

Abstract
Plasma proteomic experiments performed rapidly and economically using several of the latest high-resolution mass spectrometers were compared. Four quantitative hyperfractionated plasma proteomics experiments were analyzed in replicates by two AB SCIEX TripleTOF 5600 and three Thermo Scientific Orbitrap (Elite/LTQ-Orbitrap Velos/Q Exactive) instruments. Each experiment compared two iTRAQ isobaric-labeled immunodepleted plasma proteomes, provided as 30 labeled peptide fractions. 480 LC-MS/MS runs delivered >250 GB of data in two months. Several analysis algorithms were compared. At 1 % false discovery rate, the relative comparative findings concluded that the Thermo Scientific Q Exactive Mass Spectrometer resulted in the highest number of identified proteins and unique sequences with iTRAQ quantitation. The confidence of iTRAQ fold-change for each protein is dependent on the overall ion statistics (Mascot Protein Score) attainable by each instrument. The benchmarking also suggested how to further improve the mass spectrometry parameters and HPLC conditions. Our findings highlight the special challenges presented by the low abundance peptide ions of iTRAQ plasma proteome because the dynamic range of plasma protein abundance is uniquely high compared with cell lysates, necessitating high instrument sensitivity.
Keywords: immunodepletion, Seppro, IgY14, iTRAQ, EMMOL normalization, TripleTOF, Orbitrap, Q Exactive
INTRODUCTION
Research results are often dependent on access to analytical instruments of ever increasing capability, but these instruments are expensive and the expertise for their operation and data evaluation are significant investments. The sharing of expensive instrumentation through core resource facilities within a research center is a traditional solution, but as instruments quickly become outdated, replacing them is often unaffordable. A society that has facilitated the sharing of best-practice is the Association of Biomolecular Resource Facilities (ABRF.org), the membership of which includes core facility personnel, researchers and administrators. Since 1989, ABRF benchmarking, education, and method development has indirectly benefited numerous research projects. This report is not an official ABRF-sponsored study. Unlike the traditional ABRF-sponsored benchmarking studies, this current study was funded by individual peer-reviewed grants and designed, executed and interpreted by the principle investigators with the goal of addressing specific biological questions. In this report, “investigators” refers to the biologists with an understanding of mass spectrometry but lacking sufficient familiarity with each instrument to define what the optimal LC-MS/MS parameters for each instrument should be. Hence each core facility performed their service on “best effort” basis so that the best available mass spectrometry experience was brought to bear on the biology projects. Four large biological experiments were completed in just two months. This enabling mechanism can be easily implemented for other disciplines.
Proteomics aims to study all the proteins and their modified forms in a given tissue at the same time. We used differential isobaric chemical tags1-3 in LC-MS/MS hyper-fractionation analyses, experiments of about 30 fractions each, with each LC-MS/MS replicated in four mass spectrometers, to provide multiplexed quantitative comparison of thousands of proteins between cases and controls in search of disease biomarkers. This approach benchmarked the current performance in a core facility setting of five mass spectrometers. These included two AB SCIEX TripleTOF 56004 and Thermo Scientific Orbitraps Elite5/LTQ-Orbitrap Velos/Q Exactive6-10. Also benchmarked were four software for peak-picking: Mascot Distiller 2.4, ProteinPilot 4.5 beta11, 12, AB SCIEX MS Data Converter 1.3 beta, and Proteome Discover 1.3; and the algorithm-dependency of protein database searching for protein identification and quantitation by Mascot Server 2.4.01 and Protein Pilot Paragon13. One database, Swiss-Prot 2012_07 with 536789 sequence entries, was used for all database search algorithms.
To compare the accuracy of the instruments for determining protein fold-changes in an iTRAQ experiment, a novel method that interprets the confidence in fold-changes in a manner that is dependent on Mascot Protein Score is presented. Moreover, this study offers baseline data for planning further improvements in the methodologies for high resolution LC-MS/MS platforms for quantitative proteomics.
MATERIALS AND METHODS
Study Design
An email inquiry for fee-for-service for mass spectrometer time for several iTRAQ hyper-fractionation experiments was submitted to the ABRF Discussion Forum1 that reached 1,700 recipients. Five of the 18 core facilities that responded were selected to participate based on available technologies and price structures. Four iTRAQ-labeled hyper-fractionation experiments were performed by the investigators. The iTRAQ-labeled peptides from each immunodepleted plasma comparative proteomics experiment were divided into about 30 fractions each by IPG strip isoelectric focusing14, 15 and shipped to the five core facilities on dry ice for LC-MS/MS analysis over a period of two months. Each iTRAQ 4-plex experiment consisted of two replicates of cases and controls. Raw data was returned to the investigators for analysis. Some of the data were also analyzed by each core facility to help educate the investigators about the parameters optimized by each core facility. The results presented herein provide a view of the performance of five mass spectrometers for these studies and the impact of data analysis software. Four instruments were compared by each experiment. Normalization of two experiments, each differing in one mass spectrometer, thus allowed a comparison of all five mass spectrometers. To facilitate initial comparison using a common software platform, the raw files were provided to Matrix Science to produce optimized parameters for Mascot Distiller peak-picking for each instrument. Next the Mascot Server and the ProteinPilot Paragon algorithm were used separately to allow universal comparison of peak-picking results as judged by peptide identification and protein family identification by Mascot Server in a Swiss-Prot database. In total, four software analysis routines provided by three manufacturers were compared, in peptide identification, protein identification, and iTRAQ reporter quantitation.
Study Samples
Plasma samples were from protocols approved by Institutional Review Boards and were deidentified prior to delivery to the research laboratory. Plasma samples were collected using the BD™ P100 Blood Collection Tubes16, 17 (Becton Dickinson, Franklin Lakes, NJ) containing protease inhibitors, according to manufacturer’s instructions. Experiment #2 consisted of a pool of the plasma of six Caucasian patients with Familial Polyposis Coli syndrome18-20 (APC gene mutations verified) against a pool of the plasma of six controls matched for race, age and gender. Experiment #4 consisted of a pool of the plasma of five male and five female Caucasian patients with Lynch syndrome21 (MSH2 mutations verified) against equal number of matched controls. The use of plasma from these subjects in this study was incidental and resulted from a desire to use high quality samples in this analysis. The other two hyper-fractionation experiments performed only illustrated the total throughput of this outsource arrangement and will not be discussed further in this report.
The Immunodepletion of High-Abundance Proteins
The design for the immunodepletion methods for each of four mass spectrometry experiments is shown in Figure 1. Patient cases and a similar number of controls were pooled separately for each experiment. Each experiment consisted of two replicates of cases versus two replicates of controls to allow a measure of the technical reproducibility for protein identification and iTRAQ protein quantitation. Individual samples taken from disease and control groups were not mixed. Five 100 μL aliquots of pooled plasma were individually immunodepleted, removing about 155 plasma proteins using the Seppro IgY14+SuperMix column (Seppro Human IgY14 SuperMix LC5, catalog number SEP030-KT, Sigma-Aldrich Inc., St. Louis, MO) 22, 23. The flow through fractions were pooled and concentrated by centrifugal filtration using Amicon Ultracel 3K centrifugal filters to 0.45 mL. The concentrated proteins were immunodepleted one more time by passage through the same LC5 column once to achieve more thorough immunodepletion than one might accomplish in a single run. The immunodepleted protein solution was next concentrated by centrifugal filtration to 50 μL, its protein concentration determined by Bio-Rad protein assay, analyzed by SDS PAGE, and used for iTRAQ labeling.
Figure 1.
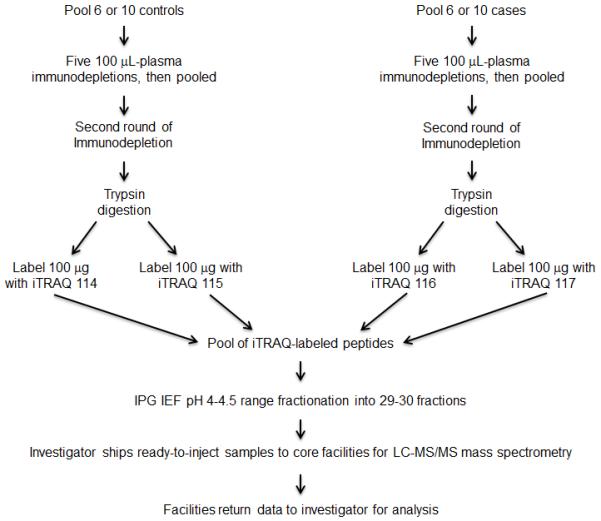
Work flow of an immunodepletion experiment. The process was done once for each of four biological experiments.
