

Abstract
An amine-N-oxide ligated palladium complex, in conjunction with a silver co-catalyst, catalyzes imidation of arenes by the reagent N-fluorobenzenesulfonimide. The reaction enables imidation of a variety of arenes at or below room temperature, requires no coordinating directing group on the substrate, and gives synthetically useful yields with only one equivalent of arene. Mechanistic data implicates an unusual mechanism devoid of commonly invoked organometallic intermediates; oxidation of the palladium catalyst occurs as the turnover-limiting step, while C–H bond functionalization occurs subsequently at a high oxidation state of the catalyst.
Transition metal catalysis holds promise as a strategy for the direct functionalization of otherwise unreactive C–H bonds. Many reported transition metal catalyzed aryl C–H functionalization approaches rely on metallation of the arene followed by functionalization of the aryl-metal ρ bond.1 To accelerate an otherwise sluggish metallation step, coordinating directing groups are commonly employed to accelerate carbon-metal bond formation; without such directing groups, often multiple equivalents or even solvent quantities of arene are required to drive the metallation.2 With few exceptions,3 catalytic C–H functionalization without the aid of directing groups remains an unmet challenge. Herein, we report a catalytic, intermolecular C–H imidation of arenes enabled by a departure from the conventional metallation-functionalization sequence. The new palladium catalyst 1 enables group transfer without the formation of conventionally targeted organometallic intermediates. Our transformation affords synthetically useful yields with the arene substrate as limiting reagent.
Historically, perhaps the most important method of introducing an amino group into arenes has been electrophilic aromatic nitration followed by reduction of the nitro group.4 However, nitration of arenes typically requires strongly acidic or oxidizing reaction conditions, which limits its use on substrates with sensitive functional groups. Alternatively, several aniline derivatives can be reliably prepared by transition-metal catalyzed directed C–H amidation,5 but the requirement for a coordinating directing group limits the potential substrate scope. Non-chelation-assisted transition-metal catalyzed C–H amination reactions,6 as well as metal free approaches involving the use of an oxidant and amine to introduce the C–N bond, have also been reported.7 However, unless the arene has particularly acidic C–H bonds, such as in perfluoroarenes or benzoxazole derivatives,6c,6d,7b these methods require a minimum of 1.5 equivalents, and not uncommonly solvent quantities of arene substrate, and yields are often based on the amidating reagent, not the arene.
Imidation of arenes by the reagent N-fluorobenzenesulfonimide (NFBS) is catalyzed by 1 and Ag(bipy)2ClO4 as shown in Figure 1. Both catalyst 1 and the silver co-catalyst are required in the reaction; control experiments in which either is omitted gave less than 10% of imidated product, although the silver co-catalyst can be replaced with Ru(bipy)3(PF6)2 with similar results (Table 1, compounds 2a and 2b). The reaction proceeds equally well in the presence or absence of light. Catalyst 1 was readily prepared from tetrakis(acetonitrile)palladium(II) triflate and the N-(2-pyridylmethyl)pyrrolidine N oxide ligand, which itself is available in two steps from commercial starting materials. The catalyst can also be generated in situ by mixing Pd(NCMe)4(OTf)2 and the ligand with nearly identical results as obtained with isolated and purified 1. Because catalyst 1 is easily prepared and stored, we have used it directly in our investigations. The unusual pyridine-N-oxide ligand motif is exceptionally effective for the catalytic imidation, and several other palladium-based catalysts supported by ligands such as bipyridine afforded product in at most 3% yield (Supporting Information). A rationale for the distinct ability of the amine-N oxide to effect arene functionalization is presented in the mechanism section of this manuscript; we propose that the reactivity of well-defined catalyst 1 induced by the amine-N-oxide ligands is also responsible for the distinct reactivity when compared to other palladium catalyzed amination reactions, such as benzylic amination with NFBS.8
Figure 1.

