

Abstract
Background. Changes in hepatosplanchnic lactate exchange are likely to contribute to hyperlactatemia in sepsis. We hypothesized that septic and cardiogenic shock have different effects on hepatosplanchnic lactate exchange and its contribution to hyperlactatemia. Materials and Methods. 24 anesthetized pigs were randomized to fecal peritonitis (P), cardiac tamponade (CT), and to controls (n = 8 per group). Oxygen transport and lactate exchange were calculated during 24 hours. Results. While hepatic lactate influx increased in P and in CT, hepatic lactate uptake remained unchanged in P and decreased in CT. Hepatic lactate efflux contributed 20% (P) and 33% (CT), respectively, to whole body venous efflux. Despite maintained hepatic arterial blood flow, hepatic oxygen extraction did not increase in CT. Conclusions. Whole body venous lactate efflux is of similar magnitude in hyperdynamic sepsis and in cardiogenic shock. Although jejunal mucosal pCO2 gradients are increased, enhanced lactate production from other tissues is more relevant to the increased arterial lactate. Nevertheless, the liver fails to increase hepatic lactate extraction in response to rising hepatic lactate influx, despite maintained hepatic oxygen consumption. In cardiac tamponade, regional, extrasplanchnic lactate production is accompanied by hepatic failure to increase oxygen extraction and net hepatic lactate output, despite maintained hepatic arterial perfusion.
1. Introduction
Hyperlactatemia is a common finding in patients with sepsis and multiple organ failure and is associated with increased morbidity and mortality [1–3]. If such patients are well fluid resuscitated, systemic blood flow and oxygen delivery to the tissues are often high. Despite this, organ dysfunction and mild to moderate hyperlactatemia may persist.
In clinical practice, hyperlactatemia, especially in combination with metabolic acidosis, is commonly interpreted as sign of tissue hypoxia [4–7]. If sepsis is associated with hyperdynamic circulation, increased lactate production due to enhanced glycolytic metabolism, decreased oxygen extraction capability, and/or lactate clearance, and pyruvate dehydrogenase inactivation may offer alternative explanations for hyperlactatemia [8–12]. However, as a consequence of regional blood flow redistribution and metabolic compartmentalization, high systemic blood flow may not necessarily guarantee sufficient oxygen delivery to all tissues [13, 14]. In addition, sepsis often alters the function of organs which usually extract lactate, such as the liver and the kidneys. This may result in hyperlactatemia even in the presence of normal or only slightly increased lactate production. In contrast, in severe low cardiac output states, hyperlactatemia is likely to result from anaerobic metabolism and impaired lactate extraction due to generalized tissue hypoperfusion [15].
The aim of this study was to evaluate the contribution of hepatosplanchnic and extrahepatic lactate exchange to arterial hyperlactatemia in fecal peritonitis and cardiac tamponade. We hypothesized that the increase in arterial lactate concentration in fecal peritonitis is the result of a generalized increase in lactate production with maintained hepatic lactate uptake.
2. Materials and Methods
2.1. Anesthesia, Monitoring, and Animal Preparation
The study was performed in accordance with the National Institutes of Health guidelines for the care and use of experimental animals and with the approval of the Animal Care Committee of the Canton of Berne, Switzerland. Here, we present data from a subset of animals from two previous publications, where effects of fluid resuscitation on mortality and organ function in sepsis [16] and oxygen transport and mitochondrial function in shock models [17] were investigated.
