

Abstract
Protection of pancreatic β cells is an approach to prevent autoimmune type 1 diabetes (T1D) and to protect transplanted islets. Reactive oxygen species (ROS) are important mediators of β cell death during the development of T1D. We have examined the role of elevated ROS dissipation in the prevention of T1D using the ALR mouse strain. The selection of ALR, for resistance against alloxan-induced free radical–mediated diabetes, led to a strain of mice with an elevated systemic as well as pancreatic ROS dissipation. Independent genetic mapping studies have identified ALR-derived diabetes protective loci. Conplastic and congenic mouse as well as cell line studies have confirmed the genetic mapping and demonstrated that the elevated ROS dissipation protects ALR β cells from autoimmune destruction. Our data support the hypothesis that elevated ROS dissipation protects β cells against autoimmune destruction and prevents T1D development.
Keywords: type 1 diabetes, reactive oxygen species, mitochondria, mouse model, genetics
Introduction
Type 1 diabetes (T1D) is a metabolic disorder resulting from the autoimmune cell-mediated destruction of pancreatic insulin-producing β cells. This disease is affected by genetic and environmental factors. The destruction of pancreatic β cells during the autoimmune attack occurs when activated cells of the immune system induce apoptosis via engaging FAS on the surface of β cells or necrosis by secreting proinflammatory cytokines, perforin and granzyme B, and reactive oxygen (ROS) and nitrogen (RNS) species.1 While these routes employ different receptors and signaling pathways, ROS are common to all of these mechanisms of cell death induction. In clinical studies, T1D patients and antibody-positive at-risk individuals showed an increase in oxidative stress compared to healthy controls or their first-degree relatives.2,3 Therefore, oxidative stress plays an important role in the pathogenesis of T1D.
Pancreatic islets exhibit greater susceptibility to damage by ROS compared to other tissues as a result of lower antioxidant defenses.4,5 This exquisite sensitivity has been proposed to play a role in the pathogenesis of T1D.6–12 Production of ROS and downregulation of antioxidant defenses characterized by a diminished level of reduced glutathione (GSH) and a progressive decline in the transcripts for catalase (CAS), superoxide dismutase (SOD), and thioredoxin (TRX) have been observed in apoptotic processes.13–15 Hence, the ability to maintain redox potential in β cells may counteract apoptotic signaling, protecting β cells and inhibiting T1D.8,9,16–19 In vitro studies have illustrated that ROS are involved in β cell damage induced by proinflammatory cytokines and that antioxidant treatment prevents the damage.20–24 Further, increasing the stress response in islets can prevent or reduce immune-mediated damage including the rejection of islet grafts.25–27 GSH, SOD, and TRX have all been shown to inhibit proapoptotic signaling by blocking the actions of apoptosis signal-regulating kinase (ASK1), AP-1, and NF-κB.28–30 Recombinant TRX has been shown to protect cells against apoptosis mediated through TNF and Fas pathways,31 and when TRX was overexpressed in β cells of NOD mice, there was a significant reduction in T1D incidence.26 The rapid rejection of islet grafts in diabetic NOD recipients has also been attributed in part to damage mediated by ROS, and treatment with ROS scavengers slowed al-lograft rejection.32,33 These data strongly suggest that antioxidative capacity modulates both necrosis and apoptosis and increases in antioxidant defenses can inhibit or slow the rate of β cell death in T1D onset, regardless of the mode by which β cell destruction occurs. Our overall goal is to discover how pancreatic islets can be rendered more resistant to immune stress, allowing for maintenance of β cell mass and for a reduction of immunosuppression after transplantation. The key to these studies is the ALR mouse.
We have been investigating the role of ROS in the pathogenesis of T1D using the NOD and ALR mouse models. While the NOD strain was bred to be a model of human T1D,34 progenitors of the ALR strain were selected for resistance to alloxan-induced free radical–mediated diabetes.35 These two strains were derived from the same outbred Jcl:ICR mouse population. NOD and ALR share approximately 85% identity genome wide, based on the testing of approximately 3,000 microsatellite and single nucleotide polymorphism (SNP) markers, including most known Idd loci. Although closely related to T1D-prone NOD, ALR mice not only are resistant to alloxan-induced diabetes, but they also resist development of spontaneous autoimmune T1D and diabetes induced by adoptive transfer of diabetogenic T cells.8 In addition, in vitro the islets of ALR mice are resistant to destruction mediated by diabetogenic cytotoxic T cells or proinflammatory cytokines.8 When compared to NOD or the co-selected alloxan-sensitive ALS strain, ALR mice show a higher antioxidative status, including elevated superoxide dismutase (SOD), glutathione peroxidase (GPX), and glutathione reductase (GSR) activities in many tissues, including the pancreatic islets.9,18,19 Alloxan is known to induce diabetes by selectively damaging pancreatic β cells by releasing free radicals inside the cells.36,37 The elevated antioxidant enzyme activity in the ALR pancreas is therefore a foundation of the alloxan-resistance. Since free radicals also act as mediators of autoimmune β cell destruction in the development of T1D, we further investigated the mechanism of T1D resistance in ALR mice.