Protein Digestion, iTRAQ Labeling, Isoelectric Focusing Fractionation and Quality Control
The iTRAQ proteomic analysis procedures used in the current study have been described in detail previously15. To avoid systematic errors, assignments of iTRAQ labels for controls versus cases were varied among experiments. For both experiment #2 and experiment #4 for which data are shown herein, the assignment of Figure 1 was followed. From above immunodepletion, 100 μg of the pooled immunodepleted plasma protein from Group 1 subjects was digested with trypsin and labeled with the iTRAQ 114 label and as a replicate by the iTRAQ 115 label. 100 μg of the pooled immunodepleted protein from Group 2 was labeled with the iTRAQ 116 label and as a replicate by the iTRAQ 117 label. These iTRAQ peptides of the four samples were pooled into an IPG strip isoelectric focusing fractionation experiment.14, 15, 24 A 24 cm IPG strip (pH 3.5-4.5 GE Healthcare) was cut down to 18 cm, effectively making the pH range 4.0-4.5. This strip was used to separate the peptides into 60 fractions as previously described in detail15. Pairs of fractions were pooled to form about 30 fractions for LC-MS/MS. This pooling decreased the cost of the mass spectrometry by 50% while the fractionation of the peptides still appeared sufficient. Quality control of the pooled labeled peptides without sample fractionation was performed using LC-MS/MS on an AB SCIEX QSTAR mass spectrometer to ascertain that the four labels were approximately stoichiometric as judged by the labeling of the high abundance proteins (Supplemental Information 1). Up to about 2 μg of peptides per fraction in 25 μL of 0.1% formic acid 5% CH3CN was aliquoted into a 100 μL Agilent microsampler vial insert (#5181-1270), capped in a 2 mL screw-top vial (Agilent #5183-2067), briefly centrifuged down at 400 x g, stored frozen at -20 C, and sent on dry ice to each mass spectrometry core facility as identical replicates.
HPLCs and Mass Spectrometers
The LC parameters and Mass spectrometer parameters for each instrument are compared in Table 1. Further details are provided below:
Table 1.
LC-MS/MS parameters performed for this study in the five core facilities.
| Parameter | TripleTOF 5600 #1 |
TripleTOF 5600 #2 |
LTQ-Orbitrap Velos |
Q Exactive | Orbitrap Elite |
|---|---|---|---|---|---|
| LC | cHiPLC Nanoflex | cHiPLC Nanoflex | UPLC | UPLC | nanoHPLC |
| Model | Eksigent | Eksigent 2D ultra plus |
Waters nanoACQUITY |
Waters nanoACQUITY |
Proxeon nEasy II |
| Peak width at half height |
17 sec | variable | <30 sec | 6 sec | variable |
| Trap | Nano cHiPLC Trap column 200 μm i.d. × 0.5 mm Eksigent 3 um (120Å) C18 |
Nano cHiPLC Trap column 200 μm i.d. × 0.5 mm Eksigent 3 um (120Å) C18 |
Integrafrit, 100 μm i.d. × 5cm of 5 μm Magic C18AQ |
Homemade 5 μm (200Å) C18 |
Homemade 5 μm (200Å) Magic C18 |
| Analytical column model |
Nano cHiPLC column Eksigent 75 μm i.d. |
Nano cHiPLC column Eksigent 75 μm i.d. |
Picofrit, 75 μm i.d. 15 μm tip |
Homemade 75 μm i.d. |
Homemade 75 μm i.d. |
| Length | 15 cm bed | 15 cm bed | 20 cm bed | 25 cm bed | 25 cm bed |
| Resin type | ChromXP C18-CL C18 on silica |
ChromXP C18-CL C18 on silica |
Magic C18AQ silica |
Magic C18AQ silica |
Magic C18AQ silica |
| Particle size | 3 μm (120Å) | 3 μm (120Å) | 3 μm (100Å) | 3 μm (100Å) | 5 μm (100Å) |
| Average pressure (psi) |
1500 | 1800 | 2500 | 3500 | 1810 |
| Sample injected | Variable volume for 200 ng |
10 μL, about < 1000 ng |
15/35 μL about < 1200 ng |
4.5 μL, about 400 ng |
12 μL, about < 1200 ng |
| Gradient length for data acquisition |
120 min | 120 min | 90 min | 90 min | 90 min |
| LC flow rate | 300 nL/min | 300 nL/min | 200 nL/min | 300 nL/min | 300 nL/min |
| Survey scan resolution |
~30,000 | ~30,000 | 30,000 | 70,000 | 120,000 |
| Survey scan m/z range |
400-1250 | 400-1250 | 395-1600 | 300-1750 | 400-1800 |
| Survey scan length | 250 msec | 250 msec | ~250 msec | Max 120 msec | 250 msec |
| Isotope exclusion ON | No | No | Yes | Yes | No |
| Fragmentation method |
CID | CID | HCD | HCD | HCD |
| Charge for MS/MS | +2 to +5 | +2 to +5 | +1 or higher | +2 to +7 | +2 or higher |
| Dynamic exclusion | 10 sec after 1 occurrence |
25 sec | Yes, 60 sec repeat count 1 |
30 sec | 100 sec |
| Max. # of MS/MS scans per period |
20 | 50 | 10 | 12 | 10 |
| MS/MS Scan Length | 100 msec | Variable for 2.8 sec period |
Max ion time 250 msec |
Max ion time 120 msec |
250 msec |
| Parent ion selection window |
0.7 Da | 0.7 Da | 1.0 Da | 1.2 Da | 1.2 Da |
| MS/MS scan resolution set |
>19,000 | ~15,000 | 7500 | 35,000 | 15,000 |
| Maximum MS/MS Fragment m/z |
2000 | 2000 | Variable | 1750 | Variable |
| Minimum MS/MS Fragment m/z |
100 | 65 | 100 | 100 | 100 |
Thermo LTQ-Orbitrap Velos
Peptide fractions (25 μL as received) were diluted with 10 μL of 0.1% formic acid (A buffer) for a final volume of 35 μL. Aliquots of 15 μL were injected under trapping conditions using a Waters nanoACQUITY UPLC, trapping at 2 % B with a flow rate of 2 μL/min for 15 minutes. After this time, flow was reduced to 200 nL/min and gradient elution began. The gradient ran from 2 % B (0.1% formic acid in acetonitrile) to 32 % B over 90 minutes, rose to 95 % B to wash the column, then re-equilibrated for 20 minutes at 2 % B for the next injection. The chromatographic system was composed of IntegraFrit (100 μm ID) and PicoFrit (75 μm ID, 15 μm tip) trapping and analytical columns (New Objective, Woburn, MA) packed with 5 cm of 5 μm and 20 cm of 3 μm Magic C18AQ media (Michrom Bioresources), respectively. Background ions were reduced using active background ion reduction (ABIRD, ESI Source Solutions, Woburn, MA). Data collection used the Orbitrap at 30K resolution for the full scan MS, then proceeded to isolate the Top 10 ions for MS/MS by HCD at 7500 resolution (38% collision energy, 1 second activation time, minimum TIC 5000 for MS/MS selection, dynamic exclusion enabled with unassigned, +1, and greater than +8 charges ignored for MS/MS selection).
Thermo LTQ-Orbitrap Elite
Peptide fractions (25 μL as received) were undiluted and aliquots of 12 μL were injected under trapping conditions using a Thermo Scientific Easy nanoLC II, trapping at 0 % B with maximum pressure of 260 Bar for 50 μL. The trap was equilibrated (washed) at a maximum pressure of 260 Bar for 6 μL followed by column equilibration at a maximum of 260 Bar for 8 μL before starting gradient elution of column. After this time, flow was reduced to 300 nL/min and gradient elution began. The gradient ran from 5 % B (0.1% formic acid in acetonitrile) to 35 % B over 90 minutes, rose to 95 % B to wash the column. The chromatographic system was composed of IntegraFrit (100 μm ID) and PicoFrit (75 μm ID, 15 μm tip) trapping and analytical columns (New Objective, Woburn, MA) packed with 2 cm of 5 μm, 200 Å and 25 cm of 5 μm, 100Å Magic C18AQ media (Michrom Bioresources), respectively. Data collection used the Orbitrap at 120K resolution for the full scan MS, then proceeded to isolate the top 10 ions for MS/MS by HCD at 15000 resolution (40% collision energy, 0.1 second activation time, minimum TIC 10000 for MS/MS selection, dynamic exclusion enabled with unassigned and +1 charges ignored for MS/MS selection).
