

Abstract
A chemical library of carboxamide-substituted tetramates designed by analogy with antibacterial natural products, a method for their rapid construction, and the evaluation of their antibacterial activity is reported.
Keywords: acylation, antibacterial, drug discovery, natural products, tetramate
Introduction
The discovery of new antibiotic families with novel modes of action is a promising way to overcome resistant or virulent bacteria, since novel modes of action might be expected to slow target-based endogenous resistance [1]. In this regard, natural products play a major role by providing a biologically validated starting point [2]. Recently discovered antibiotic lead compounds of major interest include platensimycin (a FabF inhibitor) [3], R207910 (an ATP synthase inhibitor) [4] and moiramide B (a bacterial acetyl-CoA carboxylase inhibitor) [5]. Both natural 3-acyltetramic acids, for example streptolydigin 1a (bacterial RNA polymerase (RNAP) inhibitory activity) [6] and kibdelomycin 1b (bacterial type II topoisomerase inhibitory activity) [7] and unnatural systems, such as 3-carboxamide tetramic acid 1c and 3-carboxamide piperidine-2,4-dione 1d (undecaprenyl pyrophosphate synthase (UPPS) inhibitory activity) [8] exhibit high levels of antibacterial activity (Figure 1). All these systems share a β-dicarbonyl core. A drug discovery programme inspired by these natural products, as promoted by Waldmann [9], was of interest to us. We have recently focused on the construction and evaluation of libraries derived from tetramic acid scaffolds and discovered that bicyclic 3-carboxamide 1e, bicyclic 3-acyl 1f and monocyclic 3-acyl 1g exhibit a dual targeting ability at RNAP and UPPS, while 3-acyl piperidine-2,4-dione 1h only targets UPPS [10]. Although tetramates are well-known as a core component in many natural products that continue to excite interest [11–14], we carried out a more detailed study of the synthesis, tautomeric behaviour and antibiotic activity of related monocyclic 3-carboxamide tetramic acid systems 2 and 3 (Schemes 1–3), the results of which are outlined below. In system 2, the N(1) and C(5) substituents were chosen in order to probe the effect of the length of the N-alkyl chain on antibiotic activity. The substituents of system 3 were chosen in order to probe the effect of a C(3) substituent containing a sulfur heteroatom, which we had earlier seen results in enhanced antibacterial activity compared with the oxygen counterpart [10], for two types of amide substituent and a range of C(3) carboxamides.
Figure 1.

Some antibiotic natural and unnatural tetramic acids.
Results and Discussion
Synthesis
The synthesis of the required tetramic acid systems 2a–g (Scheme 1), 2h (Scheme 2) and 3a–f (Scheme 3) was achieved by Dieckmann cyclisation of the required N-alkyl-N-malonyl glycine (readily prepared from glycine). A similar strategy for the base-mediated cyclisation of N-acetoacetylamino acid esters leading to 3-acetyltetramates has been reported, which give N–H rather than N-alkyl systems [15–17]. Tetramate 7 was obtained from amino acid 6, except that the key intermediate 5 was obtained by reductive amination (Scheme 2) of (R)-citronellal (4), which although proceeding in poor yield gave enough material with which to proceed [18]. By contrast, N-acyl derivatives could not be easily prepared by an equivalent approach because of the difficulty of a controlled double acylation on N(1). Although the synthesis of N-acetyl 3-alkoxycarbonyls from N-hydroxysuccinimide esters of N-acetylamino acids has been reported [19], we wished to exploit an alternative approach based upon C-acyloxylation of enolates followed by amine exchange, which had been shown to be very effective in a pyroglutamate series, since it offered synthetic simplicity and the potential for generality [20–21]. We found that an approach based upon direct acylation of methyl thioethers 8a,b (these were readily obtained from the required N-acylmethionine by DCC/DMAP coupling with Meldrum’s acid and cyclisation under reflux) was possible, which made use of the high acidity of the tetramate system. Thus, conversion to the n-butyloxycarbonyl derivatives N-acyl 9a,b by using 1.2 equivalents of butyl chloroformate along with 2.2 equivalents of 4-(dimethylamino)pyridine (DMAP) proceeded in good yields (over 90%) (Scheme 3) [10]. With the 3-alkoxycarbonyl tetramic acid core systems 7 and 9 in hand, conversion to 3-carboxamides 2a–g and 3a–f by direct ester–amide exchange under reflux in toluene was readily achieved, providing access to a range of amides in good to excellent yield (Schemes 1–3). This process neatly complements a strategy we had earlier used for the introduction of amine substituents in pyroglutamates by a conjugate addition of amines [22].