ALR-Derived T1D Protective Loci
The ALR mouse, with islets that are remarkably resistant to destruction by NOD-derived autoimmune effectors, provides a unique model to address the hypothesis that factors present at the islet level can prevent diabetes onset. To identify the genes that protect ALR islets from destruction, we initiated three independent genetic experiments by crossing ALR with either NOD or ALS mice. These experiments provide a unique opportunity to map novel loci that are restricted to protection at the islet level and likely associated with dissipation of ROS.
A backcross study was initiated between ALR and NOD to identify ALR-derived T1D-protective loci. Using a cohort of 227 first back-cross mice, we mapped ALR-derived T1D resistance loci to Chr. 3 (Susp), 8 (Idd22), and 17 (Idd16.2),38 as well as to the mitochondrial genome (mtDNA).39 The major histocompatibility (MHC) gene cluster locates on Chr. 17. The linkage peak on Chr. 17 mapped in the backcross is very close to the MHC,38 suggesting the participation of the immune system in ALR-derived protection. The linkage on Chr. 3 overlaps with the Susp (suppressor of superoxide production) locus that was independently mapped between the ALR and ALS strain.40 This Susp locus controls the ability of ALR neutrophils and macrophages to suppress free radical burst after phorbol mystrate acetate (PMA) stimulation.40 While previous genetic studies using the NON, C57BL/B6, or C57BL/10 strains as outcross partners to NOD mapped T1D loci to both Chr. 3 and 17,41–45 prior to our studies Chr. 8 had not been associated with autoimmune diabetes. We therefore believe that Idd22 on Chr. 8 is a unique ALR-derived locus.
ALR-Derived T1D Resistance on Chr. 3 Maps to the Susp Locus
In BC1 of NOD with C57BL/10, C57BL/6, and NON, Chr. 3 has been repeatedly identified as a source of multiple susceptibility loci.41–45Idd3, 10, 17, and 18 all map to Chr. 3. Yet, as ALR and NOD share the IL-2b allele38 and diagnostic markers for Idd10, 17 and 18 are equivalent between ALR and NOD, it was surprising that a linkage was detected onChr. 3.Of interest, the ALR-derived T1D resistance locus, at 33cM, sited between previously mapped Idd loci, completely overlapped with the Susp locus, which was independently mapped and controls the ability of ALR cells to suppress free radical burst after PMA activation.40 This was demonstrated by outcross with, and then a backcross to, the free radical–sensitive ALS strain. Unlike the suppressed oxidative burst from activated ALR or [ALSxALR] F1 hybrid neutrophils, PMA-activated neutrophils from ALS bone marrow exhibited normal oxidative burst. Testing of a backcross 1 cohort produced a 1:1 segregation of the phenotype that allowed mapping to Chr. 3 (peak linkage D3Mit241 at 33cM, χ2 = 32, P = 0.0001).
Overexpression of SOD1 alone can either dissipate the superoxide generated via NADPH oxidase (NOX) or inhibit activation of NOX leading to a greater than 80% reduction in superoxide production.46,47 Therefore, the increased SOD1 activity in ALR cells18,19,40 could rapidly detoxify the superoxide from NOX to peroxide and the previously reported increased GPX activity in ALR cells9,18 would turn the peroxide to water. Indeed, the decreased levels of neutrophil oxidative burst correlated inversely with superoxide dismutase 1 (SOD1) activity (r2 = 0.821). The overlapping positions of these two linkages suggest that the ALR locus contributing to the heightened ability to dissipate superoxide is the same locus contributing to protection from T1D.
ALR-Derived Protective Loci against Alloxan
Since ALR islets resist destruction by alloxan and NOD-derived autoimmune effectors, it is likely that some or all of the loci identified as providing T1D resistance are responsible for protection at the β cell level and are associated with elevated dissipation of free radicals. We proposed that genetic elements that provide resistance to alloxan would co-localize to regions of ALR-derived T1D resistance. F1 hybrid mice between NOD and ALR were protected only when the maternal parent is ALR,39 suggesting that a maternal factor, either X or mtDNA, plays a role in this protection. To clarify the maternal factor and to map genes contributing to alloxan resistance, F2 hybrid mice were created between NOD and ALR strains, and diabetes was induced by a single intravenous injection of alloxan, 52 mg/Kg body weight, at 6 weeks of age. A genome-wide scan was performed on this population of 646 F2 mice using 117 genetic markers. The highest incidence of alloxan-induced diabetes (45.6%) was seen in the group of male mice with both the mtDNA and Y chromosome derived from NOD. ALR mtDNA was significantly protective (χ2 = 9.5, P = 0.002). In addition, linkages were mapped to Chr. 2, 3, and 8. The linkage on Chr. 8 overlapped with the previously mapped ALR-derived T1D resistance locus, Idd22.38 However, the ALR allele on Chr. 3 contributed to a higher susceptibility to alloxan. This linkage locates proximal to the previously mapped, T1D protective Susp locus. The Chr. 2 locus shows a higher susceptibility contributed by the heterozygous type. Heterosis is not unusual in sensitivity to chemicals, possibly by altering the activity of a metabolic enzyme. An example is the previously demonstrated increased sensitivity to thiazolidinedione-induced hepatosteatosis in an F1 cross between two genetically diverse inbred strains, NON/Lt and NZO/Lt.48 For these findings we propose that the mtDNA and Chr. 8 loci control β cell resistance to ROS and thus protect against T1D.