C–H imidation catalyzed by 1 and Ag(bipy)2ClO4, and x ray structure of the cation of 1 (elipsoids drawn at 50% probability)
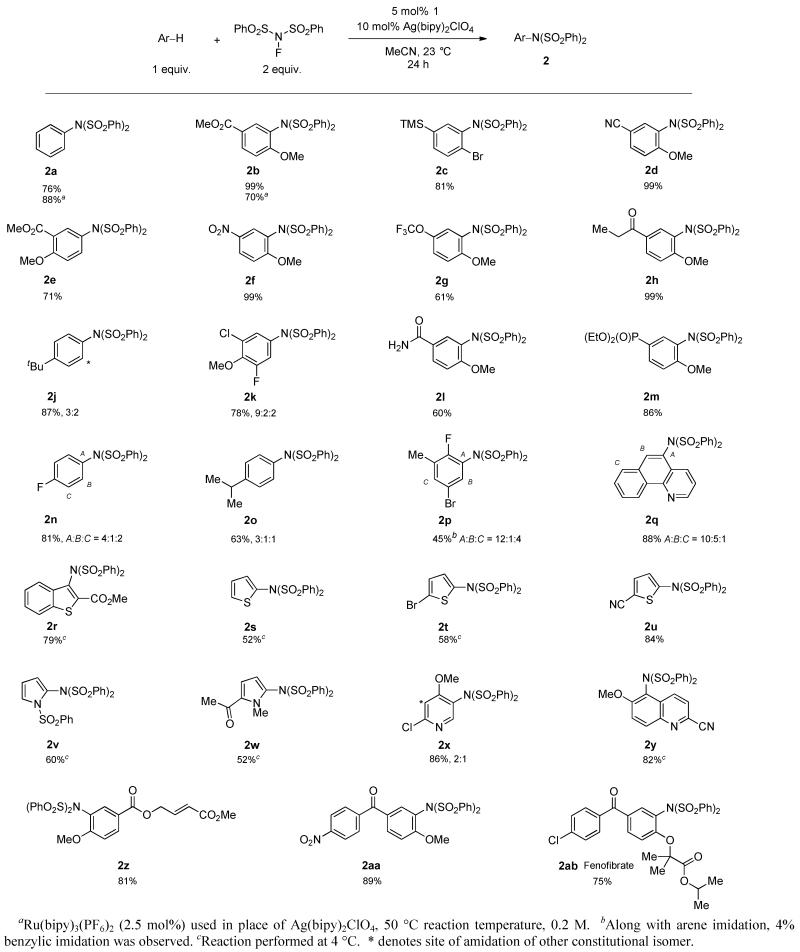
Table 1. Substrate Scope of C–H Imidation Catalyzed by 1 and Ag(bipy)2ClO4.
|
A variety of arenes, including N- and S-heteroarenes, could be efficiently imidated (Table 1). Selectivity is substrate-intrinsic; resonance donors, such as alkoxy and halogen groups, direct imidation ortho/para, similar to electrophilic aromatic substitution. Inductive donors do not direct as effectively (e.g., 2c, 2j), and substrates that lack a strong directing bias (e.g. 2n, 2o, 2p) afford mixtures of constitutional isomers. Functional groups that can cause problems for nitration such as esters and silanes are tolerated. Arenes more electron-poor than those shown in Table 1 show diminished reactivity and give lower yields. Competitive C–H fluorination and double imidation are significant side-reactions for more electron-rich arenes, though performing the reaction at lower temperature (4 °C) can substantially reduce both side reactions for some substrates (2r-t, 2v, 2w, 2y). Potential coordinating directing groups do not influence regioselectivity as they do in directed catalytic C–H functionalization reactions5 (2b, 2e, 2l, 2q). The reactions for which the data is shown in Table 1 were performed rigorously dry and air-free, but similar results were obtained for reactions performed in air with reagent quality solvents (see Supporting Information). Product 2b, synthesized on 11 g scale with this methodology, was chosen to demonstrate the removal of the sulfonyl protecting groups. Treatment of 2b with magnesium in methanol under sonication produced aniline 3 in 85% yield (eq. 1).
 |
(1) |
Mechanistic data, detailed below, unambiguously implicate a mechanism with the following noteworthy features: 1) oxidation of the bis-cationic Pd(II) complex 1 enabled by the amine-N-oxide ligands, 2) involvement of the co-catalyst in single-electron redox chemistry, 3) irreversible substrate binding prior to C–H bond functionalization, and 4) C–N bond formation occurring without C–H palladation. A plausible mechanistic proposal including these features is depicted in Scheme 1. Turnover-limiting oxidation of catalyst 1 yields Pd(IV) complex II. Single-electron reduction of II by Ag(bipy)2+ and substrate association follow to form Pd(III) intermediate III. Intermediate III formally transfers sulfonimidyl radical to the bound substrate, which expels a delocalized radical and regenerates 1. Oxidation of the radical intermediate by Ag(bipy)22+, followed by deprotonation, yields the sulfonimidated product.