Q Exactive
The run conditions follow the “sensitive” conditions recommended by Olsen et al for optimizing the operations of the Q Exactive for low abundance proteins6. Labeled peptide fractions were reconstituted in 25 μL 5% acetonitrile containing 0.2% (v/v) formic acid and separated on a nanoACQUITY (Waters) UPLC. In brief, a 4.5 μL injection was loaded in 5% acetonitrile containing 0.1% formic acid at 4.0 μL/min for 4.0 min onto a 100 μm I.D. fused- silica pre-column packed with 2 cm of 5 μm (200 Å) Magic C18AQ and eluted using a gradient at 300 nL/min onto a 75 μm I.D. analytical column packed with 25 cm of 3 μm (100Å) Magic C18AQ particles to a gravity-pulled tip. The solvents were A, water (0.1% formic acid); and B, acetonitrile (0.1% formic acid). The time program was: 0-10 min, 5-12 % B; 10-80 min, 12-30 % B; 80-90 min, 30-35 % B; 90-91 min, 35-90 % B; 91-101 min, 90% B; 101-102 min, 90-5 % B; 120 min, stop. Ions were introduced by positive electrospray ionization via liquid junction into a Q Exactive hybrid mass spectrometer. Mass spectra were acquired over m/z 300-1750 at 70,000 resolution (m/z 200) and data-dependent acquisition selected the top 12 most abundant precursor ions for tandem mass spectrometry by HCD fragmentation using an isolation width of 1.2 Da, collision energy of 30, and a resolution of 35,000. Dynamic exclusion and other data-dependent parameters in Table 1 were used to minimize redundant MS/MS collection and maximize peptide identifications. It is worth noting that HCD fragmentation efficiency on the Q Exactive has been enhanced relative to previous implementations of HCD on older Orbitrap models making HCD as effective as CID for peptide identification25-27.
5600 TripleTOF #2
Peptide fractions were injected under trapping conditions using an Eksigent nanoLC ultra 2D plus system. Trap and elute mode was used to separate each fraction using the microfluidics on a cHiPLC Nanoflex system equipped with a trap column (200 μm x 0.5 mm ChromXP C18-CL 3 μm 120 Å) and a separation column (75 μm x 15 cm C18 3 μm 120 Å). The gradient ran at 300 nL/min from 5 % B (0.1% formic acid in acetonitrile) to 35 % B over 120 minutes, rose to 85 % B to wash the column, then re-equilibrated at 5 % B for the next injection. Eluate was delivered into the mass spectrometer with a NanoSpray III source using a 10 μm ID nanospray tip (New Objective, Woburn, MA). Gas and other mass spectrometer settings used varied depending on optimization on each day, but typical values were curtain gas = 25, Gas 1= 3-4, Gas 2 = 0, an ion spray floating voltage around 2300, and a rolling collision energy voltage was used for CID fragmentation for MS/MS spectra acquisitions. Each cycle consisted of a TOF-MS spectrum acquisition for 250 ms (mass range 400-1250 Da), followed by acquisition of up to 50 MS/MS spectra (50 ms each, mass range 65-2000 Da) of MS peaks above intensity 150 taking 2.8 seconds total per full cycle. Once MS/MS fragment spectra were acquired for a particular mass; that mass was dynamically excluded for 25 seconds. Mass spectrometer recalibration was performed at the start of each sample using a β-galactosidase digest standard.
NanoOrange Peptide Quantitation for TripleTOF #1
The hardware configuration of TripleTOF 5600 #1 and #2 and their Eksigent cHiPLC front ends were identical. TripleTOF #1 operation differed from TripleTOF #2 in the quantity of peptides injected for each fraction and the settings used for the instruments (Table 1). TripleTOF #1 used NanoOrange peptide quantitation to normalize each injected sample to 200 ng. TripleTOF #2 injected a fixed sample volume containing about 1000 ng from each sample (Table 1). NanoOrange protein quantitation kit was from Invitrogen. The diluent and the fluorescent dye were prepared according to the vendor’s instruction, in thermocycler 96-well microtiter plate. Bovine serum albumin (BSA) was used as standard. Serial dilutions of BSA were performed from 5 μg/μL to 4 ng/μL. Each sample was run in duplicate. Each well was 50 μL in volume. Two μL of samples were mixed with 50 μL of dye diluent working solution. The plate was sealed with aluminum sealing film (Platemax) and incubated at 94 °C for 10 min. The plate was then centrifuged at 3000 rpm (1690 × g) at 4 °C for 10 min. Forty μL of the content of the plate was transferred to Costar 3694 black microtiter plate. The plate was read in Synergy 2 plate reader (BioTek) with fluorescence excitation at 480±20 nm and emission at 590±35 nm. Concentration and standard curves were generated using Gen5 software (BioTek).
Data Processing - General Considerations
Two of the core facilities have LIMS support for logging in of purchase orders and entry of sample details. One LIMS also supported data upload and download. Four of the five core facilities easily established electronic purchase orders and invoicing, while one core facility still relies on paper purchase orders and ink signatures. Typical raw data files were about 8 GB for each hyper-fractionation experiment. This file size can be uploaded and downloaded through the internet. The speed depended on the site, varying from several hours to about 30 hours for download of each hyper-fractionation file. One of the core facility used FedEx shipping of the data saved to an external hard drive. The total raw data for four hyper-fractionation experiments was about 250 GB. A single proteome database, Swiss-Prot 2012_07 with 536789 sequence entries was used for all software comparisons.
Data file processing into peak lists requires about two hours for individual fractions and about 40 hours for a hyper-fractionation experiment. Computer requirements appeared at least a dual- core processor with >3 GHz CPU. A Dell Precision T3400 with 18 GB RAM represents a minimum requirement, the i7 six-core CPU with 18 GB RAM was better, but the Dual-Xenon CPU with 8 cores and 12 GB RAM appeared the most stable. Each computer was configured to use Windows 7 64 bit operating system and Windows Office 2010 Professional 64 bit software. Software at 64 bit are required to open many of the large files in this study. A ProteinPilot report in Excel is about 900 MB in size and requires 20 minutes to open in a 64 bit system. Auto- calculation was turned off in Excel to avoid 20 minute duration updating of numbers upon each mouse click viewing the analyses. Proteome Discoverer, Mascot Distiller and Mascot Server are all true 64 bit software, but ProteinPilot 4.5 beta is still a 32 bit application even when it is installed on a 64 bit computer.
Peak List Generation
Five options for peak list generation used in this study are depicted in Figure 2 and indicated by numbers next to arrows.
Figure 2.
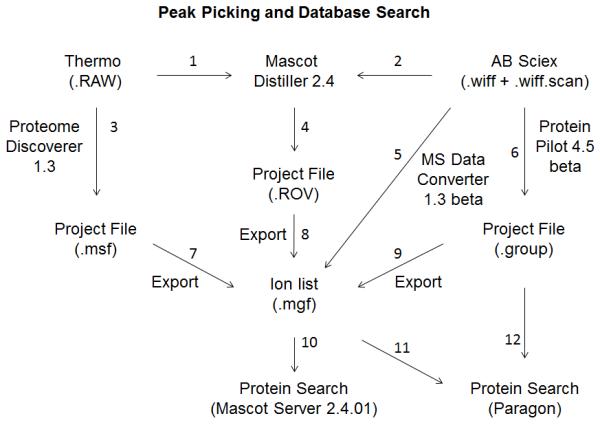
Informatics workflow for peak-picking and database searching. All paths in this figure to process raw data files of the five instruments to arrive at the results of protein search were explored. Numbers next to arrows identify specific unique procedures.
Thermo Scientific Orbitrap .RAW files were processed to peak lists by two different ways:
-
1
Raw file was processed by Thermo Scientific Proteome Discoverer 1.3 software to create an “.msf project file” then exported as a .mgf ion list. Arrows 3+7.
-
2
Raw file was processed by Mascot Distiller 2.4 to create a “.ROV project file” using new optimized parameters (Supplemental Information 2-3) for each Orbitrap optimized with the corresponding data of fraction 18 of experiment #2. The .mgf ion list files were exported from the .ROV project file. Arrows 1+4+8.
AB SCIEX TripleTOF 5600 .WIFF files were processed to peak lists three different ways:
-
3
Raw file was processed by AB SCIEX MS Data Converter 1.3 beta software to create an ion list. Arrow 5.
-
4
Raw file was processed by AB SCIEX ProteinPilot 4.5 beta software to create a “.group project file”, then exported as a .mgf ion list. Arrow 6 and 6+9.
-
5
Raw file was processed by Mascot Distiller using new optimized parameters for TripleTOF 5600 (Supplemental Information 4) optimized with the corresponding data of fraction 18 of experiment #2 to create a “.ROV project file”. The .mgf ion list files were exported from the .ROV files. Arrows 2+4+8.
Because of resource limitations, this study has not evaluated the effectiveness of other popular software for the peak-picking and database searching of data from these high resolution mass spectrometers. For example, ProteoWizard28 (open source) and Peaks 29.
Identification of Significant Sequences and Protein Families
Three different methods of searching the protein database for the identification of significant peptides and significant protein families were presented in this report.
Both Orbitrap and TripleTOF .mgf files from five paths of finding peak lists, as described above, were used to search the database using optimized parameters (Supplemental Information 5) for Mascot Server 2.4.01. Arrow 10.
Orbitrap .mgf files from two paths of finding peak lists, as described above, were used to search the database using the ProteinPilot 4.5 beta Paragon algorithm. Arrow 11. Thermo Orbitrap data include: 3+7+10, 3+7+11, 1+4+8+10, and 1+4+8+11. The paths explored for AB SCIEX TripleTOF data include: 5+10, 6+12, 6+9+10, 2+4+8+10, 2+4+8+11, and 5+11. The Orbitrap .msf files for all 30 fractions combined contained too many spectra to be imported and processed by ProteinPilot in the 3+7+11 route while AB SCIEX Wiff files do not present the same limitation.