24 pigs (median body weight 41 kg, range 38–44 kg) were deprived of food but not water 24 hrs before the experiment. They were premedicated with atropine 0.05 mg/kg of body weight, azaperone 6 xylazine 2 mg/kg, and ketamine 20 mg/kg intramuscularly, followed by cannulation of an ear vein and intravenous administration of 5–15 mg/kg pentobarbital and atropine 0.05 mg/kg followed by tracheal intubation. Anesthesia was maintained with pentobarbital 7 mg/kg hour and fentanyl 25 μg/kg hour until the end of the operation and thereafter 3 μg/kg hour. The animals were ventilated with a volume controlled ventilator with 5 cm of H2O end-expiratory pressure. FIO2 was adjusted to keep paO2 between 100 mm Hg and 150 mm Hg. Tidal volume was kept at 10 mL/kg, and the minute ventilation was adjusted to maintain paCO2 between 35 and 40 mm Hg. A pulmonary artery catheter and a femoral arterial catheter were inserted. Ringer's lactate was infused at 10 mL/kg hour. Additional fluid was administered if necessary to keep pulmonary capillary wedge pressure between 5 and 8 mm Hg. The aim was to keep the body temperature at 38 ± 0.5°C using and operating table heater and warmed fluids if necessary.
2.2. Surgical Preparation
The carotid artery was exposed, and an ultrasound flow probe was placed around the artery (Transonic Systems, Ithaca, NY, USA). A large bore catheter for fluid administration was inserted into the femoral vein. The abdominal cavity was opened by a midline incision. A drainage catheter was inserted into the urinary bladder. The superior mesenteric, hepatic, splenic, and renal artery, the celiac trunk, and the portal vein were exposed, and ultrasound flow probes were placed around the vessels. Two fluid filled catheters were inserted into a mesenteric vein. The tip of the first catheter was placed into the portal vein, while the tip of the second catheter remained in the mesenteric vein. A catheter was inserted into the splenic vein and another catheter into the hepatic artery via the left gastric artery. A hepatic vein catheter was inserted, and the location of the tip was confirmed by ultrasound and oxygen vein saturation. The catheter was withdrawn 0.5 cm from the wedged position to allow measurement of hepatic venous pressure and saturation. A central venous catheter was inserted into the pericardial space via dissection of the left diaphragm. Before closing the laparotomy with clamps, two large bore tubes were placed with their tips in the peritoneal cavity to allow later insertion of autologous feces.
2.3. Hemodynamic Monitoring
Femoral and pulmonary arterial, central, hepatic, and portal venous blood pressures and pulmonary artery occlusion pressure were recorded with quartz pressure transducers and displayed continuously on a multimodular monitor (S/5 Compact Critical Care monitor, GE, Helsinki, Finland) and recorded. All pressure transducers were zeroed to the level of the heart. Cardiac output was measured by a thermodilution technique (mean value of three measurements, cardiac output module, S/5) and by using an esophageal Doppler probe (Cardio Q, Deltex Medical limitex, Chichester, UK). Central venous blood temperature (°C) was recorded from the thermistor in the pulmonary artery catheter and peripheral temperature from a thermistor placed between the toes. Regional blood flows were recorded by ultrasound transit time flow probes (Transonic Systems, Ithaca, NY, USA). Heart rate was measured from the ECG wich was also continuously monitored.
2.4. Experimental Protocol
After surgical preparation, 12 hours were allowed for hemodynamic stabilization. The animals were randomized into three different groups: peritonitis (n = 8), cardiac tamponade (n = 8), and controls (n = 8). Peritonitis was induced by instilling 1 g/kg of autologous feces suspended in warm 5% dextrose solution through a previously placed peritoneal drainage tube. Cardiac tamponade was induced by instilling warmed hydroxyethyl starch into the pericardial space with goals of cardiac output of 60 mL/kg/min during the first 6 hours, 50 mL/kg/min from 6 to 12 hours, and 40 mL/kg/min from 12 to 18 hours. Afterwards, cardiac output was not manipulated anymore.
When clinical signs of hypovolemia developed in any group of the animals (rapidly decreasing blood pressure, increasing heart rate or urinary output <0.5 mL/kg/hr, and pulmonary artery occlusion pressure <5 mm Hg), additional aliquots of hydroxyethyl starch were infused. Glucose 50% was administered when blood glucose was <3.5 mmol/L. Animals surviving 24 hours were sacrificed with an overdose of intravenous potassium chloride.