Scheme 1. Proposed Catalytic Cycle.

Turnover-limiting oxidation of 1 by NFBS is supported by the measured rate law of the reaction, which is first order with respect to both 1 and NFBS, and zero-order with respect to arene and Ag(bipy)2ClO4. When only NFBS and 1 are combined in acetonitrile, catalytic reduction of NFBS to HN(SO2Ph)2 and HF is observed, with the reducing equivalents evidently derived from the solvent. The rate of the catalytic reduction of NFBS is identical to the rate of NSBS consumption in the imidation reaction (Scheme 2). The rate law and the identical rate for both reactions shown in Scheme 2 establish turnover-limiting oxidation of 1 by NFBS to yield a common, short-lived high-valent palladium intermediate such as II.
Scheme 2. Kinetic Study of Imidation and NFBS Reduction Catalyzed by 1.

Oxidation of Pd(II) complexes typically requires strongly ρ-donating anionic ligands such as hydrocarbyl ligands, which are absent from 1.9 DFT calculations suggest that the HOMO of 1 is an extended M–L π-antibonding orbital of dxz parentage from palladium, instead of the dz2-based orbital more typical of square-planar d8 complexes. The oxygen lone pairs of the amine-N-oxide ligand interact strongly with the Pd-based dxz orbital, driving it higher in energy than the dz2-based orbital (Figure 2). This interaction may explain how complex 1 can be oxidized by NFBS, despite its two formal positive charges.
Figure 2.

Valence orbital diagram of 1
We propose that the silver co-catalyst serves to provide access to intermediate III, which is the putative intermediate responsible for C–N bond formation. Ag(bipy)22+ was observed by EPR spectros copy during catalysis, which implicates the co-catalyst in redox reactivity. Inner-sphere reactivity of the co-catalyst can be ruled out because coordinatively saturated Ru(bipy)3(PF6)2 is also an effective co-catalyst. Furthermore, the oxidation of Ru(bipy)32+ to Ru(bipy)33+ by NFBS is substantially accelerated in the presence of 1, consistent with oxidation of 1 to II by NFBS followed by single-electron oxidation of Ru(bipy)32+ by II (see Supporting Information for details). Reduction of II by the co-catalyst to yield the C–N bond forming species III explains why for most arenes, substrate consumption is not observed in the absence of the co-catalyst.
Because oxidation of 1 by NFBS is turnover-limiting, the C–N bond forming step cannot be studied kinetically. We have therefore employed competition kinetic isotope effect (KIE) experiments to probe this step (Scheme 3). An inverse secondary intramolecular kH/kD of 0.80 ± 0.01 for imidation of 1,3,5-trideuterobenzene was measured. The measured KIE implicates rehybridization of the C–H bond from sp2 to sp3 in the product-determining transition state, consistent with C–N bond formation via inner-sphere addition of dibenzenesulfonimidyl radical to the bound arene in intermediate III. Inverse secondary KIEs are unusual for palladium-catalyzed C–H functionalization reactions; C–H palladations at Pd(II)10 and Pd(IV)11 typically display primary KIE values. Electrophilic addition would also be consistent with the observed intramolecular isotope effect. However, inverse secondary KIE values for electrophilic substitution are rare;12 most commonly, KIEs close to unity are observed for electrophilic processes due to the opposing effects of rehybridization and hyperconjugation on the zero-point vibrational energy of the affected C–H bond.13 Addition of nitrogen-based radicals to arenes is well-precedented, including catalytic imidations with NFBS which are proposed to proceed through high-valent transition-metal imidyl radical species.14 Inner-sphere attack from an intermediate such as III to form the putative aryl radical is supported by an intermolecular competition kH/kD of 1.03 ± 0.02. The absence of a KIE in this case is consistent with irreversible substrate binding prior to C–N bond formation.10,11b
Scheme 3. Intramolecular and Intermolecular Kinetic Isotope Effect.