AB SCIEX .group peak lists were also used to search the database using the ProteinPilot Paragon algorithm. Arrow 12.
Default conditions used by each software for conversion of peptide-level information into protein-level condition were used.
Normalization of iTRAQ Channels Before Quantitation
iTRAQ reporter quantification was performed by Mascot Server from the optimized peak lists. Small errors in protein determination, pipetting and iTRAQ labeling reaction can lead to significant errors in the relative intensities of the four iTRAQ channels. ProteinPilot corrects this imbalance by a setting called BIAS that uses the weighed best peptides of the whole proteome. In contrast, methods including EMMOL15 and Peaks30 weigh the normalization towards the proteins and peptides, respectively, of the highest quality to estimate the optimal ratio of the four iTRAQ channels. In the current report, we calculated the iTRAQ relative quantities for each iTRAQ channel for each protein and summed the iTRAQ channel values of the range of proteins that showed less than |log2| 0.35 change between cases and controls. We further imposed the requirement of three or more significant peptides for a protein to be in the list used for normalization. Detailed protocol is shown in Supplemental Information 6.
Mascot Protein Score Dependency of FDR
The FDR (false discovery rate) of iTRAQ quantitation varies locally from protein to protein as a function of the quality of ion statistics of the iTRAQ reporters from individual significant peptides. This important element of data interpretation has not been adequately addressed in literature. Thus we established the error of iTRAQ measurements by using the replicate pairs of iTRAQ channels in the experiments of this report. For a given protein, the 114:115 ratio is ideally 1. We calculate the absolute log2 value (|log2|) of the deviation of the 114:115 from 0. Then we averaged this value with the corresponding value for the 116:117 channels. Data scatter occurs between two technical replicates, e.g. 114 and 115, and the magnitude of error is represented by the absolute log 2 value so that the choice of which channel is the numerator or denominator becomes unimportant. Because there are fold changes for some proteins, neither the 114:115 nor the 116:117 ratio will perfectly represent a given protein. Hence we averaged the absolute log2 values of the deviations of the 114:115 and 116:117 to obtain an averaged representation of the situation of a given protein. The averaged value is plotted against the Mascot Protein Score for that protein. Table 5, Figures 3 and 4, present the reliability for fold-change determination for each protein in the proteome as a function of the Mascot Protein Score for each protein by each mass spectrometer.
Table 5.
Comparison of iTRAQ ratio compression by the four mass spectrometers. Peak lists were generated by the Proteome Discoverer for the Orbitrap and by ProteinPilot for the TripleTOF 5600. Combined fractions 6+18+24 of experiment #2 was used for this evaluation.
| Triple TOF #1 | Triple TOF #2 | LTQ-Orbitrap Velos |
Q Exactive | |
|---|---|---|---|---|
| Protein Name | |Log21 Fold- change |
|Log2| Fold- change |
|Log21 Fold- change |
|Log21 Fold- change |
| PZP_HUMAN | 1.58 | 1.78 | 1.70 | 1.58 |
| A2MG_HUMAN | 1.79 | 1.75 | 1.72 | 1.91 |
| TSP1_HUMAN | 0.77 | 0.95 | 0.88 | 0.72 |
| APOC2_HUMAN | 0.99 | 0.92 | 0.89 | 0.91 |
| APOA4_HUMAN | 0.76 | 0.82 | 0.78 | 0.77 |
| CAH1_HUMAN | 0.82 | 0.81 | 0.92 | 0.81 |
| PON1_HUMAN | 0.69 | 0.78 | 0.63 | 0.62 |
| CNDP1_HUMAN | 0.74 | 0.71 | 0.73 | 0.68 |
| CBG_HUMAN | 0.59 | 0.68 | 0.60 | 0.65 |
| FA5_HUMAN | 0.66 | 0.67 | 0.54 | 0.64 |
| PROS_HUMAN | 0.54 | 0.66 | 0.52 | 0.55 |
| ANGT_HUMAN | 0.59 | 0.64 | 0.54 | 0.53 |
| ZA2G_HUMAN | 0.58 | 0.59 | 0.49 | 0.57 |
| KAIN_HUMAN | 0.46 | 0.55 | 0.52 | 0.44 |
| SEPP1_HUMAN | 0.55 | 0.52 | 0.44 | 0.45 |
| CPN2_HUMAN | 0.32 | 0.49 | 0.31 | 0.41 |
| A1AT_HUMAN | 0.48 | 0.47 | 0.51 | 0.50 |
| PEDF_HUMAN | 0.43 | 0.46 | 0.34 | 0.38 |
| BGH3_HUMAN | 0.32 | 0.44 | 0.39 | 0.36 |
| IGHM_HUMAN | 0.27 | 0.44 | 0.45 | 0.57 |
| HEP2_HUMAN | 0.41 | 0.41 | 0.34 | 0.40 |
| C1S_HUMAN | 0.25 | 0.40 | 0.26 | 0.25 |
| THBG_HUMAN | 0.37 | 0.37 | 0.35 | 0.32 |
Figure 3.

Comparison of iTRAQ reporter quantitation reproducibility by five mass spectrometers. A: TripleTOF #1. B: TripleTOF #2. C: LTQ-Orbitrap Velos. D: Q Exactive. E: Orbitrap Elite. The number of proteins identified by each instrument combining all fractions of experiment #2 is shown in Table 4. The fold-change of a given protein is represented on the Y axis as the absolute log 2 value of fold-change determined by two technical replicates. It is correlated with the quality of identification of that protein (X-axis, Mascot protein score), as the collective confidence of identification of its peptides. Fold-changes for that protein above its threshold shown can be regarded as highly confident. The Orbitrap Elite data is from the combined fractions 7+16+24 of Experiment #4.
Figure 4.

iTRAQ protein quantitation consensus among four mass spectrometers, relationship of data tightness with decreasing Mascot Protein Score. iTRAQ fold-change ratios scatter increases with decreasing Mascot Protein Score. Red, TripleTOF #1; Blue, TripleTOF #2; Green, LTQ- Orbitrap Velos; Purple, Q Exactive. X axis represent the Mascot Protein Score from TripleTOF #2 for each protein plotted. X axis of panel A is in log. X axis of B and C are linear. Proteins with Mascot Protein Scores >100 and the absolute ratios of cases to control of |log2|>0.35 in fractions 6, 18 and 24 of experiment #2 are plotted.
RESULTS AND DISCUSSION
In 2005, 35 laboratories participated in a HUPO report describing the proteome of human plasma31. One of the difficulties experienced in that study was the diversity of database scoring algorithms and incompatible databases used by the participant laboratories, leading to numerous questionable protein identifications. Our current study has used a common data workflow to partly address some of the outstanding questions about quantitative plasma proteomics suggested by this HUPO report. All four experiments of 30 fractions each were each replicated on four mass spectrometers within a two months period. The fee-for-service volume-discounted in October 2012 was $100 to $125 per sample injection.
Peak List Generation
There are many algorithms for peak-picking of mass spectrometry data to extract information from the spectra for identifying and quantifying the ions detected prior to peptide sequence identification32. We compared peak-picking efficiency using the number of significant sequences and proteins identified among instruments (Table 2). The AB SCIEX ProteinPilot 4.5 software peak-picking has improved over the AB SCIEX MS Data Converter by including another cycle of spectra recalibration based on the abundant proteins identified. Peak-picking for the TripleTOF 5600 data was best performed by ProteinPilot 4.5 beta, as indicated by the greater number of proteins identified when compared with the peak-picking by Mascot Distiller (Table 2 B cf. A). Thus, ProteinPilot peak-picking was chosen for subsequent analyses in Tables 3-5. Peak-picking by Proteome Discoverer 1.3 for the Orbitrap instruments appeared about equal in performance to the optimized Mascot Distiller, as judged by the number of significant peptides and protein families identified by Mascot Server search of the Swiss-Prot human protein database (Table 2).
Table 2.