2.5. Blood Measurements
Arterial hemoglobin and oxygen haemoglobin saturation were analyzed with an analyzer designed for porcine blood (OSM3, Radiometer, Copenhagen, Denmark). Blood gases were measured and temperature corrected in a blood gas analyzer (ABL 520, Radiometer, Copenhagen, Denmark). Arterial and venous blood lactate were measured with a lactate analyzer (YSI 2300 Stat Plus, Yellow Springs Instruments, CA, USA).
2.6. Calculations and Statistics
Hepatic lactate influx (μmol/kg/min) = (portal venous lactate × portal vein blood flow) + (arterial lactate × hepatic arterial blood flow).
Hepatic lactate efflux (μmol/kg/min) = hepatic venous lactate × (portal venous + hepatic arterial blood flow).
Hepatic lactate uptake (μmol/kg/min) = hepatic lactate influx − hepatic lactate efflux.
Hepatic lactate extraction (%) = hepatic lactate efflux/hepatic lactate influx.
Other regional lactate exchanges: regional lactate influx − regional lactate efflux.
Area under arterial lactate concentration-time curve (mmol/L): sum of products of average of two consecutive lactate concentrations and time between the two measurements, divided by total time of experiment (mmol/L).
Whole body venous efflux: cardiac output × mixed venous lactate concentration. Extrahepatic organ lactate efflux: (cardiac output × mixed venous lactate concentration) − hepatic lactate efflux.
For statistical analysis, the SPSS software package (SPSS version 14.1, SPSS Schweiz AG, Zurich, Switzerland) was used. In each group, nonparametric ANOVA for repeated measurements (Friedman test) was used to evaluate evolution over time. There was significant 24 hours mortality in peritonitis and tamponade groups. To ensure that the same number of measurements from each animal was available, only the last of the remaining measurements between 12 hours and 24 hours was used for analysis. For between-group comparison of variables with only one measurement, Kruskal Wallis test was employed. Statistical significance was set at P = 0.05. All results are presented as median and range.
3. Results
In both study groups, 4 animals died before 24 hours.
3.1. Systemic Hemodynamics and Oxygen Transport
In both peritonitis and cardiac tamponade, shock ensued (Table 1). There was a transient increase in cardiac output in peritonitis and a decrease in cardiac tamponade. Systemic oxygen consumption was maintained in all groups (Table 1).
Table 1.
Systemic hemodynamics and oxygen transport.
| N | Baseline | 3 hours | 6 hours | 12 hours | Last measurement | P* | ||
|---|---|---|---|---|---|---|---|---|
| Cardiac output (mL/kg/min) | Controls | 8 | 89 (69–116) | 91 (48–117) | 98 (56–121) | 92 (66–58) | 108 (67–127) | 0.344 |
| Peritonitis | 8 | 90 (55–107) | 87 (63–154) | 114 (74–151) | 82 (49–133) | 102 (80–146) | 0.060 | |
| Tamponade | 7 | 73 (54–101) | 60 (38–76) | 57 (53–76) | 53 (47–81) | 46 (38–62) | 0.004 | |
