additives (preincubation for 5–30 min before insulin). The experiment was terminated by removing the medium, washing with ice-cold PBS, and adding 200 μL/dish cell lysis buffer (Biosource) supplemented with 1 mM PMSF, 50 mM protease inhibitor set III (Sigma), and 2 mM sodium orthovanadate as the tyrosine phosphatase inhibitor on ice for 10 min. Lysates were centrifuged at 12,000 rpm at 4°C for 12 min. In each CGN lysate, amounts of autophosphorylated receptors were measured as described by the manufacturer. For each experiment, values were normalized to the total amounts of insulin receptor β-subunit (IR) measured using the insulin receptor (β-subunit) ELISA kit (Calbiochem) and normalized to scale between 0 and 1 using Equation 1:
| (1) |
where Y i is an original receptor autophosphorylation value, Normalized (Y i ) the normalized autophosphorylation value, Y CTR the mean autophosphorylation value in neurons exposed to vehicle, and Y INS the mean autophosphorylation value in neurons exposed to 100 nM insulin.
Monitoring of Rhodamine 123 fluorescence
Mitochondrial depolarization changes in individual neurons within CGN cultures in response to additives were measured via fluorescence of Rhodamine 123, as described earlier [75]. CGN cultures were equilibrated with 10 μg/ml Rhodamine 123 in Hepes-buffered salt solution for 10 min at 20°C. Cells were washed with HBSS before the experiment (excitation, 488 nm; emission, 530 nm). CGN cultures were exposed to insulin, rotenone, malonate or FCCP for 10 min, and fluorescence measured. Data were expressed as the F/F0 ratio of the fluorescence signal measured after exposure of neurons to additives to that measured at baseline.
Curve fitting
Experimental data were fitted by non-linear regression to the Hill equation to provide the parameter values in Equation 2:
| (2) |
where Y is a parameter value, X a variable, K the concentration of the variable producing half the effect, and n H the Hill coefficient.
Statistical analysis
Data were analyzed for statistical significance with one-way analysis of variance (ANOVA). Values are presented as means ± SEM. Differences were considered significant at P<0.05.
Abbreviations
CGN: Cerebellar granule neurons; DPI: Diphenyleneiodonium; FCCP: Carbonyl cyanide-4-(trifluoromethoxy)-phenylhydrazone; HBSS: Hanks’ buffered salt solution; HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; NAC: N-acetylcysteine; PBS: Phosphate-buffered saline; PMSF: Phenylmethylsulfonyl fluoride; PTX: Pertussis toxin; PTPs: Protein tyrosine phosphatases; SEM: Standard error of mean.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NAP carried out the in vitro studies with CGN cultures and data analysis. TPS carried out the in vitro studies with CGN cultures and data analysis. YES carried out the in vitro studies with CGN cultures and data analysis. LRG carried out the DPI experiments in CGN cultures and data analysis; VGP participated in the design of the in vitro studies with CGN cultures, and manuscript evaluation/critique. IAP conceived, designed and coordinated the study, and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Nadezhda A Persiyantseva, Email: nadushka99@rambler.ru.
Tatiana P Storozhevykh, Email: tstor@mail.ru.
Yana E Senilova, Email: ysenilova@mail.ru.
Lubov R Gorbacheva, Email: lyubovgorbacheva@gmail.com.
Vsevolod G Pinelis, Email: pinelis@mail.ru.
Igor A Pomytkin, Email: ipomytkin@mail.ru.
Acknowledgements
This work was funded by Biosignal Ltd., Moscow, Russia.