Comparison of the number of significant peptides and protein families identified by five peak-picking algorithms. The fractions shown in this table were from Experiment #2. N/A = not performed because it was not recommended. MS Data Converter is an older version of current algorithm found in ProteinPilot 4.5 beta, and found lower numbers of peptides and proteins (data not shown). Mascot Server was used to search the Swiss-Prot Database. Queries, Families and Significant Sequences are defined for the Mascot Server at the Matrix Science web site1. The sequence query is a search in which one or more peptide molecular masses are combined with sequence, composition and fragment ion data. Peptides identified with 95% confidence are considered “significant sequences” by Mascot Server. We further applied a filter ion score of 20 and a requirement of 2 significant sequences for confident protein identification. Proteins discovered are grouped into Families by means of shared peptide matches and, within each family, hierarchical clustering is used to illustrate which proteins are closely related and which are more distant2.
| A.Proteome Discoverer for Orbitrap Peak List / ProteinPilot for TripleTOF 5600 Peak List | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TripleTOF #1 | TripleTOF #2 | LTQ-Orbitrap Velos | Q Exactive | Orbitrap Elite | |||||||||||
| Fraction | Queries | Families | Significant Sequences | Queries | Families | Significant Sequences |
Queries | Families | Significant Sequences |
Queries | Families | Significant Sequences |
Queries | Families | Significant Sequences |
| 6 | 12,963 | 170 | 227 | 20,512 | 179 | 248 | 16,318 | 173 | 302 | 16,203 | 242 | 424 | 12,492 | 181 | 315 |
| 18 | 12,442 | 305 | 998 | 26,174 | 322 | 1109 | 19,542 | 344 | 1384 | 21,765 | 563 | 2,023 | 16,781 | 420 | 1,505 |
| 24 | 11,799 | 292 | 1,109 | 26,789 | 291 | 1,125 | 18,232 | 358 | 1696 | 21,323 | 557 | 2,206 | 16,440 | 416 | 1,641 |
| B. Mascot Distiller Peak-picking | |||||||||||||||
| 6 | N/A* | N/A* | 15,102 | 165 | 283 | 15,793 | 215 | 385 | 12,305 | 148 | 274 | ||||
| 18 | 25,622 | 195 | 594 | 18,099 | 334 | 1322 | 20,992 | 555 | 1,966 | 16,356 | 382 | 1,398 | |||
| 24 | 26,161 | 166 | 582 | 15,605 | 346 | 1622 | 20,612 | 560 | 2,188 | 16,060 | 386 | 1,556 | |||
Table 3.
Comparison of significant peptide yield and protein family identification by ProteinPilot Paragon algorithm for searching the Swiss-Prot database at 99% confidence. Peak lists were generated by the Proteome Discoverer for the Orbitrap and by ProteinPilot for the TripleTOF 5600, then imported into ProteinPilot. A, distinct peptides = peptides of all qualities. B. Distinct peptides identified by ProteinPilot at high stringency of 1% FDR using ProteinPilot Data FIT. C. High confidence proteins identified by ProteinPilot at high stringency of 1% FDR using ProteinPilot Data FIT. “\” indicates no report from ProteinPilot.
| A.Distinct peptides and proteins identified by ProteinPilot from peak lists | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TripleTOF #1 | TripleTOF #2 | LTQ-Orbitrap Velos | Q Exactive | Orbitrap Elite | |||||||||||
| Fraction | Proteins Detected |
Proteins before Grouping |
Distinct Peptides |
Proteins Detected |
Proteins before Grouping |
Distinct Peptides |
Proteins Detected |
Proteins before Grouping |
Distinct Peptides |
Proteins Detected |
Proteins before Grouping |
Distinct Peptides |
Proteins Detected |
Proteins before Grouping |
Distinct Peptides |
| 6 | 126 | 133 | 1644 | 137 | 154 | 1848 | 131 | 153 | 1550 | 182 | 220 | 2720 | 124 | 194 | 1501 |
| 18 | 194 | 221 | 2557 | 219 | 251 | 3607 | 235 | 582 | 3042 | 386 | 550 | 5640 | 264 | 486 | 3112 |
| 24 | 194 | 222 | 2663 | 205 | 233 | 3565 | 283 | 452 | 3905 | 396 | 493 | 5579 | 271 | 436 | 3078 |
| B. Distinct peptide level FDR Analysis Globa FDR from FIT- number of peptides identified | |||||||||||||||
| 10% | 5% | 1% | 10% | 5% | 1% | 10% | 5% | 1% | 10% | 5% | 1% | 10% | 5% | 1% | |
| 6 | 1753 | 1665 | 1493 | 1917 | 1784 | 1572 | 1681 | 1584 | 1380 | 3077 | 2885 | 2461 | 1,682 | 1,573 | 1317 |
| 18 | 2672 | 2531 | 2297 | 3485 | 3239 | 2853 | 3247 | 3076 | 2759 | 6185 | 5860 | 5294 | 3,381 | 3,195 | 2832 |
| 24 | 2786 | 2620 | 2245 | 3425 | 3169 | 2708 | 4038 | 3786 | 3077 | 6005 | 5689 | 5026 | 3,283 | 3,102 | 2689 |
| C. Protein Level FDR Analysis- Global FDR from FIT- number of proteins detected | |||||||||||||||
| 10% | 5% | 1% | 10% | 5% | 1% | 10% | 5% | 1% | 10% | 5% | 1% | 10% | 5% | 1% | |
| 6 | \ | \ | \ | \ | \ | 273 | \ | \ | \ | \ | \ | 396 | \ | \ | 266 |
| 18 | \ | \ | 309 | \ | \ | \ | 390 | 369 | 320 | \ | \ | 563 | \ | \ | 378 |
| 24 | \ | \ | 307 | \ | \ | 307 | 422 | 397 | 359 | \ | \ | 592 | \ | \ | 397 |
Identification of Significant Sequences and Proteins
Proteome Discoverer 1.3 search of the Swiss-Prot database produced information substantially similar to Mascot Server 2.4 (data not shown), but Proteome Discoverer 1.3 was not able to import peak lists from ProteinPilot to enable broader comparison. Proteome Discoverer database search was not used further in the following studies. Mascot Server was chosen as the common reference to facilitate the comparison of the performance of these five mass spectrometers. From a peak list, Mascot Server first generates a peptide list that contains ions of varying quality and confidence (Table 2, Supplemental Information 7 and 8). Peptides identified with 95% confidence are considered “significant sequences” by Mascot Server. When a filter of ion score of 20 and a confidence cut-off of 95% are imposed, the list of peptides decreases substantially. The homologs of proteins identified are grouped into protein families using algorithms explained by the Matrix Science web site. A reason for TripleTOF #1 to have identified a smaller number of significant sequences and protein families than TripleTOF #2 (Table 2A) may be because the amount of peptides loaded was 200 ng/fraction (quantified by NanoOrange binding and normalized in quantity before injection) for TripleTOF #1 compared with up to 1.2 microgram per fraction for TripleTOF #2. Compared with the TripleTOFs, the Q Exactive identified for each fraction at least 40% higher number of significant sequences and protein families as TripleTOF #2, using only about 500 ng of peptides per fraction. The LTQ-Orbitrap Velos identified slightly more peptides and proteins than TripleTOF #2 while Orbitrap Elite performance was comparable to TripleTOF #2.
We have included the peptide list of each instruments used for the experiments #2 (Supplemental Information 7) and #4 (Supplemental Information 8) to cover all 5 instruments. Each instrument has about 80,000 -110,000 peptides, thus only the non-redundant peptides with the best score are presented. Peptide probability based scores, iTRAQ ratios, peptide overlap and non-overlap among instruments are shown.
Table 3 used ProteinPilot Paragon algorithm for searching the Swiss-Prot Database to address the impact of software innovations. The ProteinPilot analyses supported the conclusions of the Mascot Server analyses. Protein Pilot 4.5 beta recalibrates the spectra using the theoretical masses of the high precision peptides sequences detected for the high abundance proteins33. The software also fits non-identified ions to potential modified-peptides of the high abundance proteins during protein identification. Both the Mascot “significant sequences” and ProteinPilot “1% FDR (False Discovery Rate) “confident distinct peptides” used a randomized Swiss-Prot protein database as decoy to determine the discovery significance and FDR. Instead of “significant sequences” from Mascot Server, ProteinPilot considers each modified peptide a “distinct peptide”, thus reporting higher numbers than Mascot. “1% Global FDR with data fitting” represents the “high confidence” peptides identified in ProteinPilot Paragon algorithm13. For example, after choosing high confidence, the number of distinct peptides in Table 3 for fraction 24 for TripleTOF #2 decreased from 3,565 in Table 3A to 2,708 in Table 3B. However, Table 3 shows that the TripleTOF #2 “distinct peptides” identified proteins about 2/3 as effectively as the “distinct peptides” of the Q Exactive. The “distinct peptides” 3,565 for TripleTOF #2 in Table 3A dropped 30% to a 2,708 in B, half as much (19%) for TripleTOF #1, and only 10% for the Q Exactive. TripleTOF #1 identified more peptides and proteins than TripleTOF #2 (identical hardware) in spite of injecting only 20% as much samples, perhaps because it specified ten scans maximum per period of 2.8 seconds instead of 25 scans for TripleTOF #2. Thus benchmarking can readily lead to testable improvements even for these advanced instruments.
Combining the Data of 30 Fractions for Peptide and Protein Identification and Quantitation
Table 4 summarizes the outcome of database searching of each 30 fraction hyper- fractionation experiment. All 30 fractions of experiment #2 were run on the LTQ-Orbitrap Velos and the Q Exactive as well as both Triple-TOF 5600s. Similarly, all 30 fractions of experiment #4 were run on the Elite and the Q Exactive as well as both Triple-TOF 5600s. We found the Q Exactive and the LTQ-Orbitrap Velos continued to identify the largest number of peptides.
Table 4.