Role of ALR Mitochondrial Genome in the Protection of β Cells against Free Radical–Mediated Damage
The mitochondrial genome was mapped to be protective against both alloxan-induced and autoimmune diabetes. Sequence analysis of the mitochondrial genome revealed a novel SNP distinguishing ALR from other closely related mouse strains, including NOD.39 The mitochondrial genomes of the two strains are identical except this SNP, which is in mt-Nd2. mt-Nd2 encodes a subunit of NADH:ubiquinone dehydrogenase, the first enzyme complex of the mitochondrial electron transport chain and the major source of cellular ROS production.49 A nucleotide difference, C in NOD or A in ALR, results in an amino acid residue of leucine or methionine, respectively.39 In humans, there is a corresponding mt-ND2 C/A polymorphism resulting in the same amino acid replacement. The human mt-ND2a allele is reported to be linked with lower incidence of a series of diseases associated with oxidative stress,50–53 including T1D.54
To study the effect of the mt-Nd2a, reciprocal conplastic mice were created.17 Basal function of mitochondria isolated from the liver of conplastic mice did not show any difference when compared to the parental strains.17 However, mitochondria isolated from mice encoding mt-Nd2a demonstrated lower ROS production than did mice encoding mt-Nd2c.16,17 While NOD.mtALR mice exhibited lower mitochondrial free radical production, this reduction in oxidative burden did not change the course of spontaneous T1D nor were NOD.mtALR mice resistant to alloxan-induced diabetes. In contrast, when ALR.mtNOD were challenged with different doses of alloxan, they showed an increased susceptibility compared to ALR (Fig. 1A). The increased ROS production by ALR.mtNOD mitochondria likely results in a decrease in the amount of exogenous ROS required to destroy the β cells of this mouse strain.
Figure 1.
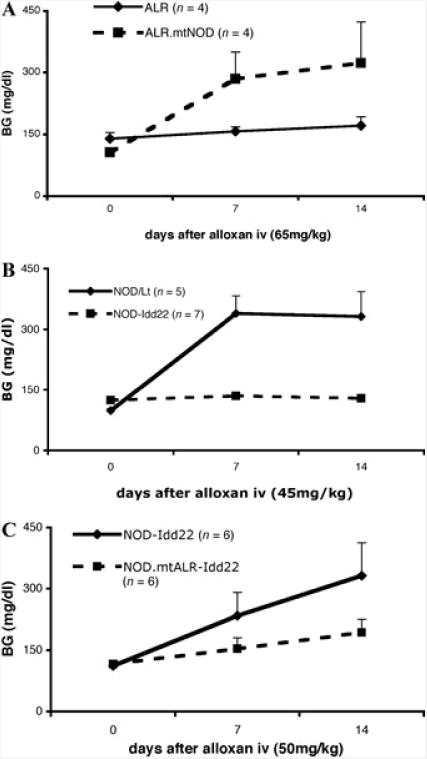
(A) Diabetes was induced in ALR.mtNOD but not ALR mice by i.v. administration of 65 mg/kg alloxan (6-week-old males, n = 4 in each group). (B) Diabetes was induced in NOD/Lt but not NOD-Idd22 mice by 45 mg/kg alloxan i.v. (6-week-old males, n = 5 for NOD, n = 7 for NOD-Idd22). (C) When challenged with 50 mg/kg alloxan intravenously, NOD.mtALR-Idd22 mice (6-week-old males, n = 9) showed higher resistance than did NOD-Idd22 mice (6-week-old males, n = 6).
The amount of islet β cells harvested from mice is limited. For mitochondrial function studies, large numbers of cells are needed to isolate fresh mitochondria. Therefore, we created β cell lines containing the NOD nuclear genome and ALR mt-Nd2a allele. These cells, NIT-4, are genetically identical to NIT-1 except for the SNP in mt-Nd2. NIT-4 cells were created by mating female conplastic NOD.mtALR mice to NOD.Cg-Tg(Ins2-TAg) 1 Lt Prkdcscid/DvsJ male mice, isolating islets from F1 hybrids and culturing islet cells to develop to an immortalized cell line. The insulin secretory capabilities stimulated by glucose and arginine were comparable between NIT-1 and NIT-4 cells (data not shown). Under oxidative stress exerted either by alloxan or H2O2, NIT-4 cells showed a significantly higher viability than NIT-1 cells (Table 1). The strong resistance of NIT-4 cells to free radical damage supports a protective role of mt-Nd2a at the β cell level in T1D.
Table 1. Cell Viability under Oxidative Stress.
| Cell Type | ||
|---|---|---|
|
|
||
| Agent | NIT-1 | NIT-4 |
| H2O2 (200 μM) | 50.97 ± 2.76 | 95.09 ± 2.95* |
| Alloxan (100 mM) | 26.83 ± 7.62 | 65.41 ± 10.11* |
NIT-1 and NIT-4 cells were treated with either H2O2 for 3 hours or Alloxan for 10 minutes. After washing, cell survival was assayed with the MTT assay. Results are displayed as percent of untreated cells ± standard deviation.
P < 0.05.