Scheme 1.

Synthesis of simple 3-carboxamide tetramic acids. Reaction conditions: (a) triethylamine (2.0 equiv), 1-bromohexane (0.5 equiv), EtOH, reflux; (b) monoethyl malonate (1.1 equiv), DCC (1.1 equiv), CH2Cl2, rt; (c) NaOMe (1.1 equiv), benzene, EtOH, reflux; (d) RNH2 (1.0 equiv), toluene, reflux.
Scheme 2.

Synthesis of N-alkyl 3-carboxamide tetramic acid. Reaction conditions: (a) 1. glycine methyl ester∙HCl (1.0 equiv), Et3N (1.2 equiv), MgSO4 (2.0 equiv), THF, rt. 2. NaBH4 (2.0 equiv), MeOH, rt; (b) ethyl malonyl chloride (1.05 equiv), Et3N (1.2 equiv), CH2Cl2, rt; (c) KOt-Bu (1.1 equiv), THF, reflux; (d) amine (1.0 equiv), toluene, reflux; (e) butyl chloroformate (1.2 equiv), DMAP (2.2 equiv), CH2Cl2, rt.
Scheme 3.

Synthesis of C(5)-alkyl 3-carboxamide tetramic acids. Reaction conditions: (a) butyl chloroformate (1.2 equiv), DMAP (2.2 equiv), CH2Cl2, rt; (b) RNH2 (1.0 equiv), toluene, reflux.
Tautomerism
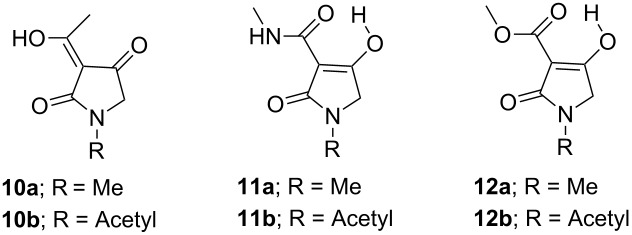
Tautomerism in tricarbonyl 3-acyltetramate systems is known to be complex and strongly dependent on the identity of the side chain acyl group [23]. 3-Acyl (X = CH2) [10,24–25], 3-carboxamide (X = NH) [8,10] and 3-alkoxycarbonyl (X = O) tetramic acids (Figure 2) have been found to exist as a pair of external conformers (AB and CD) in slow equilibrium (AB CD), each consisting of a pair of internal tautomers in rapid equilibrium (AB and CD). The tautomerisation of 3-acyltetramic acids has been shown to be mainly affected by substitution on N(1) rather than the functionalities on the 3-acyl and C(5) positions. Thus, the dominant tautomer of N-unsubstituted and N-alkyltetramates is D, while N-acyltetramates exist as a mixture of external tautomers AB and D in approximately equal ratio [23]. By contrast, it was found that the tautomerisation of 3-carboxamides and 3-alkoxycarbonyls was not affected by substitution on N(1). Therefore, the dominant tautomer of 3-carboxamide tetramates is tautomer A (over 80%) with a minor contribution of tautomer D, while 3-alkoxycarbonyltetramates exist as only tautomer A (>99%). In order to understand this phenomenon, the ground state energy of simplified 3-carboxamides 11a,b and 3-alkoxycarbonyls 12a,b was calculated and compared with that of 3-acyl derivatives 10a,b (Table 1). To the best of our knowledge, such a detailed analysis has not been previously reported. In the calculation for N-alkyl 3-acyltetramate 10a, the ground-state energies of tautomers B and D, which shows preference for tautomer D, is considerably lower than those of tautomers A and C. This outcome supports experimental NMR observations (>80% of tautomer D and <20% of tautomer B, Table 1, entry 1). On the other hand, the ground state energy of tautomer C of N-acyl 10b is considerably higher than that of tautomers A, B and D, also supporting the NMR observations (tautomer AB:D = about 50:50, Table 1, entry 2). However, tautomer A of 3-carboxamides was the most stable for both of N-alkyl tetramate 11a and N-acyl tetramate 11b (>80% of tautomer A and <20% of tautomer D, Table 1, entry 3 and entry 4), while for 3-alkoxycarbonyltetramates 12a,b, tautomer A was the most stable, and significantly more stable than tautomers B–D. These finding support the conclusion that 3-alkoxycarbonyls exist only as tautomer A (Table 1, entry 5 and entry 6).