We have described the first aryl C–H imidation reaction which gives synthetically useful yields with only one equivalent of arene and does not require coordinating directing groups. The transformation is made possible by a departure from the most common strategies: we have developed a catalyst supported by amine-N-oxide ligands, which enable oxidation of the doubly cationic Pd(II) complex prior to substrate activation. C–H functionalization proceeds from a high oxidation state complex without the formation of conventional organometallic intermediates. We anticipate that our distinct approach may find utility in other C–H functionalization reactions.
Supplementary Material
ACKNOWLEDGMENT
Funding was provided by NIH-NIGMS (GM088237) and the NSF (CHE-0952753). G.B.B. thanks NSF GRFP (DGE0644491) for a graduate fellowship. We thank S. E. Parker and M. G. Campbell for helpful discussions, M. G. Campbell for X-Ray analysis, and F. Bastuck for 5 g scale synthesis of catalyst 1.
Footnotes
Notes
A provisional patent application on catalysts and methods presented herein has been filed through Harvard.
ASSOCIATED CONTENT
Detailed experimental procedures, spectroscopic characterization for all new compounds, mechanistic data, and crystallographic information files. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Engle KM, Mei T-S, Wasa M, Yu J-Q. Acc. Chem. Res. 2012;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Neufeldt SR, Sanford MS. Acc. Chem. Res. 2012;45:936–946. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kuhl N, Hopkinson MN, Wencel-Delord J, Glorius F. Angew. Chem., Int. Ed. 2012;51:10236–10254. doi: 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]
- (3).(a) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem. Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (b) Nagib DA, MacMillan DWC. Nature. 2011;480:224–228. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Olah GA, Ripudaman M, Narang SC. Nitration: Methods and Mechanisms. VCH Publishers, Inc.; New York: 1989. [Google Scholar]
- (5).(a) Thu HY, Yu W-Y, Che C-M. J. Am. Chem. Soc. 2006;128:9048–9049. doi: 10.1021/ja062856v. [DOI] [PubMed] [Google Scholar]; (b) Ng K-H, Chan ASC, Yu W-Y. J. Am. Chem. Soc. 2010;132:12862–12864. doi: 10.1021/ja106364r. [DOI] [PubMed] [Google Scholar]; (c) Xiao B, Gong T-J, Xu J, Liu ZJ, Liu L. J. Am. Chem. Soc. 2011;133:1466–1474. doi: 10.1021/ja108450m. [DOI] [PubMed] [Google Scholar]; (d) Sun K, Li Y, Xiong T, Zhang J, Zhang Q. J. Am. Chem. Soc. 2011;133:1694–1697. doi: 10.1021/ja1101695. [DOI] [PubMed] [Google Scholar]; (e) Yoo EJ, Ma S, Mei T-S, Chan KSL, Yu J-Q. J. Am. Chem. Soc. 2011;133:7652–7655. doi: 10.1021/ja202563w. [DOI] [PubMed] [Google Scholar]; (f) Ng K-H, Zhou Z, Yu W-Y. Org. Lett. 2012;14:272–275. doi: 10.1021/ol203046n. [DOI] [PubMed] [Google Scholar]; (g) Grohmann C, Wang H, Glorius F. Org. Lett. 2012;14:656–659. doi: 10.1021/ol203353a. [DOI] [PubMed] [Google Scholar]