References
- White MF, Shoelson SE, Keutmann H, Kahn CR. A cascade of tyrosine autophosphorylation in the β-subunit activates the phosphotransferase of the insulin receptor. J Biol Chem. 1988;263:2969–2980. [PubMed] [Google Scholar]
- Rosen OM, Herrera R, Olowe Y, Petruzzelli LM, Cobb MH. Phosphorylation activates the insulin receptor tyrosine protein kinase. Proc Natl Acad Sci USA. 1983;80:3237–3240. doi: 10.1073/pnas.80.11.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wie L, Hubbard SR, Hendrickson WA, Ellis L. Expression, characterization, and crystallization of the catalytic core of the human insulin receptor protein-tyrosine Kinase domain. J Biol Chem. 1995;270:8122–8130. doi: 10.1074/jbc.270.14.8122. [DOI] [PubMed] [Google Scholar]
- White MF, Kahn CR. The insulin signaling system. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- Mukherjee SP, Lane RH, Lynn WS. Endogenous hydrogen peroxide and peroxidative metabolism in adipocytes in response to insulin and sulfhydryl reagents. Biochem Pharmacol. 1978;27(22):2589–2594. doi: 10.1016/0006-2952(78)90332-5. [DOI] [PubMed] [Google Scholar]
- May JM, de Haen C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J Biol Chem. 1979;254(7):2214–2220. [PubMed] [Google Scholar]
- Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- Pomytkin IA, Kolesova OE. Key role of succinate dehydrogenase in insulin-induced inactivation of protein tyrosine phosphatases. Bull Exp Biol Med. 2002;133:568–570. doi: 10.1023/A:1020229724717. [DOI] [PubMed] [Google Scholar]
- Schmid E, El Benna J, Galter D, Klein G, Dröge W. Redox priming of the insulin receptor beta-chain associated with altered tyrosine kinase activity and insulin responsiveness in the absence of tyrosine autophosphorylation. FASEB J. 1998;12(10):863–870. doi: 10.1096/fasebj.12.10.863. [DOI] [PubMed] [Google Scholar]
- Schmid E, Hotz-Wagenblatt A, Hacj V, Dröge W. Phosphorylation of the insulin receptor kinase by phosphocreatine in combination with hydrogen peroxide: the structural basis of redox priming. FASEB J. 1999;13(12):1491–1500. doi: 10.1096/fasebj.13.12.1491. [DOI] [PubMed] [Google Scholar]
- Abbott MA, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J Neurosci. 1999;19:7300–7308. doi: 10.1523/JNEUROSCI.19-17-07300.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SL, Cline HT. Insulin receptor signaling in the development of neuronal structure and function. Neural Dev. 2010;5:7. doi: 10.1186/1749-8104-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: more than just a peripheral hormone. J Aging Res. 2012;2012:384017. doi: 10.1155/2012/384017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammeltoft S, Fehlmann M, Van Obberghen E. Insulin receptors in the mammalian central nervous system: binding characteristics and subunit structure. Biochimie. 1985;67(10–11):1147–1153. doi: 10.1016/s0300-9084(85)80113-9. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Flier JS, Yokota A, Benecke H, Backer JM, Moller DE. Functional properties of two naturally occurring isoforms of the human insulin receptor in Chinese hamster ovary cells. Endocrinology. 1991;129(4):2058–2066. doi: 10.1210/endo-129-4-2058. [DOI] [PubMed] [Google Scholar]
- Storozhevykh TP, Senilova YE, Persiyantseva NA, Pinelis VG, Pomytkin IA. Mitochondrial respiratory chain is involved in insulin-stimulated hydrogen peroxide production and plays an integral role in insulin receptor autophosphorylation in neurons. BMC Neurosci. 2007;8:84. doi: 10.1186/1471-2202-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–126. doi: 10.1016/S0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- Goldbeter A, Koshland DE Jr. An amplified sensitivity arising from covalent modification in biological systems. Proc Natl Acad Sci USA. 1981;78(11):6840–6844. doi: 10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE Jr. Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996;21:460–466. doi: 10.1016/S0968-0004(96)20026-X. [DOI] [PubMed] [Google Scholar]
- Pardee AB, Potter VR. Inhibition of succinic dehydrogenase by oxalacetate. J Biol Chem. 1948;176:1085–1094. [PubMed] [Google Scholar]
- Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem. 2001;276:23357–23361. doi: 10.1074/jbc.M100320200. [DOI] [PubMed] [Google Scholar]