|
| ||||||||
| Central venous pressure (mmHg) | Controls | 8 | 4 (2–6) | 4 (2–7) | 5 (3–8) | 5 (3–7) | 8 (5–9) | 0.001 |
| Peritonitis | 8 | 4 (0–5) | 3 (1–5) | 4 (2–6) | 6 (3–9) | 7 (4–10) | <0.001 | |
| Tamponade | 7 | 5 (3–9) | 7 (4–9) | 7 (6–10) | 11 (7–15) | 13 (12–16) | <0.001 | |
|
| ||||||||
| Mean arterial pressure (mm Hg) | Controls | 8 | 72 (61–82) | 70 (52–91) | 71 (56–89) | 78 (64–81) | 74 (50–91) | 0.623 |
| Peritonitis | 8 | 67 (61–92) | 76 (54–92) | 63 (54–88) | 66 (49–89) | 42 (34–90) | 0.048 | |
| Tamponade | 7 | 80 (52–88) | 66 (57–93) | 64 (52–84) | 56 (38–88) | 36 (33–64) | <0.001 | |
|
| ||||||||
| Systemic oxygen consumption (mL/kg/min) | Controls | 8 | 4.76 (3.66–5.45) | 4.42 (3.26–5.75) | 4.70 (3.25–6.49) | 4.78 (3.37– 11.31) | 4.56 (3.22–5.45) | 0.861 |
| Peritonitis | 8 | 4.96 (2.50–5.98) | 5.38 (4.20–7.86) | 5.99 (4.39–6.88) | 5.29 (3.19–6.14) | 5.59 (3.55–6.43) | 0.199 | |
| Tamponade | 7 | 4.17 (3.31–5.35) | 4.07 (2.75–5.63) | 3.87 (3.39–5.82) | 4.35 (3.80–6.67) | 4.17 (2.85–7.12) | 0.966 | |
|
| ||||||||
| Systemic oxygen extraction | Controls | 8 | 0.43 (0.34–0.54) | 0.41 (0.29–0.61) | 0.47 (0.31–0.54) | 0.46 (0.29–0.54) | 0.42 0.28–0.51) | 0.995 |
| Peritonitis | 8 | 0.48 (0.36–0.59) | 0.39 (0.31–0.42) | 0.35 (0.29–0.50) | 0.44 (0.33–0.66) | 0.50 (0.32–0.66) | 0.053 | |
| Tamponade | 7 | 0.48 (0.39–0.59) | 0.54 (0.45–0.67) | 0.59 (0.43–0.67) | 0.62 (0.55–0.75) | 0.82 (0.73–0.94) | <0.001 | |
*Friedman test.
3.2. Lactate Concentrations
Arterial and all regional lactate concentrations increased in peritonitis and in cardiac tamponade (Table 2). The area under the lactate concentration-time curve was 0.6 (0.5–1.1) mmol/L, 1.3 (0.9–1.7) mmol/L, and 1.3 (0.5–2.4) mmol/L for control, peritonitis, and cardiac tamponade groups, respectively (P = 0.005 for the comparison between all three groups).
Table 2.
Arterial, mixed and regional venous lactate concentrations.
| N | Baseline | 3 hours | 6 hours | 12 hours | Last measurement | P* | ||
|---|---|---|---|---|---|---|---|---|
| Arterial lactate concentration | Controls | 8 | 0.557 (0.439–0.999) |
0.505 (0.160–0.766) |
0.555 (0.451–1.880) |
0.611 (0.453–0.863) |
0.658 (0.450–0.993) |
0.793 |
| Peritonitis | 7 | 0.586 (0.391–0.960) |
1.150 (0.679–2.270) |
1.315 (0.614–2.200) |
1.037 (0.721–2.010) |
1.250 (0.972–2.720) |
0.002 | |
| Tamponade | 7 | 0.586 (0.503–0.720) |
0.600 (0.420–0.800) |
0.650 (0.473–1.890) |
0.935 (0.526–3.640) |
3.560 (1.030–7.000) |
0.001 | |
|
| ||||||||
| Mixed venous lactate concentration | Controls | 8 | 0.531 (0.443–0.754) |
0.504 (0.443–0.729) |
0.587 (0.470–1.220) |
0.638 (0.507–0.808) |
0.681 (0.461–1.050) |
0.645 |
| Peritonitis | 6 | 0.660 (0.417–1.390) |
1.195 (0.757–2.120) |
1.140 (0.797–2.370) |
1.110 (0.714–2.090) |
1.425 (0.884–2.750) |
0.015 | |
| Tamponade | 7 | 0.600 (0.481–0.829) |
0.619 (0.471–0.800) |
0.753 (0.520–1.750) |