Genes on Chromosome 8 Protect at β Cell Level
The ALR allele on Chr. 8 (Idd22) was mapped to be protective against both alloxan diabetes65 and T1D,38 and the peak linkages overlap with each other. Chr. 8 congenic mice, NODcALR(D8Mit205-D8Mit33) [NOD-Idd22], were created as previously described,17,55 to further study the role of and identify T1D protective genes in this region. Spontaneous T1D was completely eliminated in the homozygous congenic stock at N10. Compared to NOD mice, these congenic mice showed decreased susceptibility to alloxan (Fig. 1B). The protection contributed by Idd22 is very likely at the β cell level. The common effect of β cell damage in both autoimmune and alloxan-induced diabetes again suggests that this protection may be due to elevated ROS dissipation, as previously observed in ALR mice.18 Given the role of oxidative stress in the pathogenesis in both types of β cell damage, a plausible candidate gene in this region would be Prdx2, located at 87.6MB. This gene product belongs to peroxiredoxins, which are a ubiquitous family of antioxidant enzymes. Another peroxiredoxin-related locus was previously mapped on Chr.8, called Prdx1-rs2 (peroxiredoxin 1, related sequence2), at 32cM.56 Another type of candidate in this region is Kruppel-like factor 2 (Klf2). Klf2, which is elevated 4-fold in ALR islets and 3.6-fold in NOD-Idd22 compared to NOD, is capable of both upregulating antioxidant gene expression57 as well as inhibiting NF-κB activation.58 NF-κB plays a key role in cytokine-induced β cell death.9 In fact, ALR islets are cytokine-resistant and showed defective nuclear translocation of NF-κB P65 subunit after cytokine treatment, correlating with reduced kinetics of IκB degradation and suppressed iNOS induction.9
Interactions of Mitochondrial and Nuclear Genome Determine Susceptibility/Resistance
As shown above, the ALR-derived mt-Nd2a allele alone did not affect the susceptibility of NOD mice to either T1D or alloxan. Although the NOD-derived mt-Nd2c allele did increase the susceptibility of ALR mice to alloxan when compared to unmanipulated ALR mice, ALR.mtNOD conplastic mice still resist the development of spontaneous T1D. Likewise, the Chr. 8 allele from ALR increased the resistance of NOD mice to alloxan, but compared to ALR mice, the protection is only partial. When the linkages mapped in the above-mentioned F2 crosses were stratified by the allele of mt-Nd2 inherited, some linkages were dependent of the mt-Nd2c or mt-Nd2a allele. These data suggest that interactions between genes on nuclear chromosomes and in mitochondrial genome determine the contribution to the phenotype.
The effect of the interaction between Chr.8 and the mtDNA was studied in the congenic-conplastic stock, NOD.mtALR.ALR-(D8mit205-D8mit33) [NOD.mtALR-Idd22]. These mice have both the Idd22 locus from ALR and the mt-Nd2a allele on the NOD background. When compared to NOD-Idd22 mice, these NOD.mtALR-Idd22 mice show an even higher resistance to alloxan (Fig. 1C), suggesting that the interaction of ALR genes on Chr.8 and mt-Nd2a allele contributes the resistance. The distal peak linkage mapped in the F2 cross, marked by the D8mit107 at 89.52 MB, overlaps the linkage mapped for T1D resistance with the peak D8mit80 at 90.99 MB. A clear link between the candidates discussed above, Prdx2 or Klf2, and the mitochondria might not be obvious. A plausible explanation may be that these gene products may not interact, by work additively to both reduce ROS production and dissipation, resulting in an increase in the ROS threshold for β cell death. Nuclear mitochondrial genes could also interact directly with mt-Nd2 to either reduce or enhance ROS production. Within the 95% confidence interval, two subunits of Complex I are encoded, Ndufa13 and Ndufb7. Because Complex I is the major site of intracellular ROS production during normal cellular metabolism, the interaction of mt-Nd2a with a second protective allele within this enzyme complex could result in more efficient enzyme function and a reduction in any electron leak that would lead to ROS production.
Genes on Chromosome 3 Protect Islets through Elevated ROS Dissipation
The overlapping of Susp with the T1D protective locus on Chr. 3 suggests elevated SOD1 activity and the resulting decrease in superoxide from NOX may be involved in ALR-derived T1D protection.38,40 A previous study had mapped Susp in an outcross of ALR with ALS. In order to study the contributions of Susp in T1D resistance in a NOD model system, Chr. 3 congenic mice were developed. NOD cells exhibit a robust oxidative burst upon PMA stimulation that is on average 5 times greater than that of ALR. ALRxNOD F1 hybrid mice have a burst phenotype equal to [ALRxALS] F1 hybrids, which is approximately half of NOD or ALS. The ALR.NODc3(D3Mit167-D3Mit174) congenic mice had NOD contributing genome from 32.4 to 83.3 cM (Fig. 2A). This region contains the Susp locus. When these congenic mice were assessed for neutrophil and macrophage superoxide production after stimulation with PMA, a high burst capability comparable to that of NOD (Fig. 2B) was observed.
Figure 2.