CD), each consisting of a pair of internal tautomers in rapid equilibrium (AB and CD). The tautomerisation of 3-acyltetramic acids has been shown to be mainly affected by substitution on N(1) rather than the functionalities on the 3-acyl and C(5) positions. Thus, the dominant tautomer of N-unsubstituted and N-alkyltetramates is D, while N-acyltetramates exist as a mixture of external tautomers AB and D in approximately equal ratio [23]. By contrast, it was found that the tautomerisation of 3-carboxamides and 3-alkoxycarbonyls was not affected by substitution on N(1). Therefore, the dominant tautomer of 3-carboxamide tetramates is tautomer A (over 80%) with a minor contribution of tautomer D, while 3-alkoxycarbonyltetramates exist as only tautomer A (>99%). In order to understand this phenomenon, the ground state energy of simplified 3-carboxamides 11a,b and 3-alkoxycarbonyls 12a,b was calculated and compared with that of 3-acyl derivatives 10a,b (Table 1). To the best of our knowledge, such a detailed analysis has not been previously reported. In the calculation for N-alkyl 3-acyltetramate 10a, the ground-state energies of tautomers B and D, which shows preference for tautomer D, is considerably lower than those of tautomers A and C. This outcome supports experimental NMR observations (>80% of tautomer D and <20% of tautomer B, Table 1, entry 1). On the other hand, the ground state energy of tautomer C of N-acyl 10b is considerably higher than that of tautomers A, B and D, also supporting the NMR observations (tautomer AB:D = about 50:50, Table 1, entry 2). However, tautomer A of 3-carboxamides was the most stable for both of N-alkyl tetramate 11a and N-acyl tetramate 11b (>80% of tautomer A and <20% of tautomer D, Table 1, entry 3 and entry 4), while for 3-alkoxycarbonyltetramates 12a,b, tautomer A was the most stable, and significantly more stable than tautomers B–D. These finding support the conclusion that 3-alkoxycarbonyls exist only as tautomer A (Table 1, entry 5 and entry 6).
Figure 2.

Tautomerism of tetramates.
Table 1.
Calculated energy of the ground state of 3-acyltetramic acids 10a,b, 3-carboxamide tetramic acids 11a,b and 3-alkoxycarbonyl tetramic acids 12a,b.
 | |||||
| Entry | Compound | Calcd relative energy (kcal/mol)a,b | |||
| Form A | Form B | Form C | Form D | ||
| 1 | 10a | +4.16 | +1.61 | +4.30 | 0 |
| 2 | 10bc | +1.59 | +0.17 | +5.62 | 0 |
| 3 | 11a | −0.51 | +0.42 | +1.74 | 0 |
| 4 | 11b | −0.91 | −0.15 | +3.82 | 0 |
| 5 | 12a | −3.58 | +1.75 | −1.41 | 0 |
| 6 | 12b | −6.24 | −0.64 | +3.20 | 0 |
aThe energy difference between each tautomer related to tautomer D. bCalculated by using DFT B3LYP (6-31G*) in Spartan 02. cReported in our previous paper [10].