0.972 (0.532–3.360) |
3.510 (1.000–8.300) |
0.001 | |
|
| ||||||||
| Hepatic vein lactate concentration | Controls | 8 | 0.275 (0.170–0.440) |
0.270 (0.130–0.380) |
0.325 (0.220–0.540) |
0.310 (0.220–0.370) |
0.375 (0.210–0.750) |
0.502 |
| Peritonitis | 7 | 0.420 (0.180–0.540) |
0.860 (0.300–1.220) |
0.840 (0.290–1.510) |
0.745 (0.330–3.240) |
0.910 (0.630–3.240) |
0.010 | |
| Tamponade | 7 | 0.295 (0.200–0.894) |
0.305 (0.180–0.643) |
0.405 (0.250–1.170) |
0.460 (0.200–3.090) |
3.850 (0.640–8.400) |
0.001 | |
|
| ||||||||
| Portal vein lactate concentration | Controls | 8 | 0.607 (0.493–0.792) |
0.626 (0.521–0.815) |
0.666 (0.545–1.280) |
0.682 (0.599–0.859) |
0.793 (0.535–1.030) |
0.273 |
| Peritonitis | 6 | 0.822 (0.557–0.954) |
1.350 (1.210–2.010) |
1.390 (0.817–2.250) |
1.390 (0.970–2.040) |
1.650 (1.170–3.100) |
0.001 | |
| Tamponade | 6 | 0.699 (0.556–0.894) |
0.731 (0.582–1.000) |
0.800 (0.532–1.900) |
1.110 (0.716–3.430) |
3.400 (1.140–6.700) |
0.004 | |
|
| ||||||||
| Mesenteric vein lactate concentration | Controls | 8 | 0.622 (0.427–0.807) |
0.697 (0.504–0.777) |
0.665 (0.553–1.250) |
0.685 (0.590–0.823) |
0.753 (0.591–1.030) |
0.434 |
| Peritonitis | 6 | 0.946 (0.586–1.380) |
1.250 (0.882–1.840) |
1.330 (0.816–3.510) |
1.325 (1.020–2.360) |
1.730 (1.020–2.970) |
0.005 | |
| Tamponade | 8 | 0.729 (0.655–1.000) |
0.846 (0.394–1.100) |
0.780 (0.569–1.910) |
1.140 (0.797–3.580) |
2.715 (0.974–5.110) |
0.002 | |
|
| ||||||||
| Kidney vein lactate concentration | Controls | 7 | 0.641 (0.505–1.000) |
0.602 (0.468–0.965) |
0.660 (0.504–1.690) |
0.706 (0.541–0.948) |
0.801 (0.393–1.510) |
0.273 |
| Peritonitis | 7 | 0.682 (0.390–1.290) |
1.270 (0.903–2.050) |
1.590 (0.974–2.510) |
0.928 (0.762–3.670) |
1.210 (1.140–2.660) |
<0.001 | |
| Tamponade | 6 | 0.475 (0.409–0.919) |
0.418 (0.372–0.800) |
0.713 (0.418–1.860) |
0.830 (0.401–2.830) |
2.290 (0.634–7.000) |
0.004 | |
|
| ||||||||
| Femoral vein lactate concentration | Controls | 8 | 0.919 (0.506–1.480) |
0.765 (0.567–1.120) |
0.830 (0.621–2.510) |
0.852 (0.715–1.530) |
1.245 (0.643–2.210) |
0.141 |
| Peritonitis | 5 | 0.834 (0.595–2.010) |
1.690 (0.966–3.490) |
1.935 (1.080–2.530) |
1.720 (1.110–2.410) |
1.980 (1.220–2.610) |
0.014 | |
| Tamponade | 5 | 0.865 (0.549–2.380) |
0.935 (0.769–1.050) |
1.030 (0.783–3.180) |
1.900 (1.310–3.960) |
3.420 (1.520–6.600) |
0.005 | |
|
| ||||||||
| Spleen vein lactate concentration | Controls | 8 | 0.702 (0.551–0.780) |
0.758 (0.543–0.968) |
0.759 (0.528–1.650) |
0.699 (0.583–1.360) |
0.742 (0.489–1.320) |
0.393 |
| Peritonitis | 4 | 0.718 (0.674–0.874) |
1.720 (0.985–2.260) |
1.650 (0.878–2.590) |
1.645 (1.070–2.220) |
1.860 (1.490–3.600) |
0.007 | |
| Tamponade | 6 | 0.714 (0.569–1.000) |
0.740 (0.708–1.140) |
0.810 (0.640–1.960) |
1.100 (0.636–3.770) |
3.410 (1.550–8.300) |
0.002 | |
All concentrations are in mmol/L. *Friedman Test.