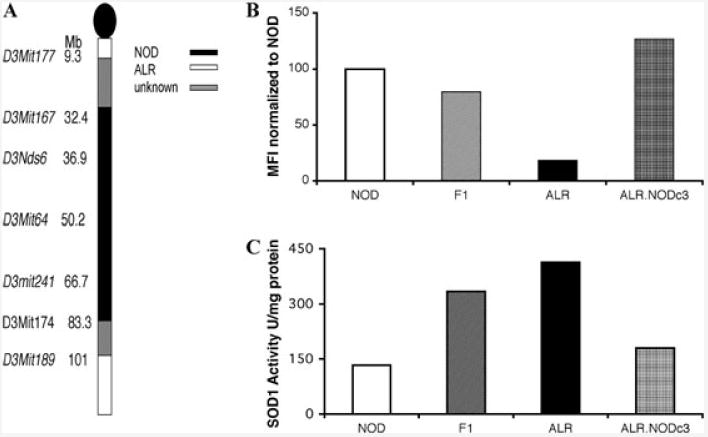
(A) Chromosome 3 congenic mice were developed by mating ALR females to NOD males and the offspring were bred to ALR. The progeny were then genotyped to determine ALR and NOD contributing genome on each chromosome. Mice with NOD contributions on Chr. 3 were chosen to breed with ALR mice. This process continued until NOD genome contributions were present only on Chr. 3, leading to the development of ALR.NODc3 congenic mice that had NOD contributing genome between D3Mit177 and D3Mit189. (B) Respiratory burst was evaluated in neutrophils and macrophages isolated from the bone marrow of the NOD, [NODxALR]F1 hybrids, ALR, and ALR.NODc3 mice. (C) SOD1 activity was measured in homogenized liver tissue from NOD, [NODxALR]F1 hybrids, ALR, and ALR.NODc3 mice using the Cayman Chemical SOD Assay Kit (Ann Arbor, MI, USA) following the manufacturer's directions.
The SOD1 activity in ALR is elevated compared not only to ALS, but also to NOD (Fig. 2C). F1 hybrids between NOD and ALR have a SOD1 activity that is intermediate comparing ALR to NOD (Fig. 2C). When homozygous ALR.NODc3(D3Mit167-D3Mit174) were tested, SOD1 activity was lower, consistent with a NOD phenotype (Fig. 2C). This confirms the mapping of Susp located on Chr. 3 as responsible in controlling the ALR-derived changes in oxidative status.
Shifts in oxidative status have been described in the ALR and hypothesized to play a role in T1D resistance. This shift may serve two purposes: by conferring protection at the level of the immune system and to the β cells. CD4+ T cells are principal mediators of autoimmune destruction of the β cell, and studies of diabetogenic T cell clones have described important roles for IFN-γ and TNF-α in T1D pathogenesis.59 ROS have also been proposed to be critical cellular signaling molecules60,61 and recent work has implicated NOX activity as an important component in T cell activation. Disruption of NOX subunits gp91phox and p47phox leads to a skewing of the T helper response and altering the secreted cytokine profile.62 When p47phox-deficient C57BL/6 mice were infected with A. fumigatus, CD3+ T cells were found to produce decreased IFN-γ and increased IL-17 compared to wild-type mice, suggesting that superoxide was important for the initiation of a Th1 response during an inflammatory response.62 The inherent lack of superoxide in the ALR due to increased dissipation by SOD1 may be downregulating the initiation of a pro-inflammatory Th1 response and limiting T1D.
While the immune cells of the ALR have shown decreased superoxide production, the β cell has also demonstrated changes in ROS production. β cells are known to have low ROS scavenging capabilities, yet the ALR increases in antioxidant activity were shown to extend to the β cell. Previous reports have described a NOX complex in the β cell that is activated by glucose, suggesting that superoxide is also important to β cell function and intracellular signaling.63,64 Islets exposed to pro-inflammatory cytokines, including IFN-γ and TNF-α, were shown to increase superoxide production via NOX within the β cell.64 ROS produced within the β cell in the context of a pro-inflammatory setting contributes to cell death. ALR β cells, however, fail to increase ROS production after exposure to proinflammatory cytokines,9 again demonstrating resistance to changes in redox balance on account of increased dissipation by SOD1, and reduced production of superoxide is protecting the β cell. The role of Susp in T1D protection is likely to be global, suggesting that improvements in systemic antioxidant capacity can protect against onset of T1D.
ALR-Derived Idd16.2 MHC-Linked T1D Protection Associated with H2-Ddx
The major histocompatibility complex (MHC) is located on Chr. 17. ALR and NOD share the majority of this cluster, the only difference being at the distal part of class III, extending to class I, where ALR is Ddx and NOD is Db. The linkage peak on Chr.17, termed Idd16, mapped in the backcross was very close to the MHC.38 Analysis of two lines of NOD mice congenic for the MHC of ALR (H2-Ddx) demonstrated that diabetes was suppressed.55 The ALR-derived MHC-linked gene on this chromosome is likely the Class I MHC Ddx allele and has not yet been associated with dissipation of free radicals.55
Summary
ROS are thought to be involved in several pathways in the autoimmune destruction of pancreatic β cells during the development of T1D. The ALR mouse strain, with its elevated endogenous ROS dissipation, provides a good model to study the protective role of ROS dissipation on β cells. The protective roles of ALR genes are mapped on Chr. 3 and 8 and the mtDNA provides either enhanced antioxidant defenses or a reduction in basal ROS production. Our data clearly show that elevated ROS dissipation protects islet β cells from autoimmune insults and prevents the development of T1D, and that this protection is exerted through the interaction of genes in the mitochondrial and nuclear genomes.