Antibacterial activity
The antibiotic activities of tetramates 2a–h and 3a–f along with analogues 1c and 1d (reported by Novartis [8]) were determined against 4 species of Gram-positive bacteria, consisting of 4 strains of Staphylococcus aureus, including a methicillin-resistant strain (MRSA, S2), vancomycin susceptible Enterococcus faecalis (VSE, E1), vancomycin resistant E. faecium (VanA VRE, E2)), and 2 strains of Streptococcus pneumoniae, including multi-drug resistant strain (MDRSP, P9), as well as 3 species of Gram-negative bacteria, consisting of Pseudomonas aeruginosa and 2 strains of Haemophilus influenzae, including an efflux-negative strain, and 2 strains of Escherichia coli, including an efflux-negative strain (Table 2). Relevant physicochemical properties of the analogues are also shown in Table 3. These were used to elaborate structure–activity relationships (SAR) [26]. In the assay against Gram-negative bacteria, no activity against E. coli and P. aeruginosa was found (MIC > 64 µg/mL, data not shown), consistent with the inactivity of 3-acyl and 3-carboxamide tetramic acids against these strains seen earlier [7–8,10]. This result is most likely explained by their poor cell permeability as a result of their hydrophobic character [14,27]. However, activities against another Gram-negative bacterium, H. influenzae, were found, the magnitude of which depended on the substituent. An SAR consistent with transportation by the efflux pump was found [28]. Analogues 2e,h and 1c,d, for which activity differences between efflux-positive (H3) and negative (H4) H. influenza strains are large, are more lipophilic compared with other active analogues (rel-PSA < 13.5, c log P > 2.79 and c log D (7.4) > 1.41). On the other hand, analogues 2 and 3 show a broad-spectrum activity against Gram-positive bacteria, although the activity depends on the substituent identities. Importantly, the variation of antibacterial activity for analogues 2 and 3 was less pronounced in the resistant strains MRSA, VRE and MDRSP (normally less than 8 times variation, with the exception of analogues 2e and 3b), while the activities of Novartis analogue 1c against MRSA [8], amoxicillin against MDRSP and ciprofloxacin against MRSA and VRE were significantly lower (more than 250 times compared with the activity against the most sensitive strain). Among the various substituent groups, N-alkyl phenyl derivatives 2a,b,h were active, while N-acetyl phenyl derivatives 3e,f were inactive or only very weakly active. This SAR might be accounted for by their physicochemical properties: the less lipophilic character of N-acetyl 3e,f (rel-PSA > 17.0%, c log P < 0.80 and c log D (7.4) < −1.20) compared with those of N-alkyl 2a,b,h (rel-PSA < 15.0%, c log P > 1.77 and c log D (7.4) > 0.41) might make penetration of the bacterial cell membrane more difficult. However, 3-carboxamides 2c–g and 3a–d, all possessing alkyl substituents, including a benzyl group on the amide function, are also active to Gram-positive strains. Furthermore, the activities of n-alkyl 2d–f depended on the chain length, with a marked drop-off in activity for the longer chain 2f. In addition, the activity of the more lipophilic analogues 2a,d–f,h (PSA ≤ 13.7%, c log P ≥ 1.69 and c log D (7.4) ≥ 0.23) in the presence of 2.5% horse blood was shifted to high MICs even though that of less lipophilic analogues 2c,g and 3a–e (PSA ≥ 13.7%, c log P ≤ 1.84 and c log D (7.4) ≤ −0.04) was maintained. This serum-protein binding significantly affected the activity against S. aureus S26, since almost all analogues showed inactivity (MIC > 64 µg/mL) in the presence of 10% human serum with the exception of 2a (32 µg/mL) and 2b (64 µg/mL) (data not shown).