- Moser MD, Matsuzaki S, Humphries KM. Inhibition of succinate-linked respiration and complex II activity by hydrogen peroxide. Arch Biochem Biophys. 2009;488:69–75. doi: 10.1016/j.abb.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackrell BA, Kearney EB, Edmondson D. Mechanism of the reductive activation of succinate dehydrogenase. J Biol Chem. 1975;250:7114–7119. [PubMed] [Google Scholar]
- Gutman M, Kearney EB, Singer TP. Control of succinate dehydrogenase in mitochondria. Biochemistry. 1971;10:4763–4770. doi: 10.1021/bi00801a025. [DOI] [PubMed] [Google Scholar]
- Gutman M, Kearney EB, Singer TP. Multiple control mechanisms for succinate dehydrogenase in mitochondria. Biochem Biophys Res Commun. 1971;44:526–532. doi: 10.1016/S0006-291X(71)80114-6. [DOI] [PubMed] [Google Scholar]
- Gutman M, Kearney EB, Singer TP. Activation of succinate dehydrogenase by electron flux from NADH and its possible regulatory function. Biochem Biophys Res Commun. 1971;42:1016–1023. doi: 10.1016/0006-291X(71)90005-2. [DOI] [PubMed] [Google Scholar]
- de Gómez-Puyou MT, Chávez E, Freites D, Gómez-Puyou A. On the regulation of succinate dehydrogenase in brain mitochondria. FEBS Lett. 1972;22:57–60. doi: 10.1016/0014-5793(72)80218-7. [DOI] [PubMed] [Google Scholar]
- Singer TP, Gutman M, Kearney EB. On the need for regulation of succinate dehydrogenase. FEBS Lett. 1971;17:11–13. doi: 10.1016/0014-5793(71)80550-1. [DOI] [PubMed] [Google Scholar]
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- Gyulkhandanyan AV, Pennefather PS. Shift in the localization of sites of hydrogen peroxide production in brain mitochondria by mitochondrial stress. J Neurochem. 2004;90:405–421. doi: 10.1111/j.1471-4159.2004.02489.x. [DOI] [PubMed] [Google Scholar]
- Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2004;279:4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/S0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A. Assessment of mitochondrial polarization status in living cells based on analysis of the spatial heterogeneity of rhodamine 123 fluorescence staining. Pflugers Arch. 2000;440:941–947. doi: 10.1007/s004240000390. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem. 2001;276(52):48662–48669. doi: 10.1074/jbc.M105061200. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Wu X, Motoshima H, Goldstein BJ. Integration of multiple downstream signals determines the net effect of insulin on MAP kinase vs. PI 3′-kinase activation: potential role of insulin-stimulated H(2)O(2) Cell Signal. 2004;16(3):323–331. doi: 10.1016/j.cellsig.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Biswas S, Gupta MK, Chattopadhyay D, Mukhopadhyay CK. Insulin-induced activation of hypoxia-inducible factor-1 requires generation of reactive oxygen species by NADPH oxidase. Am J Physiol Heart Circ Physiol. 2007;292(2):H758–H766. doi: 10.1152/ajpheart.00718.2006. [DOI] [PubMed] [Google Scholar]
- Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab. 2008;9(8):686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- Majander A, Finel M, Wikstrom M. Diphenyleneiodonium inhibits reduction of iron-sulfur clusters in the mitochondrial NADH-ubiquinone oxidoreductase (Complex I) J Biol Chem. 1994;269(33):21037–21042. [PubMed] [Google Scholar]
- Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253(2):295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger-Brauer HI, Medda PK, Kather H. Insulin-induced activation of NADPH-dependent H2O2 generation in human adipocyte plasma membranes is mediated by Galphai2. J Biol Chem. 1997;272:10135–10143. doi: 10.1074/jbc.272.15.10135. [DOI] [PubMed] [Google Scholar]
- Moxham CM, Malbon CC. Insulin action impaired by deficiency of the G-protein subunit G ialpha2. Nature. 1996;379(6568):840–844. doi: 10.1038/379840a0. [DOI] [PubMed] [Google Scholar]
- Chen JF, Guo JH, Moxham CM, Wang HY, Malbon CC. Conditional, tissue-specific expression of Q205L G alpha i2 in vivo mimics insulin action. J Mol Med (Berl) 1997;75(4):283–289. doi: 10.1007/s001090050113. [DOI] [PubMed] [Google Scholar]
- Song X, Zheng X, Malbon CC, Wang H. Galpha i2 enhances in vivo activation of and insulin signaling to GLUT4. J Biol Chem. 2001;276(37):34651–34658. doi: 10.1074/jbc.M105894200. [DOI] [PubMed] [Google Scholar]