Acknowledgments
This work was supported by the Juvenile Diabetes Research Foundation and National Institutes of Health Grants AI56374 and DK74656.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- 1.Kawasaki E, Abiru N, Eguchi K. Prevention of type 1 diabetes: from the view point of beta cell damage. Diabetes Res Clin Pract. 2004;66(Suppl 1):S27–S32. doi: 10.1016/j.diabres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Rocic B, et al. Total plasma antioxidants in first-degree relatives of patients with insulin-dependent diabetes. Exp Clin Endocrinol Diabetes. 1997;105:213–217. doi: 10.1055/s-0029-1211754. [DOI] [PubMed] [Google Scholar]
- 3.Varvarovska J, et al. Aspects of oxidative stress in children with type 1 diabetes mellitus. Biomed Pharmacother. 2004;58:539–545. doi: 10.1016/j.biopha.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- 5.Tiedge M, et al. Relation between antioxidant enzyme gene-expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- 6.Gale EA. Molecular mechanisms of beta-cell destruction in IDDM: the role of nicotinamide. Horm Res. 1996;45(Suppl 1):39–43. [PubMed] [Google Scholar]
- 7.Horio F, et al. Reactive oxygen intermediates in autoimmune islet cell destruction of the NOD mouse induced by peritoneal exudate cells (rich in macrophages) but not T cells. Diabetologia. 1994;37:22–31. doi: 10.1007/BF00428773. [DOI] [PubMed] [Google Scholar]
- 8.Mathews CE, et al. Unusual resistance of ALR/Lt mouse beta cells to autoimmune destruction: role for beta cell-expressed resistance determinants. Proc Natl Acad Sci USA. 2001;98:235–240. doi: 10.1073/pnas.98.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathews CE, et al. Mechanisms underlying resistance of pancreatic islets from ALR/Lt mice to cytokine-induced destruction. J Immunol. 2005;175:1248–1256. doi: 10.4049/jimmunol.175.2.1248. [DOI] [PubMed] [Google Scholar]
- 10.Nerup J, et al. Mechanisms of panbreatic β cell destruction in type 1 diabetes. Diabetes Care. 1988;11:16–23. [PubMed] [Google Scholar]
- 11.Suarez-Pinzon WL, et al. An inhibitor of inducible nitric oxide synthase and scavenger of peroxynitrite prevents diabetes development in NOD mice. J Autoimmun. 2001;16:449–455. doi: 10.1006/jaut.2001.0507. [DOI] [PubMed] [Google Scholar]
- 12.Tabatabaie T, et al. COX-2 inhibition prevents insulin-dependent diabetes in low-dose streptozotocin-treated mice. Biochem Biophys Res Commun. 2000;273:699–704. doi: 10.1006/bbrc.2000.2959. [DOI] [PubMed] [Google Scholar]
- 13.Briehl MM, Baker AF. Modulation of the anti-oxidant defence as a factor in apoptosis. Cell Death Differ. 1996;3:63–70. [PubMed] [Google Scholar]
- 14.Briehl MM, I, Cotgreave A, Powis G. Downregulation of antioxidant defence during glucocorticoid-mediated apoptosis. Cell Death Differ. 1995;2:41–46. [PubMed] [Google Scholar]
- 15.Buttke TM, Sandstrom PA. Oxidative stress as a mediator of apoptosis. Immunol Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 16.Gusdon AM, Votyakova TV, Mathews CE. mt-Nd2a suppresses reactive oxygen species production by mitochondrial complexes I and III. J Biol Chem. 2008;283:10690–10697. doi: 10.1074/jbc.M708801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gusdon AM, et al. Nuclear and mitochondrial interaction involving mt-Nd2 leads to increased mitochondrial reactive oxygen species production. J Biol Chem. 2007;282:5171–5179. doi: 10.1074/jbc.M609367200. [DOI] [PubMed] [Google Scholar]
- 18.Mathews CE, Leiter EH. Constitutive differences in antioxidant defense status distinguish alloxan-resistant and alloxan-susceptible mice. Free Radic Biol Med. 1999;27:449–455. doi: 10.1016/s0891-5849(99)00084-2. [DOI] [PubMed] [Google Scholar]
- 19.Mathews CE, Leiter EH. Resistance of ALR/Lt Islets to free radical mediated diabetogenic stress is inherited as a dominant trait. Diabetes. 1999;48:2189–2196. doi: 10.2337/diabetes.48.11.2189. [DOI] [PubMed] [Google Scholar]
- 20.Corbett JA, et al. Interleukin-1beta induces the formation of nitric oxide by beta-cells purified from rodent islets of Langerhans. J Clin Invest. 1992;90:2384–2391. doi: 10.1172/JCI116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suarez-Pinzon WL, Strynadka K, Rabinovitch A. Destruction of rat pancreatic islet beta-cells by cytokines involves the production of cytotoxic aldehydes. Endocrinology. 1996;137:5290–5296. doi: 10.1210/endo.137.12.8940348. [DOI] [PubMed] [Google Scholar]
- 22.Lortz S, et al. Protection of insulin-producing RINm5F cells against cytokine-mediated toxicity through overexpression of antioxidant enzymes. Diabetes. 2000;49:1123–1130. doi: 10.2337/diabetes.49.7.1123. [DOI] [PubMed] [Google Scholar]