Table 2.
In vitro antibiotic activity (MIC, µg/mL) of tetramic acids.a,b
| S1 | S26 | S4 | S2 | E1 | E2 | P1 | P9 | P9B | H3 | H4 | |
| 2ac | 4 | 8 | 8 | 8 | 8 | 8 | 16 | 8 | 32 | 16 | 4 |
| 2bc | 1 | 1 | 1 | 2 | 2 | 4 | 4 | 4 | −d | 2 | 0.5 |
| 2cc | 8 | 8 | 8 | 8 | 8 | 8 | 16 | 8 | 8 | 4 | 0.25 |
| 2dc | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 1 | 4 | 1 | 0.12 |
| 2ec | 2 | 16 | 16 | 16 | 8 | 0.5 | 4 | 1 | 4 | >64 | ≤0.1 |
| 2f | >64 | >64 | >64 | >64 | >64 | >64 | 16 | 8 | 16 | >64 | >64 |
| 2g | 8 | 8 | 8 | 8 | 16 | 8 | 64 | 16 | 8 | 4 | 0.25 |
| 2h | 2 | 2 | 2 | 2 | 2 | 2 | 8 | 4 | >64 | >64 | 8 |
| 3a | >64 | >64 | 32 | >64 | 16 | 16 | 8 | 8 | 8 | >64 | >64 |
| 3b | 4 | 8 | 2 | 8 | 1 | 2 | 0.5 | 0.5 | 0.5 | 8 | 1 |
| 3c | 8 | 32 | 32 | 16 | 8 | 16 | 8 | 8 | 8 | 32 | 4 |
| 3d | 8 | 64 | 32 | 32 | 8 | 16 | 8 | 8 | 4 | 64 | 4 |
| 3e | >64 | >64 | >64 | >64 | >64 | >64 | 64 | 64 | 64 | >64 | 64 |
| 3f | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 1cc | 0.1 | 0.12 | 1 | 64 | ≤0.1 | ≤0.1 | 2 | 2 | 2 | >64 | 0.5 |
| 1d | 8 | 64 | 32 | 64 | 32 | 4 | 64 | 64 | >64 | >64 | 16 |
| Linec | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 0.5 | 0.5 | 16 | 4 |
| Cip | 0.1 | 0.5 | 0.12 | 16 | 1 | 32 | 1 | 1 | 1 | 0.5 | ≤0.1 |
| Amox | −d | −d | −d | −d | −d | −d | >0.03 | 8 | −d | −d | −d |
| Caz | 8 | 16 | 16 | −d | −d | −d | −d | −d | −d | −d | −d |
aAbbreviation; S1; S. aureus 1, ATCC13709 in vivo (methicillin sensitive), S26; S. aureus 26, ATCC25923 (vancomycin susceptible), S4; S. aureus 4, Oxford, S2; S. aureus 2, (MRSA in vivo), E1; E. faecalis 1, ATCC29212 VanS (vancomycin susceptible), E2; E. faecium 1, VanA (vancomycin resistant), P1; S. pneumonia 1, ATCC49619 (erythromycin susceptible), P9; S. pneumonia 9, (multi-drug resistant), P9B; S. pneumonia 9 in presence of 2.5% horse blood, H3; H. influenzae 3, ATCC31517 MMSA, H4; H. influenzae 4, LS2 Efflux knock out, Line; linezolid, Cip; ciprofloxacin, Amox; amoxicillin, Caz; ceftazidime. bAll analogues are inactive (MIC > 64 µg/mL) against E. coli 1, ATCC25922 (non pathogenic strain), E. coli 50, Ec49 No efflux and P. aeruginosa 1, ATCC27853. cThe activity was reported in our previous publication [10]. dNot determined.
Table 3.
Physicochemical properties of 3-carboxamide tetramic acids.a,b
| MW | MSA | PSA | rel-PSA | c log P | c log D (7.4) | HD/HA | RB | |
| 2ac | 387 | 599 | 82.1 | 13.7 | 1.77 | 0.41 | 2/5 | 8 |
| 2bc | 302 | 462 | 69.6 | 15.1 | 1.85 | 0.64 | 2/3 | 7 |
| 2cc | 308 | 509 | 69.6 | 13.7 | 1.28 | −0.15 | 2/3 | 7 |
| 2dc | 310 | 540 | 69.6 | 12.9 | 1.69 | 0.23 | 2/3 | 11 |
| 2ec | 353 | 632 | 69.6 | 11.0 | 2.88 | 1.42 | 2/3 | 14 |
| 2f | 395 | 725 | 69.6 | 9.6 | 4.07 | 2.61 | 2/3 | 17 |
| 2g | 316 | 492 | 69.6 | 14.1 | 1.47 | −0.04 | 2/3 | 8 |
| 2h | 442 | 691 | 82.1 | 11.9 | 2.79 | 1.41 | 2/5 | 9 |
| 3a | 427 | 662 | 95.9 | 14.5 | 0.90 | −1.32 | 2/5 | 10 |
| 3b | 409 | 613 | 86.7 | 14.1 | 1.84 | −0.19 | 2/4 | 6 |
| 3c | 371 | 590 | 86.7 | 14.7 | 0.84 | −1.13 | 2/4 | 7 |
| 3d | 383 | 586 | 86.7 | 14.8 | 1.06 | −0.96 | 2/4 | 5 |
| 3e | 377 | 530 | 90.0 | 17.0 | 0.80 | −1.20 | 2/5 | 6 |
| 3f | 419 | 577 | 99.2 | 17.2 | 0.46 | −1.54 | 2/6 | 6 |
| 1c | 390 | 568 | 69.6 | 12.3 | 3.51 | 2.03 | 2/3 | 5 |
| 1d | 405 | 591 | 78.4 | 13.3 | 3.63 | 2.96 | 3/3 | 5 |
aMW; molecular weight, MSA; molecular surface area, PSA; polar surface area, %PAS; relative polar surface area = (PSA/MSA) × 100, c log P; calculated partition coefficient, c log D (7.4); calculated distribution coefficient at pH 7.4, HD; hydrogen-bond donor count, HA; hydrogen-bond acceptor count, RB; rotatable bond count. bTautomer A was selected for the calculation. cReported in our previous publication [10].