- (6).(a) Díaz Requejo MM, Belderraín TR, Nicasio MC, Trofimenko S, Pérez PJ. J. Am. Chem. Soc. 2003;125:12078–12079. doi: 10.1021/ja037072l. [DOI] [PubMed] [Google Scholar]; (b) Li Z, Capretto DA, Rahaman RO, He C. J. Am. Chem. Soc. 2007;129:12058–12059. doi: 10.1021/ja0724137. [DOI] [PubMed] [Google Scholar]; (c) Zhao H, Wang M, Su W, Hong M. Adv. Synth. Catal. 2010;352:1301–1306. [Google Scholar]; (d) Guo S, Qian B, Xie Y, Xia C, Huang H. Org. Lett. 2011;13:522–525. doi: 10.1021/ol1030298. [DOI] [PubMed] [Google Scholar]; (e) Froehr T, Sindlinger CP, Kloeckner U, Finkbeiner P, Nachtsheim BJ. Org. Lett. 2011;13:3754–3757. doi: 10.1021/ol201439t. [DOI] [PubMed] [Google Scholar]; (f) Shrestha R, Mukherjee P, Tan Y, Litman ZC, Hartwig JF. J. Am. Chem. Soc. 2013;135:8480–8483. doi: 10.1021/ja4032677. [DOI] [PubMed] [Google Scholar]
- (7).(a) Antonchick AP, Samanta R, Kulikov K, Lategahn J. Angew. Chem., Int. Ed. 2011;50:8605–8608. doi: 10.1002/anie.201102984. [DOI] [PubMed] [Google Scholar]; (b) Froehr T, Sindlinger CP, Kloeckner U, Finkbeiner P, Nachtsheim BJ. Org. Lett. 2011;13:3754–3757. doi: 10.1021/ol201439t. [DOI] [PubMed] [Google Scholar]; (c) Kim HJ, Kim J, Cho SH, Chang S. J. Am. Chem. Soc. 2011;133:16382–16385. doi: 10.1021/ja207296y. [DOI] [PubMed] [Google Scholar]; (d) Samanta R, Bauer JO, Strohmann C, Antonchick AP. Org. Lett. 2012;14:5518–5521. doi: 10.1021/ol302607y. [DOI] [PubMed] [Google Scholar]
- (8).Iglesias A, Alvarez R, de Lera A, Muñiz K. Angew. Chem., Int. Ed. 2012;51:2225–2228. doi: 10.1002/anie.201108351. [DOI] [PubMed] [Google Scholar]
- (9).(a) Oloo W, Zavalij PY, Zhang J, Khaskin E, Vedernikov AN. J. Am. Chem. Soc. 2010;132:14400–14402. doi: 10.1021/ja107185w. [DOI] [PubMed] [Google Scholar]; (b) McCall AS, Wang H, Desper JM, Kraft S. J. Am. Chem. Soc. 2011;133:1832–1848. doi: 10.1021/ja107342b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McCall AS, Kraft S. Organometallics. 2012;31:3527–3538. [Google Scholar]
- (10).Simmons EM, Hartwig JF. Angew. Chem., Int. Ed. 2012;51:3066–3072. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- (11).(a) Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J. Am. Chem. Soc. 2009;131:9488–9489. doi: 10.1021/ja9031659. [DOI] [PubMed] [Google Scholar]; (b) Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J. Am. Chem. Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]; (c) Racowski JM, Ball ND, Sanford MS. J. Am. Chem. Soc. 2011;133:18022–18025. doi: 10.1021/ja2051099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Olah GA, Kuhn SJ, Flood SH. J. Am. Chem. Soc. 1961;83:4571–4580. [Google Scholar]; (b) Olah GA. Acc. Chem. Res. 1971;4:240–248. [Google Scholar]; (c) Nakane R, Kurihara O, Takematsu A. J. Org. Chem. 1971;36:2753–2756. [Google Scholar]
- (13).(a) Streitwieser A, Jagow RH, Fahey RC, Suzuki S. J. Am. Chem. Soc. 1958;80:2326–2332. [Google Scholar]; (b) Melander L. Isotope Effects on Reaction Rates. The Ronald Press Company; New York: 1960. p. 112. [Google Scholar]; (c) Taylor R. Electrophilic Aromatic Substitution. John Wiley & Sons; West Sussex, England: 1990. p. 33. [Google Scholar]
- (14).(a) Stella L. Angew. Chem., Int. Ed. Engl. 1983;22:337–422. [Google Scholar]; (b) Ni Z, Zhang Q, Xiong T, Zheng Y, Li Y, Zhang H, Zhang J, Liu Q. Angew. Chem., Int. Ed. 2012;51:1244–1247. doi: 10.1002/anie.201107427. [DOI] [PubMed] [Google Scholar]; (c) Zhang H, Pu W, Xiong T, Li Y, Zhou X, Sun K, Liu Q, Zhang Q. Angew. Chem., Int. Ed. 2013;52:2529–2533. doi: 10.1002/anie.201209142. [DOI] [PubMed] [Google Scholar]; (d) Kaneko K, Yoshino T, Matsunaga S, Kanai M. Org. Lett. 2013;15:2502–2508. doi: 10.1021/ol4009848. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.