Total peptide identification and protein identification of two experiments each from combining all 30 fractions. This data is for subsequent evaluation of the iTRAQ quantitation performance. Peak lists were generated by the Proteome Discoverer for the Orbitrap and by ProteinPilot for the TripleTOF 5600. Mascot Server was used for Swiss-Prot database searching. The data was not normalized among the mass spectrometers. N/A indicates not performed. Legends are the same as in Table 2.
| TripleTOF #1 | TripleTOF #2 | LTQ-Orbitrap Velos | Q Exactive | Orbitrap Elite | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All 30 fractions |
Queries | Families | Significant Sequences |
Queries | Families | Significant Sequences |
Queries | Families | Significant Sequences |
Queries | Families | Significant Sequences |
Queries | Families | Significant Sequences |
| Exp #2 |
387,417 | 1,192 | 5,960 | 665,934 | 1,169 | 5,997 | 506,793 | 1,141 | 7,703 | 566,927 | 1,689 | 10,530 | N/A | N/A | N/A |
| Exp #4 |
402,165 | 1,273 | 5,735 | 748,273 | 1,243 | 5,278 | N/A | N/A | N/A | 559,067 | 1,571 | 8,274 | 436,126 | 1,299 | 6,650 |
Tables 2-4 showed that peptide and protein identification was not proportional to the number of queries submitted, suggesting that some MS/MS scans were not optimally acquired. In fact the strategy of using NanoOrange peptide quantitation for TripleTOF #1 to normalize sample quantities for each injection improved coverage for some fractions (data not shown). Alternatively, injecting larger quantity of peptides for all fractions by TripleTOF #2 may have been an equally successful strategy.
Normalization of iTRAQ channels Before Quantitation
To compare the relative performance of the data from different mass spectrometers, one must decide whether to normalize the four iTRAQ channels to compensate for errors introduced by pipetting, protein determination and iTRAQ label efficiency variations. ProteinPilot Paragon algorithm BIAS normalization is ON for Paragon analyses in this study. iTRAQ reporter intensity quantitation is only as precise as the ion statistics of the best peptides selected to represent a given protein. We used a variation of the EMMOL method to weigh the normalization towards the high ion statistics of the higher abundance proteins when using Mascot Server, which does not perform iTRAQ channel normalization. To avoid bias, the data of each instrument was used for its own normalization. The stoichiometry of the observed labeling efficiencies of the four channels of experiment #2 for each mass spectrometer is shown in Supplemental Information 6.
Evaluation of iTRAQ Quantitation - Reproducibility Depends on Signal Collection
The evaluation of iTRAQ quantitation performance has received much attention in literature. Major concerns are potential iTRAQ underestimation of fold-change due to mixed MS/MS of contaminating background peptide ions34, 35, the possibility of iTRAQ 8-plex suffering from the confusion of the 120 m/z iTRAQ reporter ion with any contaminating phenylalanine immonium ion26, and the presence of variance heterogeneity where higher error is observed for data with lower signal intensities.
While the importance of ion statistics is well appreciated, how it limits the confidence of fold-change determination on different platforms has seldom been discussed. Literature reports traditionally use a p value for each protein by itself, which allowed chance to lead to false confidence in trusting lower p values than the instrument is capable of delivering. In a typical iTRAQ proteome analysis, a subjective threshold will often be selected to qualify a subset of proteins that changed as technically relevant for the conditions of the study. For example, the threshold may be defined as the value of the log2 iTRAQ ratio at which 95% of all proteins had no deviation from each other36. In literature, a variance-stabilization normalization method has been proposed to stabilize the variance across the entire intensity range35. Instead of a single number representing global confidence for iTRAQ quantified fold-change in a given dataset (for example, defined with a given peptide quality threshold), we introduce the notion that the confidence in the fold-change determination by iTRAQ may be best expressed with respect to the ion statistics, or conveniently, the Mascot Protein Score of each given protein. This consideration addresses the problem of variance heterogeneity. In practice, the cases and controls of our experiments were run as technical replicates using two iTRAQ channels each. These replicates can be used to establish the precision of the iTRAQ reporter ion statistics for each protein for a given instrument as shown in Figure 3. The X axis represents the Mascot Protein Score from TripleTOF #2 for each protein plotted. However, the protein score for a given protein will be different for different mass spectrometers as a result of having different ion statistics obtained. Protein identification ion statistics may differ from those for reporter ion quantitation. A similar analysis of experiment #4 allows another visualization of the performance differences (Supplemental Information 9). The level of ion statistics for the detection of an iTRAQ reporter ion impacts quantitation sensitivity. A mass spectrometer run with less ion statistics than the more sensitive mass spectrometer may have to raise the threshold of reliable fold-change determination of case/control in order to use the proteins with lower Mascot Protein Scores (Figure 3).
We observed that the confidence in the fold-change determination is not coupled to the performance of protein identification. Longer scan time is important for getting good iTRAQ reporter signals. All three Orbitrap instruments tested, as operated in these core facilities, produced lower data scatter that allowed more proteins to be accepted at lower fold-change thresholds for differential proteomes than the data obtained from the TripleTOF 5600 instruments. Whether this observation is correct needs to be considered in light of the phenomenon of iTRAQ Ratio Compression discussed in the following section. The LTQ- Orbitrap Velos and Elite instruments allowed the use of similar low fold-changes as the Q Exactive, but fewer proteins for quantitation. The reason may be that no quantitation is possible if a protein were not identified in the first place. Variations prominent among proteins with Mascot Protein Scores lower than 220 may be due to variability of peptide sequences detected or due to variations in the amount of iTRAQ ratio suppression discussed below.
Concerns about iTRAQ Ratio Compression
iTRAQ ratio compression is a phenomenon that if not accounted for can undermine the fold-change conclusions in an iTRAQ comparison experiment. iTRAQ ratio compression refers to an artifact of attenuated protein-fold-change values caused by the presence of non-targeted iTRAQ- labeled background ions other than the intended parent peptide ion selected for fragmentation34. These non-targeted peptides often have no differential expression, hence their iTRAQ ratios of 1:1:1:1 will add to the iTRAQ reporter ions from the intended peptides, causing the observed iTRAQ reporter ratios to approach unity. Each mass spectrometer may use a different m/z window for selecting parent peptide ions for MS/MS fragmentation and for iTRAQ reporter ion quantitation. An instrument that uses one m/z unit window will have about three times less background ion as another mass spectrometer that uses a window of three m/z units. Having the larger window could potentially lead to significant iTRAQ reporter ion compression, leading to the appearance of lower data scatter for a given Mascot Protein Score. This concern has been demonstrated true for early mass spectrometer designs, but may be less significant in the experiments in this study as will be shown below.
Our study did not include spike-in of standard peptides with defined fold differences at different parts of the LC gradient to determine the absolute amounts of iTRAQ ratio compression in different parts of the LC elution. This omission was intentional because of concern over possible interference of the proteome investigation by the material spiked in, and because absolute fold-change values are less important to pathway-change analysis than the precision and the direction of change. We used exhaustive immunodepletion and hyper-fractionation of the iTRAQ-labeled peptides to minimize the number of peptide ions that co-eluted and co-selected for MS/MS. In place of spiked-in controls, our proteome comparison contained many proteins with large differential expression. The ratios of (116+117)/(114+115) for some of these proteins with the best ion statistics for fractions 6+18+24 are shown in Table 5. These proteins showed about the same fold-changes across the four instruments, as seen in Figure 4 A & B (the Mascot Protein Score of data from TripleTOF #2 is used as reference in this comparison). Essentially, for the proteins with Mascot Protein scores above 225, all four instruments showed about the same fold-change for each protein. Other than for some individual proteins in Figure 4C, proteins with Mascot Protein Score <220 (whose peptide representation cannot be assured to be identical from different mass spectrometers), overall, the determination of protein fold-change appeared reproducible among the four instruments, indicating little difference in their iTRAQ ratio compression. A similar analysis of fractions 7+18+24 of experiment #4 supports the same conclusion is shown in Supplement Information 9.
Potential Limitations of the Study Comparison
Individual components in a mass spectrometer system can significant impact overall performance and influence instrument comparison. This report did not compare the mass spectrometer portion of each system using exactly the same HPLC support, sample sizes, column matrix, length and diameter, spray needle, and LC-MS/MS protocol. These important factors can be complementary and were considered in each mass spectrometry facility. Each system was operated as optimized as it could be at that time. Sample size is a factor for a given LC-MS/MS system but apparently not the most important factor in system comparison because the Q Exactive was twice as sensitive as the TripleTOF using sample sizes halfway between those used for the two TripleTOFs (Table 1). The number of plasma proteins identified by the Q Exactive doubled those by other Orbitrap mass spectrometers even when the LC used were almost identical. The sample size used on the Q Exactive was only about 30% that used on the other Orbitrap mass spectrometers. Interestingly, TripleTOF #1 was comparable to TripleTOF #2 in the number of proteins identified although the two systems differed 5 fold in the sample quantities injected, suggesting tolerance for user-introduced variations.