After screening to find the most active compound, tetramate 2b was selected for a detailed investigation and its antibiotic activity against various drug-resistant strains was further evaluated with reference antibiotics (Table 4). Tetramate 2b was found to be active against virulent and resistant strains, such as methicillin and fluoroquinolone-resistant S. aureus and penicillin and/or erythromycin-resistant S. pneumoniae. Remarkably, tetramate 2b maintained the activities against all the strains and was highly effective against erythromycin resistant S. pneumoniae Pn31 (MIC = 0.125 µg/mL). By way of comparison, the activities of moxifloxacin (a fourth generation fluoroquinolone) to S. aureus Sa18 (MIC = 32 µg/mL), amoxicillin (β-lactam) to S. pneumoniae Pn7, Pn10 and Pn11 (MIC = 4 µg/mL), erythromycin to S. pneumoniae Pn7, Pn10, Pn19, Pn21 and Pn31 (MIC ≥ 4 µg/mL) and ceftazidime (a third generation cephalosporin) to S. aureus Sa5, Sa18 and Sa40 (MIC ≥ 16 µg/mL) were substantially decreased.
Table 4.
Antibiotic activity of tetramic acid 2b.a
| Strains | Phenotype | MIC (µg/mL) | |||||
| 2b | Moxi | Amox | Ery | Vanco | Caz | ||
| S. pneumoniae Pn7 | EryR | 2 | 0.125 | 4 | 16 | 0.25 | −c |
| S. pneumoniae Pn10 | PenR, EryR | 2 | 0.125 | 4 | >32 | 0.5 | −c |
| S. pneumoniae Pn11 | PenR | 2 | 0.125 | 4 | <0.03 | 0.25 | −c |
| S. pneumoniae Pn19 | EryR | 2 | 0.06 | 0.06 | >32 | 0.5 | −c |
| S. pneumoniae Pn21 | EryR | 2 | 0.06 | 0.125 | 4 | 0.25 | −c |
| S. pneumoniae Pn31 | EryR | 0.13 | <0.03 | <0.03 | 16 | 0.5 | −c |
| S. aureus Sa5 | ermR PK2b | 1 | ≤0.03 | −c | −c | −c | 32 |
| S. aureus Sa18 | FQR | 1 | 32 | −c | −c | −c | 16 |
| S. aureus Sa40 | mecAb | 1 | ≤0.03 | −c | −c | −c | 32 |
aAbbreviation: EryR; erythromycin resistant, PenR; penicillin resistant, FQR; fluoroquinolone resistant, Moxi; moxifloxacin, Amox; amoxicillin, Ery; erythromycin, Caz; ceftazidime. bMethicillin-resistant strain. cNot determined.
Lipophilic efficiency (LipE) has been used to assess the suitability of drug candidates as CB agonists [29], and the usage of a similar calculation for the data presented in this work (with LipE = pMIC(nM) − c Log P) (see Table S1 and Figure S1 in Supporting Information File 1; pMIC values were calculated according to a literature protocol [30]), facilitated the identification of compounds with potential for optimisation. According to Figure S1, strongly active compounds can be found at c log P values of 1–2, 3 or 4, and for the highly susceptible strain H4, for example, compounds of interest would be 2b–e and 2g.
Conclusion
We have prepared monocyclic 3-carboxamide tetramic acids from 3-alkoxycarbonyl tetramic acids based on a direct ester–amide exchange by using butyl chloroformate with DMAP, thereby providing a general access to this type of system. The tautomerization of 3-alkoxycarbonyl and 3-carboxamide tetramic acids compared to 3-acyltetramic acids has been investigated. It has been found that 3-alkoxycarbonyl and 3-carboxamide tetramic acids prefer tautomer A, while the preference of 3-acyltetramic acids depends on the N(1)-functionality. Of particular interest is that 3-carboxamide analogues, especially 2b, have shown bioactivity against various Gram-positive bacteria including clinically resistant strains such as MRSA, fluoroquinolone-resistant S. aureus, MDRSP, penicillin and erythromycin-resistant S. pneumonia and VRE as well as Gram-negative H. influenzae. Further optimisation, especially for overcoming high plasma-protein binding, is warranted but these compounds may be suitable for an evaluation for topical use [31–32]. Significantly, these results suggest that a drug discovery approach based upon deconstruction–reconstruction inspired by suitable natural products with demonstrable biological activity provides a route for the rapid assembly of compound libraries, which, even if not fully optimized, provide a useful starting point for further elaboration.