- Tao J, Malbon CC, Wang HY. Galpha(i2) enhances insulin signaling via suppression of protein-tyrosine phosphatase 1B. J Biol Chem. 2001;276(43):39705–39712. doi: 10.1074/jbc.M105216200. [DOI] [PubMed] [Google Scholar]
- Kreuzer J, Nurnberg B, Krieger-Brauer HI. Ligand-dependent autophosphorylation of the insulin receptor is positively regulated by Gi-proteins. Biochem J. 2004;380(Pt 3):831–836. doi: 10.1042/BJ20031659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan ZU, Gutierrez A. Distribution of C-terminal splice variant of G alpha i2 in rat and monkey brain. Neuroscience. 2004;127(4):833–843. doi: 10.1016/j.neuroscience.2004.05.017. [DOI] [PubMed] [Google Scholar]
- Montmayeur JP, Borrelli E. Targeting of G alpha i2 to the Golgi by alternative spliced carboxyl-terminal region. Science. 1994;263(5143):95–98. doi: 10.1126/science.8272874. [DOI] [PubMed] [Google Scholar]
- Wedegaertner PB. Characterization of subcellular localization and stability of a splice variant of G alpha i2. BMC Cell Bio. 2002;3:12. doi: 10.1186/1471-2121-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DW, Mudd L, Boyd FT Jr, Fields M, Raizada MK. Insulin is released from rat brain neuronal cells in culture. J Neurochem. 1986;47:831–836. doi: 10.1111/j.1471-4159.1986.tb00686.x. [DOI] [PubMed] [Google Scholar]
- Wei LT, Matsumoto H, Rhoads DE. Release of immunoreactive insulin from rat brain synaptosomes under depolarizing conditions. J Neurochem. 1990;54:1661–1665. doi: 10.1111/j.1471-4159.1990.tb01219.x. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Flohé L, Loschen G, Günzler WA, Eichele E. Glutathione peroxidase, V. The kinetic mechanism. Hoppe Seylers Z Physiol Chem. 1972;353:987–999. doi: 10.1515/bchm2.1972.353.1.987. [DOI] [PubMed] [Google Scholar]
- Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- Cox AG, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem J. 2009;425:313–325. doi: 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- McClung JP, Roneker CA, Mu W, Lisk DJ, Langlais P, Liu F, Lei XG. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc Natl Acad Sci USA. 2004;101:8852–8857. doi: 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, Stepto N, Wu B, Mitchell CA, Tonks NK, Watt MJ, Febbraio MA, Crack PJ, Andrikopoulos S, Tiganis T. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenov MY, Tucker HM, Nair P, Aksenova MV, Butterfield DA, Estus S, Markesbery WR. The expression of key oxidative stress-handling genes in different brain regions in Alzheimer’s disease. J Mol Neurosci. 1998;11:151–164. doi: 10.1385/JMN:11:2:151. [DOI] [PubMed] [Google Scholar]
- Aksenov MY, Markesbery WR. Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci Lett. 2001;302:141–145. doi: 10.1016/S0304-3940(01)01636-6. [DOI] [PubMed] [Google Scholar]
- Kim SH, Fountoulakis M, Cairns N, Lubec G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J Neural Transm Suppl. 2001;61:223–235. doi: 10.1007/978-3-7091-6262-0_18. [DOI] [PubMed] [Google Scholar]
- Cumming RC, Dargusch R, Fischer WH, Schubert D. Increase in expression levels and resistance to sulfhydryl oxidation of peroxiredoxin isoforms in amyloid beta-resistant nerve cells. J Biol Chem. 2007;282:30523–30534. doi: 10.1074/jbc.M700869200. [DOI] [PubMed] [Google Scholar]
- Krapfenbauer K, Engidawork E, Cairns N, Fountoulakis M, Lubec G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003;967:152–160. doi: 10.1016/S0006-8993(02)04243-9. [DOI] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Lee HG, Casadesus G, Smith MA, Perry G. Oxidative stress signalling in Alzheimer’s disease. Brain Res. 2004;1000:32–39. doi: 10.1016/j.brainres.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Zhu X, Liu Q, Honda K, Siedlak SL, Harris PL, Smith MA, Perry G. Compensatory responses induced by oxidative stress in Alzheimer disease. Biol Res. 2006;39:7–13. doi: 10.4067/s0716-97602006000100002. [DOI] [PubMed] [Google Scholar]
- Andreeva N, Khodorov B, Stelmashook E, Cragoe E Jr, Victorov I. Inhibition of Na+/Ca2+ exchange enhances delayed neuronal death elicited by glutamate in cerebellar granule cell cultures. Brain Res. 1991;548:322–325. doi: 10.1016/0006-8993(91)91141-M. [DOI] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Lett. 1996;397:230–234. doi: 10.1016/S0014-5793(96)01139-8. [DOI] [PubMed] [Google Scholar]