- 23.Rabinovitch A, et al. Expression of calbindin-D(28k) in a pancreatic islet beta-cell line protects against cytokine-induced apoptosis and necrosis. Endocrinology. 2001;142:3649–3655. doi: 10.1210/endo.142.8.8334. [DOI] [PubMed] [Google Scholar]
- 24.Lortz S, Tiedge M. Sequential inactivation of reactive oxygen species by combined over-expression of SOD isoforms and catalase in insulin-producing cells. Free Radic Biol Med. 2003;34:683–688. doi: 10.1016/s0891-5849(02)01371-0. [DOI] [PubMed] [Google Scholar]
- 25.Bertera S, et al. Gene transfer of manganese superoxide dismutase extends islet graft function in a mouse model of autoimmune diabetes. Diabetes. 2003;52:387–393. doi: 10.2337/diabetes.52.2.387. [DOI] [PubMed] [Google Scholar]
- 26.Hotta M, et al. Pancreatic beta cell-specific expression of thioredoxin, an antioxidative and antiapoptotic protein, prevents autoimmune and streptozotocin-induced diabetes. J Exp Med. 1998;188:1445–1451. doi: 10.1084/jem.188.8.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiedge M, et al. Complementary action of antioxidant enzymes in the protection of bioengineered insulin-producing RINm5F cells against the toxicity of reactive oxygen species. Diabetes. 1998;47:1578–1585. doi: 10.2337/diabetes.47.10.1578. [DOI] [PubMed] [Google Scholar]
- 28.Ho E, Medeiros DM, Bray TM. Glutathione (GSH) inhibits activation of NF-kB, an oxidative stress response element in the pancreas and protects against alloxan-induced diabetes. FASEB J. 1998;12:A815. [Google Scholar]
- 29.Lizard G, et al. Glutathione is implied in the control of 7-ketocholesterol-induced apoptosis, which is associated with radical oxygen species production. FASEB J. 1998;12:1654–1663. doi: 10.1096/fasebj.12.15.1651. [DOI] [PubMed] [Google Scholar]
- 30.Saitoh M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuda M, et al. Protective activity of adult T cell leukemia-derived factor (ADF) against tumor necrosis factor-dependent cytotoxicity on U937 cells. J Immunol. 1991;147:3837–3841. [PubMed] [Google Scholar]
- 32.Nomikos IN, et al. Combined treatment with nicotinamide and desferrioxamine prevents islet al-lograft destruction in NOD mice. Diabetes. 1986;35:1302–1304. doi: 10.2337/diab.35.11.1302. [DOI] [PubMed] [Google Scholar]
- 33.Nomikos I, Wang Y, Lafferty K. Involvement of O2 radicals in autoimmune diabetes. Immunol Cell Biol. 1989;67:85–87. doi: 10.1038/icb.1989.12. [DOI] [PubMed] [Google Scholar]
- 34.Makino S, et al. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 35.Ino T, et al. Selection of mouse strains showing high and low incidences of alloxan-induced diabetes. Jikken Dobutsu. 1991;40:61–67. doi: 10.1538/expanim1978.40.1_61. [DOI] [PubMed] [Google Scholar]
- 36.Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. 2008;51:216–226. doi: 10.1007/s00125-007-0886-7. [DOI] [PubMed] [Google Scholar]
- 37.Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50:537–546. [PubMed] [Google Scholar]
- 38.Mathews CE, et al. Genetic analysis of resistance to type-1 diabetes in ALR/Lt mice, a NOD-related strain with defenses against autoimmune-mediated diabetogenic stress. Immunogenetics. 2003;55:491–496. doi: 10.1007/s00251-003-0603-8. [DOI] [PubMed] [Google Scholar]
- 39.Mathews CE, et al. mt-Nd2 Allele of the ALR/Lt mouse confers resistance against both chemically induced and autoimmune diabetes. Diabetologia. 2005;48:261–267. doi: 10.1007/s00125-004-1644-8. [DOI] [PubMed] [Google Scholar]
- 40.Mathews CE, et al. Genetic control of neutrophil superoxide production in diabetes-resistant ALR/Lt mice. Free Radic Biol Med. 2002;32:744–751. doi: 10.1016/s0891-5849(02)00747-5. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh S, et al. Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nature Genetics. 1993;4:404–409. doi: 10.1038/ng0893-404. [DOI] [PubMed] [Google Scholar]
- 42.McAleer M, et al. Crosses of NOD mice with the related NON strain: a polygenic model for type I diabetes. Diabetes. 1995;44:1168–1195. doi: 10.2337/diab.44.10.1186. [DOI] [PubMed] [Google Scholar]
- 43.Prins JB, et al. Linkage on chromosome 3 of autoimmune diabetes and defective Fc receptor for IgG in NOD mice. Science. 1993;260:695–698. doi: 10.1126/science.8480181. [DOI] [PubMed] [Google Scholar]
- 44.Todd JA, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature. 1991;351:542–547. doi: 10.1038/351542a0. [DOI] [PubMed] [Google Scholar]