Experimental
General. Melting points were determined with a Stuart Scientific SMP1 melting point device and are uncorrected. The 1H and 13C NMR spectra were obtained by using a Bruker Avance AV400 (400 MHz and 100 MHz, respectively) with residual solvent peaks as the internal reference. Mass spectra (MS) and high-resolution mass spectra (HRMS) were obtained with Micro Mass LCT and GCT spectrometers under the conditions of electrospray ionization (ESI) and chemical ionization (CI) respectively, and values were reported as a ratio of mass to charge in Daltons.
Synthesis. The synthesis of monocyclic precursor tetramate compounds from glycine has been reported in our previous publication [33].
Calculations. Density Functional B3LYP (6-31G*) in Spartan 02 was used for the calculation of the energy in equilibrium geometry at ground state. MarvinSketch Version 5.3.8. (http://www.chemaxon.org) was used for the calculation of the van der Waals molecular surface area (MSA), the polar surface area (PSA), the calculated partition coefficient (c log P) under VG method, the calculated distribution coefficient at pH 7.4 [c log D (7.4)] under VG method, the hydrogen-bond donor count, the hydrogen-bond acceptor count, and the rotatable bond count.
Antibacterial activity. Antibiotic activity was measured by using standard methodology (Clinical and Laboratory Standards Institute. Methods for antimicrobial susceptibility test for bacteria that grow aerobically, approved standard M7-A7, 7th ed., CLSI, Wayne, PA, 2006): compounds were diluted in DMSO to obtain 2.56 mg/mL, then 100 µL were diluted in Mueller-Hinton broth to 0.256 mg/mL and assayed against the bacterial panel by incubation in 96-well microplates at 37 °C for 24 h. The MIC was determined by visually reading the first concentration where no growth (no turbidity) appeared.
Supporting Information
Experimental details, calculated energies (Spartan) for selected compounds and NMR spectra.
Acknowledgments
We are particularly grateful for valuable input by Drs Phil Dudfield and John Lowther, and for funding by Galapagos SASU (France).
This article is part of the Thematic Series "Natural products in synthesis and biosynthesis".
References
- 1.Donadio S, Maffioli S, Monciardini P, Sosio M, Jabes D. J Antibiot. 2010;63:423–430. doi: 10.1038/ja.2010.62. [DOI] [PubMed] [Google Scholar]
- 2.von Nussbaum F, Brands M, Hinzen B, Weigand S, Häbich D. Angew Chem, Int Ed. 2006;45:5072–5129. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Soisson S M, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang Y S, Cummings R, et al. Nature. 2006;441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 4.Andries K, Verhasselt P, Guillemont J, Göhlmann H W H, Neefs J-M, Winkler H, Gestel J V, Timmerman P, Zhu M, Lee E, et al. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 5.Freiberg C, Brunner N A, Schiffer G, Lampe T, Pohlmann J, Brands M, Raabe M, Häbich D, Ziegelbauer K. J Biol Chem. 2004;279:26066–26073. doi: 10.1074/jbc.M402989200. [DOI] [PubMed] [Google Scholar]
- 6.Tuske S, Sarafianos S G, Wang X, Hudson B, Sineva E, Mukhopadhyay J, Birktoft J J, Leroy O, Ismail S, Clark A D, et al. Cell. 2005;122:541–552. doi: 10.1016/j.cell.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips J W, Goetz M A, Smith S K, Zink D L, Polishook J, Onishi R, Salowe S, Wiltsie J, Allocco J, Sigmund J, et al. Chem Biol. 2011;18:955–965. doi: 10.1016/j.chembiol.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Peukert S, Sun Y, Zhang R, Hurley B, Sabio M, Shen X, Gray C, Dzink-Fox J, Tao J, Cebula R, et al. Bioorg Med Chem Lett. 2008;18:1840–1844. doi: 10.1016/j.bmcl.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Kumar K, Waldmann H. Angew Chem, Int Ed. 2009;48:3224–3242. doi: 10.1002/anie.200803437. [DOI] [PubMed] [Google Scholar]