3.3. Lactate Exchange
Mesenteric lactate exchange—which was slightly negative at baseline (lactate release)—became early and transiently less negative in peritonitis (P = 0.031) and turned positive (lactate uptake) in the end in two thirds of the animals with tamponade (n.s.; ES-Figure 1 available online at http://dx.doi.org/10.1155/2013/251084). Renal lactate exchange was variable and did not change in either group (ES-Figure 2).
Hepatic lactate influx increased early in peritonitis and late in cardiac tamponade (Figure 1; P = 0.001, both). Hepatic lactate uptake remained unchanged in control and peritonitis groups but turned to lactate release in all but two animals with cardiac tamponade (P = 0.006; Figure 2). Hepatic lactate efflux increased significantly in cardiac tamponade (P < 0.001) and tended to increase in peritonitis (P = 0.057; ES-Figure 3). Extrahepatic organ lactate efflux (Figure 3) and whole body venous lactate efflux (ES-Figure 4) increased both in peritonitis and in cardiac tamponade (all P < 0.012).
Figure 1.
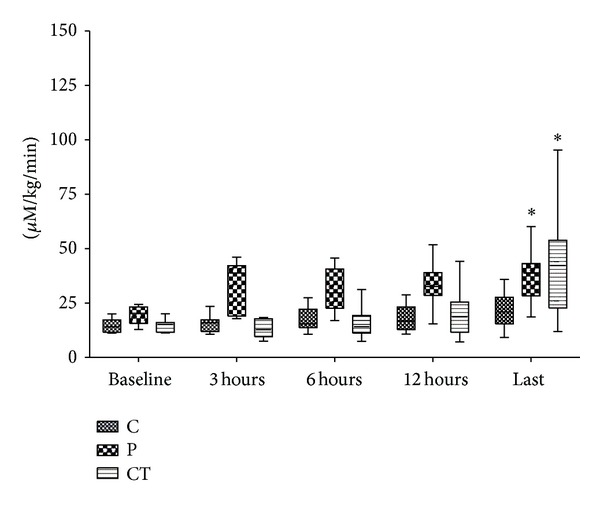
Hepatic lactate influx. *Friedmann test.
Figure 2.
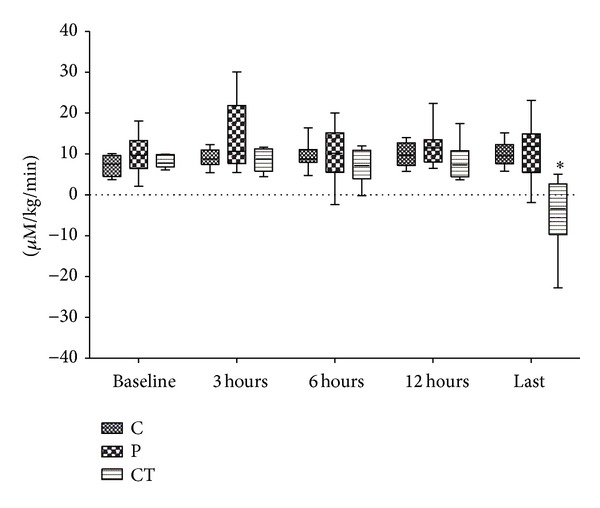
Hepatic lactate uptake. *Friedmann test.
Figure 3.