- 45.Wicker LS, et al. Resistance alleles at two non-MHC-linked insulin dependent diabetes loci on chromosome 3, Idd3 and Idd10, protect NOD mice from diabetes. J Exp Med. 1994;180:1705–1713. doi: 10.1084/jem.180.5.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wheeler MD, Thurman RG. Up-regulation of CD14 in liver caused by acute ethanol involves oxidant-dependent AP-1 pathway. J Biol Chem. 2003;278:8435–8441. doi: 10.1074/jbc.M212076200. [DOI] [PubMed] [Google Scholar]
- 47.Lin SJ, et al. Superoxide dismutase inhibits the expression of vascular cell adhesion molecule-1 and intracellular cell adhesion molecule-1 induced by tumor necrosis factor-alpha in human endothelial cells through the JNK/p38 pathways. Arterioscler Thromb Vasc Biol. 2005;25:334–340. doi: 10.1161/01.ATV.0000152114.00114.d8. [DOI] [PubMed] [Google Scholar]
- 48.Pan HJ, et al. Adverse hepatic and cardiac responses to rosiglitazone in a new mouse model of type 2 diabetes: relation to dysregulated phosphatidyl-choline metabolism. Vascul Pharmacol. 2006;45:65–71. doi: 10.1016/j.vph.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 49.Grivennikova VG, Vinogradov AD. Generation of superoxide by the mitochondrial Complex I. Biochim Biophys Acta. 2006;1757:553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 50.Takagi K, et al. Association of a 5178C–>A (Leu237Met) polymorphism in the mitochondrial DNA with a low prevalence of myocardial infarction in Japanese individuals. Atherosclerosis. 2004;175:281–286. doi: 10.1016/j.atherosclerosis.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 51.Kokaze A, et al. Longevity-associated mitochondrial DNA 5178 A/C polymorphism and blood pressure in the Japanese population. J Hum Hypertens. 2004;18:41–45. doi: 10.1038/sj.jhh.1001632. [DOI] [PubMed] [Google Scholar]
- 52.Lal S, Madhavan M, Heng CK. The association of mitochondrial DNA 5178 C > a polymorphism with plasma lipid levels among three ethnic groups. Ann Hum Genet. 2005;69:639–644. doi: 10.1111/j.1529-8817.2005.00192.x. [DOI] [PubMed] [Google Scholar]
- 53.Kokaze A, et al. Longevity-associated NADH dehydrogenase subunit-2 polymorphism and serum electrolyte levels in middle-aged obese Japanese men. Mech Ageing Dev. 2005;126:705–709. doi: 10.1016/j.mad.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 54.Uchigata Y, et al. A mitochondrial genotype associated with the development of autoimmune-related type 1 diabetes. Diabetes Care. 2002;25:2106. doi: 10.2337/diacare.25.11.2106. [DOI] [PubMed] [Google Scholar]
- 55.Pomerleau DP, et al. Major histocompatibility complex-linked diabetes susceptibility in NOD/Lt mice: subcongenic analysis localizes a component of Idd16 at the H2-D end of the diabetogenic H2(g7) complex. Diabetes. 2005;54:1603–1606. doi: 10.2337/diabetes.54.5.1603. [DOI] [PubMed] [Google Scholar]
- 56.Lyu MS, et al. Genetic mapping of six mouse peroxiredoxin genes and fourteen peroxiredoxin related sequences. Mamm Genome. 1999;10:1017–1019. doi: 10.1007/s003359901150. [DOI] [PubMed] [Google Scholar]
- 57.Ali F, et al. Statin-mediated cytoprotection of human vascular endothelial cells: a role for Kruppel-like factor 2-dependent induction of heme oxygenase-1. J Thromb Haemost. 2007;5:2537–2546. doi: 10.1111/j.1538-7836.2007.02787.x. [DOI] [PubMed] [Google Scholar]
- 58.Das H, et al. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci USA. 2006;103:6653–6658. doi: 10.1073/pnas.0508235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cantor J, Haskins K. Effector function of diabetogenic CD4 Th1 T cell clones: a central role for TNF-alpha. J Immunol. 2005;175:7738–7745. doi: 10.4049/jimmunol.175.11.7738. [DOI] [PubMed] [Google Scholar]
- 60.Grisham MB. Reactive oxygen species in immune responses. Free Radic Biol Med. 2004;36:1479–1480. doi: 10.1016/j.freeradbiomed.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 61.Griffiths HR. ROS as signalling molecules in T cells–evidence for abnormal redox signalling in the autoimmune disease, rheumatoid arthritis. Redox Rep. 2005;10:273–280. doi: 10.1179/135100005X83680. [DOI] [PubMed] [Google Scholar]
- 62.Romani L, et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–215. doi: 10.1038/nature06471. [DOI] [PubMed] [Google Scholar]
- 63.Oliveira HR, et al. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes. 2003;52:1457–1463. doi: 10.2337/diabetes.52.6.1457. [DOI] [PubMed] [Google Scholar]
- 64.Morgan D, et al. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia. 2007;50:359–369. doi: 10.1007/s00125-006-0462-6. [DOI] [PubMed] [Google Scholar]
- 65.Chen J, et al. Commonalities of genetic resistance to spontaneous autoimmune and free radical-mediated diabetes. Free Radic Biol Med. 2008;45:1263–1270. doi: 10.1016/j.freeradbiomed.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]