- 10.Jeong Y-C, Anwar M, Bikadi Z, Hazai E, Moloney M G. Chem Sci. 2013;4:1008–1015. doi: 10.1039/c2sc21713a. [DOI] [Google Scholar]
- 11.Royles B J L. Chem Rev. 1995;95:1981–2001. doi: 10.1021/cr00038a009. [DOI] [Google Scholar]
- 12.Schobert R, Schlenk A. Bioorg Med Chem. 2008;16:4203–4221. doi: 10.1016/j.bmc.2008.02.069. [DOI] [PubMed] [Google Scholar]
- 13.Barnickel B, Bayliffe F, Diestel R, Kempf K, Laschat S, Pachali S, Sasse F, Schlenk A, Schobert R. Chem Biodiversity. 2010;7:2830–2845. doi: 10.1002/cbdv.201000179. [DOI] [PubMed] [Google Scholar]
- 14.Athanasellis G, Igglessi-Markopoulou O, Markopoulos J. Bioinorg Chem Appl. 2010:315056. doi: 10.1155/2010/315056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuki H, Tohira Y, Aoki B, Kano T, Takama S, Yamazaki T. Chem Pharm Bull. 1967;15:1107–1111. doi: 10.1248/cpb.15.1107. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki S, Sano F, Yuki H. Chem Pharm Bull. 1967;15:1120–1122. doi: 10.1248/cpb.15.1120. [DOI] [PubMed] [Google Scholar]
- 17.Toda S, Nakagawa S, Naito T, Kawaguchi H. J Antibiot. 1980;33:173–181. doi: 10.7164/antibiotics.33.173. [DOI] [PubMed] [Google Scholar]
- 18.Yendapally R, Hurdle J G, Carson E I, Lee R B, Lee R E. J Med Chem. 2008;51:1487–1491. doi: 10.1021/jm701356q. [DOI] [PubMed] [Google Scholar]
- 19.Petroliagi M, Igglessi-Markopoulou O. Tetrahedron: Asymmetry. 1999;10:1873–1875. doi: 10.1016/S0957-4166(99)00192-5. [DOI] [Google Scholar]
- 20.Bailey J H, Cherry D T, Dyer J, Moloney M G, Bamford M J, Keeling S, Lamont R B. J Chem Soc, Perkin Trans 1. 2000:2783–2792. doi: 10.1039/B001999M. [DOI] [Google Scholar]
- 21.Hill T J, Kocis P, Moloney M G. Tetrahedron Lett. 2006;47:1461–1463. doi: 10.1016/j.tetlet.2005.12.068. [DOI] [Google Scholar]
- 22.Chan P W H, Cottrell I F, Moloney M G. Tetrahedron Lett. 1997;38:5891–5894. doi: 10.1016/S0040-4039(97)01312-9. [DOI] [Google Scholar]
- 23.Yamaguchi T, Saito K, Tsujimoto T, Yuki H. J Heterocycl Chem. 1976;13:533–537. doi: 10.1002/jhet.5570130323. [DOI] [Google Scholar]
- 24.Jeong Y-C, Moloney M G. J Org Chem. 2011;76:1342–1354. doi: 10.1021/jo102304y. [DOI] [PubMed] [Google Scholar]
- 25.Detsi A, Micha-Screttas M, Igglessi-Markopoulou O. J Chem Soc, Perkin Trans 1. 1998:2443–2449. doi: 10.1039/A801896K. [DOI] [Google Scholar]
- 26.O’Shea R, Moser H E. J Med Chem. 2008;51:2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 27.Gänzle M G. Appl Microbiol Biotechnol. 2004;64:326–332. doi: 10.1007/s00253-003-1536-8. [DOI] [PubMed] [Google Scholar]
- 28.Fernandes J, Gattass C R. J Med Chem. 2009;52:1214–1218. doi: 10.1021/jm801389m. [DOI] [PubMed] [Google Scholar]
- 29.Ryckmans T, Edwards M P, Horne V A, Correia A M, Owen D R, Thompson L R, Tran I, Tutt M F, Young T. Bioorg Med Chem Lett. 2009;19:4406–4409. doi: 10.1016/j.bmcl.2009.05.062. [DOI] [PubMed] [Google Scholar]
- 30.Selvaraj C, Tripathi S K, Reddy K K, Singh S K. Curr Trends Biotechnol Pharm. 2011;5:1104–1109. [Google Scholar]
- 31.Mc Chlery S, Ramage G, Bagg J. Periodontology 2000. 2009;49:151–165. doi: 10.1111/j.1600-0757.2008.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fogo A, Kemp N, Morris-Jones R. BMJ [Br Med J] 2011;343:d5343. doi: 10.1136/bmj.d5343. [DOI] [PubMed] [Google Scholar]
- 33.Jeong Y-C, Moloney M G. Synlett. 2009:2487–2491. doi: 10.1055/s-0029-1217745. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details, calculated energies (Spartan) for selected compounds and NMR spectra.