Overlap of the Proteomes Identified by Different Mass Spectrometers
706 proteins with iTRAQ reporter ratios were found to be in common among the proteomes discovered at high confidence by each of four mass spectrometers in experiment #2. TripleTOF #1 and TripleTOF #2 overlapped by 674 protein family identifications (Figure 5). Compared with the proteome described by the Q Exactive, which was the largest, the two TripleTOFs together contributed 164 proteins not found in the proteome obtained by the Q Exactive, while overlapping by 825 proteins. The LTQ-Orbitrap Velos overlapped 812 proteins with the Q Exactive, while bringing 105 proteins not found in the latter. Of these, 71 proteins were not found by the two TripleTOFs either. Thus each instrument contributed to the 1,584 protein families with iTRAQ quantitation for experiment #2, and 2,366 protein families identified in total (minimum of two peptides with one being significant sequence) (Figure 5) in the plasma proteome. A similar Venn diagram can be seen for Experiment 4 (including Orbitrap Elite) in Supplemental Information 10. While 782 proteins that were identified had no iTRAQ labeling for comparison of their expression levels in cases and controls, they can be further examined if some of their known functions are of interest to the biology under consideration. Methods that are able to target the comparison of these proteins include, but are not limited to ELISA, triplequad SRM mass spectrometry, and immunoaffinity capture before mass spectrometry. This comparison illustrates the benefit of technical replicates for obtaining reproducible exhaustive coverage of a proteome.
Figure 5.

Area-proportional Venn diagrams37 (Biovenn2) of overlap in the protein identification by four instruments for experiment #2. A and C, protein identification with iTRAQ quantitation. B, proteins identified with no iTRAQ quantitation. The data of two TripleTOFs were combined in C as surrogate replicate runs.
In 2011, Farrah et al combined multiple datasets to arrive at a number of 1929 highly non- redundant protein sequences of plasma at an estimated 1% FDR33. By comparison, Experiment #2 and #4 of this report, at the stringent peptide significance of 0.01 and Mascot peptide ion score cut off of 20, mapped over 1073 plasma proteins to the plasma proteome of Farrah et al. The 1073 figure did not include 64 immunoglobulins and 33 proteins found in the Farrah proteome which we removed by immunodepletion. Of the 1073 figure, 874 were found in the Q Exactive data which used a single 30 fraction analysis without replicates. This number represents the low limit of the overlap because, for example, at least 71 proteins in the proteome described by the Q Exactive, with Mascot Protein Score over 400, were not found in the Farrah et al proteome. This discovery efficiency illustrates the efficacy of exhaustive immunodepletion using a novel approach of two cycles of immunodepletion and the recent significant improvements in mass spectrometers.
The outsourcing-benchmarking approach described in this report can significantly improve the productivity of research for proteomics or other disciplines that are traditionally supported by local core facilities. Smaller institutions can invest in local core facility personnel that provide consultation, sample preparation, quality control, networking guidance and data analysis without investing in the high cost of frequent instrument upgrade. The cost of mass spectrometry time at less than $125 per injection is competitive with in-house chargeback. By benchmarking at multiple sites at the same time, a researcher avoids the chance of having his or her research program limited by an instrument not optimized for a particular application and thus gets the most out of the precious samples without delay.
Potential for Improving the Performance of the Mass Spectrometers
Some of the advantages of a mass spectrometer may be intrinsic in its design while others may be in its operation. The TripleTOF 5600 units were shown to be highly reproducible. They should be able to use their fast scan speed to sample more peptide sequences for identification than other slower platforms when sensitivity is not limiting. Both the TripleTOF and the Q Exactive collected an average of 20 spectra queries for each unique peptide in this study; there may be room to utilize their duty cycles more effectively. From the comparison in Table 1, it appears that longer LC column (25 cm), utilization of UPLC, and the use of “isotope peak exclusion” to prevent wasting data acquisition on the isotope peaks, correlated with the success of the Q Exactive and LTQ-Orbitrap Velos. Other factors, including the column resin, the back pressure, the gradient length, the duration of the dynamic exclusion, can also allow LC peaks to contain distinct subsets of peptides. Another important parameter for the TripleTOF 5600 may be the threshold for parent ion selection. Higher thresholds may automatically allow longer MS/MS ion accumulation to fit into the period after each full scan of parent ions. While the ion traps can wait for the traps to be filled while collecting a low abundance ion, this strategy is more difficult to implement in the TripleTOF design. However, ideally the instrument control software should set the threshold and ion accumulation for each parent ion based on its ion statistics. Furthermore, after the first MS/MS data acquisition of a given peptide, the software ideally should determine the parameters for its second data acquisition. Because the LC column used by TripleTOF #1 can handle higher peptide loads than 200 ng, injecting 500 ng of peptide may provide more peptide and protein coverage. The Orbitrap Elite was the only Orbitrap to have the “Exclude Isotope Peaks” setting turned off as a result of this setting not being helpful in earlier generations. Turning on this setting may be a potential adjustment to improve the Elite’s performance. On the positive side, having this setting turned off may have led to better reporter ion statistics (Figure 3E) by including the isotope peaks of a given peptide.
In summary, this report demonstrated that in iTRAQ quantitation, as the protein score decreases with decreasing protein abundance, the diminishing ion statistics can lead to a gradual decrease in the fold-change threshold that can be used reliably. Instead of using an arbitrary threshold for significant fold-changes, we believe even small fold-changes that are highly precise may reflect important effects on biology and should be included in pathway analysis. This report demonstrates how the performance of the mass spectrometry can affect interpretation in biological research.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge John Cottrell for optimizing the peak-picking parameters for Mascot Distiller for this study. This work was partially supported by NIH grant RC2 HL101713, CA138017, the NCI Work Assignment #16 of N01-CN-43309, the Driskill Foundation, PA Dept. of Health, and Institutional Core Grants P30 CA06927 and P30 CA16058. We acknowledge the FCCC Biorepository, and Darshna Bhatt, Hopethe Hubbard, and Danielle J. Culbert of OSU, for assistance in the collection of some of the plasma samples.
The abbreviations used are
- iTRAQ
Isobaric Tag for Relative and Absolute Quantitation
- IEF
Isoelectric Focusing
- IPG
Immobilized Polyacrylamide Gel
- FDR
False Discovery Rate
Footnotes
Conflicts of interests
The authors declared no conflict of interest. The authors received free demo licenses and advice from Thermo Scientific, AB SCIEX, and Matrix Science using their software.
S This article contains Supporting Information for Publication.
1. QSTAR LC-MSMS parameters.pdf
2. Orbi-Elite and Velos_Expt4_Frac17_optimized_Distiller_parameters.txt
3. Q-Exactive_Expt2_Frac18_optimized_Distiller_parameters.txt
4. ABSCIEX-5600_Expt2_Frac18_optimized_Distiller_parameters.txt
6. Experiment #2 normalization Protocol and ratios.xls
7. Peptide lists for experiment #2.xls
8. Peptide lists for experiment #4.xls
9. Comparison of iTRAQ reporter quantitation of Exp #4 by 4 instruments.pdf
References
- 1.DeSouza L, Diehl G, Rodrigues MJ, Guo J, Romaschin AD, Colgan TJ, Siu KW. Search for cancer markers from endometrial tissues using differentially labeled tags iTRAQ and cICAT with multidimensional liquid chromatography and tandem mass spectrometry. J Proteome Res. 2005;4(2):377–86. doi: 10.1021/pr049821j. [DOI] [PubMed] [Google Scholar]
- 2.Chong PK, Gan CS, Pham TK, Wright PC. Isobaric tags for relative and absolute quantitation (iTRAQ) reproducibility: Implication of multiple injections. J Proteome Res. 2006;5(5):1232–40. doi: 10.1021/pr060018u. [DOI] [PubMed] [Google Scholar]
- 3.Boehm AM, Putz S, Altenhofer D, Sickmann A, Falk M. Precise protein quantification based on peptide quantification using iTRAQ. BMC Bioinformatics. 2007;8:214. doi: 10.1186/1471-2105-8-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raman SP, Kawamoto S, Blackford A, Hruban RH, O’Brien-Lennon AM, Wolfgang CL, Rezaee N, Edil B, Fishman EK. Histopathologic Findings of Multifocal Pancreatic Intraductal Papillary Mucinous Neoplasms on CT. AJR Am J Roentgenol. 2013;200(3):563–9. doi: 10.2214/AJR.12.8924. [DOI] [PubMed] [Google Scholar]
- 5.Talar-Wojnarowska R, Pazurek M, Durko L, Degowska M, Rydzewska G, Smigielski J, Janiak A, Olakowski M, Lampe P, Grzelak P, Stefanczyk L, Malecka-Panas E. Pancreatic cyst fluid analysis for differential diagnosis between benign and malignant lesions. Oncol Lett. 2013;5(2):613–616. doi: 10.3892/ol.2012.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shimizu Y, Yamaue H, Maguchi H, Yamao K, Hirono S, Osanai M, Hijioka S, Hosoda W, Nakamura Y, Shinohara T, Yanagisawa A. Predictors of Malignancy in Intraductal Papillary Mucinous Neoplasm of the Pancreas: Analysis of 310 Pancreatic Resection Patients at Multiple High-Volume Centers. Pancreas. 2013 doi: 10.1097/MPA.0b013e31827a7b84. [DOI] [PubMed] [Google Scholar]
- 7.Atef E, El Nakeeb A, El Hanafy E, El Hemaly M, Hamdy E, El-Geidie A. Pancreatic cystic neoplasms: predictors of malignant behavior and management. Saudi J Gastroenterol. 2013;19(1):45–53. doi: 10.4103/1319-3767.105927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt CM. Pancreatic cyst cytology: optimization of cancer risk profiling. Cancer Cytopathol. 2013;121(1):2–3. doi: 10.1002/cncy.21267. [DOI] [PubMed] [Google Scholar]
- 9.Thornton GD, McPhail MJ, Nayagam S, Hewitt MJ, Vlavianos P, Monahan KJ. Endoscopic ultrasound guided fine needle aspiration for the diagnosis of pancreatic cystic neoplasms: a meta-analysis. Pancreatology: official journal of the International Association of Pancreatology. 2013;13(1):48–57. doi: 10.1016/j.pan.2012.11.313. [DOI] [PubMed] [Google Scholar]
- 10.Lim LG, Lakhtakia S, Ang TL, Vu CK, Dy F, Chong VH, Khor CJ, Lim WC, Doshi BK, Varadarajulu S, Yasuda K, Wong JY, Chan YH, Nga ME, Ho KY. The Asian, E. U. S. C., Factors Determining Diagnostic Yield of Endoscopic Ultrasound Guided Fine-Needle Aspiration for Pancreatic Cystic Lesions: A Multicentre Asian Study. Digestive diseases and sciences. 2013 doi: 10.1007/s10620-012-2528-2. [DOI] [PubMed] [Google Scholar]
- 11.Zhan XB, Wang B, Liu F, Ye XF, Jin ZD, Li ZS. Cyst fluid carcinoembryonic antigen concentration and cytology by endosonography-guided fine needle aspiration in predicting malignant pancreatic mucinous cystic neoplasms. J Dig Dis. 2013;14(4):191–5. doi: 10.1111/1751-2980.12027. [DOI] [PubMed] [Google Scholar]
- 12.Rockacy MJ, Zahid M, McGrath KM, Fasanella KE, Khalid A. Association Between KRAS Mutation, Detected in Pancreatic Cyst Fluid, and Long-term Outcomes of Patients. Clin Gastroenterol Hepatol. 2013;11(4):425–9. doi: 10.1016/j.cgh.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, Keating SP, Hunter CL, Nuwaysir LM, Schaeffer DA. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007;6(9):1638–55. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Lengqvist J, Uhlen K, Lehtio J. iTRAQ compatibility of peptide immobilized pH gradient isoelectric focusing. Proteomics. 2007;7(11):1746–52. doi: 10.1002/pmic.200600782. [DOI] [PubMed] [Google Scholar]
- 15.Kim PD, Patel BB, Yeung AT. Isobaric Labeling and Data Normalization without Requiring Protein Quantitation. Journal of biomolecular techniques: JBT. 2012;23(1):11–23. doi: 10.7171/jbt.12-2301-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Yi J, Schuchard MD, Mehigh RJ, Cockrill SL, Scott GB, Tammen H, Schulz-Knappe P, Speicher DW, Vitzthum F, Haab BB, Siest G, Chan DW. HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics. 2005;5(13):3262–77. doi: 10.1002/pmic.200401245. [DOI] [PubMed] [Google Scholar]
- 17.Yi J, Kim C, Gelfand CA. Inhibition of intrinsic proteolytic activities moderates preanalytical variability and instability of human plasma. J Proteome Res. 2007;6(5):1768–81. doi: 10.1021/pr060550h. [DOI] [PubMed] [Google Scholar]
- 18.Yeung AT, Patel BB, Li XM, Seeholzer SH, Coudry RA, Cooper HS, Bellacosa A, Boman BM, Zhang T, Litwin S, Ross EA, Conrad P, Crowell JA, Kopelovich L, Knudson A. One-hit effects in cancer: altered proteome of morphologically normal colon crypts in familial adenomatous polyposis. Cancer Res. 2008;68(18):7579–86. doi: 10.1158/0008-5472.CAN-08-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. doi: 10.1186/1750-1172-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157–62. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 21.Lynch HT, Smyrk T, Lynch J. An update of HNPCC (Lynch syndrome) Cancer Genet Cytogenet. 1997;93(1):84–99. doi: 10.1016/s0165-4608(96)00290-7. [DOI] [PubMed] [Google Scholar]
- 22.Qian WJ, Kaleta DT, Petritis BO, Jiang H, Liu T, Zhang X, Mottaz HM, Varnum SM, Camp DG, 2nd, Huang L, Fang X, Zhang WW, Smith RD. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Molecular & cellular proteomics: MCP. 2008;7(10):1963–73. doi: 10.1074/mcp.M800008-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel BB, Barrero CA, Braverman A, Kim PD, Jones KA, Chen DE, Bowler RP, Merali S, Kelsen SG, Yeung AT. Assessment of Two Immunodepletion Methods: Off-Target Effects and Variations in Immunodepletion Efficiency May Confound Plasma Proteomics. J Proteome Res. 2012;11:5947–5958. doi: 10.1021/pr300686k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eriksson H, Lengqvist J, Hedlund J, Uhlen K, Orre LM, Bjellqvist B, Persson B, Lehtio J, Jakobsson PJ. Quantitative membrane proteomics applying narrow range peptide isoelectric focusing for studies of small cell lung cancer resistance mechanisms. Proteomics. 2008;8(15):3008–18. doi: 10.1002/pmic.200800174. [DOI] [PubMed] [Google Scholar]
- 25.Farrah T, Deutsch EW, Aebersold R. Using the Human Plasma PeptideAtlas to study human plasma proteins. Methods Mol Biol. 2011;728:349–374. doi: 10.1007/978-1-61779-068-3_23. [DOI] [PubMed] [Google Scholar]
- 26.Evans C, Noirel J, Ow SY, Salim M, Pereira-Medrano AG, Couto N, Pandhal J, Smith D, Pham TK, Karunakaran E, Zou X, Biggs CA, Wright PC. An insight into iTRAQ: where do we stand now? Analytical and bioanalytical chemistry. 2012;404(4):1011–27. doi: 10.1007/s00216-012-5918-6. [DOI] [PubMed] [Google Scholar]
- 27.Pichler P, Kocher T, Holzmann J, Mohring T, Ammerer G, Mechtler K. Improved precision of iTRAQ and TMT quantification by an axial extraction field in an Orbitrap HCD cell. Anal Chem. 2011;83(4):1469–74. doi: 10.1021/ac102265w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012;30(10):918–20. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, Zhang W, Zhang Z, Lajoie GA, Ma B, PEAKS DB. de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics. 2012;11(4):M111. doi: 10.1074/mcp.M111.010587. 010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacerda CM, Xin L, Rogers I, Reardon KF. Analysis of iTRAQ data using Mascot and Peaks quantification algorithms. Brief Funct Genomic Proteomic. 2008;7(2):119–26. doi: 10.1093/bfgp/eln017. [DOI] [PubMed] [Google Scholar]
- 31.Agusti A, MacNee W, Donaldson K, Cosio M. Hypothesis: does COPD have an autoimmune component? Thorax. 2003;58(10):832–4. doi: 10.1136/thorax.58.10.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung KH, Ryu JK, Oh HS, Seo JY, Jin E, Lee DH, Kim YT, Yoon YB. Pancreatic pseudocyst after endoscopic ultrasound-guided fine needle aspiration of pancreatic mass. Clin Endosc. 2012;45(4):431–4. doi: 10.5946/ce.2012.45.4.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seymour S, Hunter CL. ProteinPilot Descriptive Statistics Template. 2013. [Google Scholar]
- 34.Ow SY, Salim M, Noirel J, Evans C, Rehman I, Wright PC. iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly”. J Proteome Res. 2009;8(11):5347–55. doi: 10.1021/pr900634c. [DOI] [PubMed] [Google Scholar]
- 35.Karp NA, Huber W, Sadowski PG, Charles PD, Hester SV, Lilley KS. Addressing accuracy and precision issues in iTRAQ quantitation. Molecular & cellular proteomics: MCP. 2010;9(9):1885–97. doi: 10.1074/mcp.M900628-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Qiang X, Owsiany K, Zhang S, Thannhauser TW, Li L. Evaluation of different multidimensional LC-MS/MS pipelines for isobaric tags for relative and absolute quantitation (iTRAQ)-based proteomic analysis of potato tubers in response to cold storage. J Proteome Res. 2011;10(10):4647–60. doi: 10.1021/pr200455s. [DOI] [PubMed] [Google Scholar]
- 37.Matrix Science on Mascot Protein Families.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.