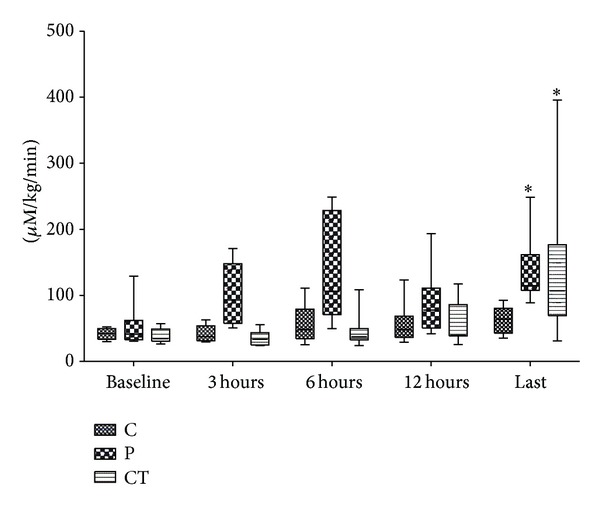
Extrahepatic organ lactate efflux. *Friedmann test.
3.4. Regional Blood Flows and Oxygen Transport
In peritonitis, renal blood flow tended to decrease (P = 0.052), while all other regional flows were maintained (Table 3). In tamponade, celiac trunk and hepatic arterial blood flow were maintained, while all other regional flows decreased (Table 3). In peritonitis, all regional VO2 were maintained (ES-Table 1). In tamponade, oxygen extraction increased in all regions/organs but the liver. Only renal VO2 decreased significantly (ES-Table 1).
Table 3.
Regional blood flows.
| N | Baseline | 3 hours | 6 hours | 12 hours | Last measurement | P* | ||
|---|---|---|---|---|---|---|---|---|
| Superior mesenteric artery blood flow | Controls | 8 | 17.64 (10.12–25.15) |
19.01 (8.77–26.34) |
17.68 (9.12–28.60) |
18.74 (11.54–26.34) |
19.68 (11.02–35.61) |
0.772 |
| Peritonitis | 8 | 16.06 (10.08–21.61) |
14.32 (11.30–17.80) |
16.12 (12.21–19.88) |
15.23 (11.78–19.39) |
16.37 (6.73–22.66) |
0.897 | |
| Tamponade | 7 | 17.29 (12.21–26.59) |
14.51 (12.59–18.25 ) |
15.69 (11.17–16.60) |
13.40 (10.70–14.06) |
11.90 (9.46–14.27 ) |
0.050 | |
|
| ||||||||
| Portal vein blood flow | Controls | 8 | 21.66 (13.00–26.37) |
23.48 (12.31–29.77) |
20.13 (12.51–33.94) |
19.86 (14.21– 32.41) |
24.53 (8.91–35.68) |
0.974 |
| Peritonitis | 8 | 21.91 (15.06–29.55) |
19.55 (10.49–31.14) |
20.66 (8.95–25.23) |
17.71 (8.31–30.23) |
18.23 (7.31–27.27) |
0.628 | |
| Tamponade | 7 | 17.26 (14.50–30.57) |
14.89 (10.68–20.75) |
15.52 (9.93–19.41) |
11.74 (6.40–16.18) |
10.49 (7.67–15.21) |
0.001 | |
|
| ||||||||
| Renal artery blood flow | Controls | 7 | 5.02 (3.11–8.10) |
4.97 (2.75–8.24) |
4.66 (2.53–9.22) |
4.63 (3.30–10.74) |
5.93 (2.42–9.87) |
0.767 |
| Peritonitis | 8 | 4.99 (3.65–8.06) |
4.41 (3.51–7.00) |
4.07 (2.72–5.50) |
4.39 (0.51–6.29) |
3.82 (0.51–7.65) |
0.052 | |
| Tamponade | 7 | 5.05 (3.85–6.37) |
3.85 (3.08–4.78) |
3.75 (3.56–5.28) |
3.13 (2.69–5.47) |
1.53 (0.72–4.80) |
0.001 | |
|
| ||||||||
| Celiac trunk blood flow | Controls | 8 | 6.32 (2.26–14.58) |
5.73 (2.26–20.80) |
7.40 (2.89–18.93) |
8.93 (4.11–19.85) |
9.10 (6.56–16.93) |
0.008 |
| Peritonitis | 8 | 7.70 (3.12–16.28) |
8.20 (4.90–16.23) |
8.53 (4.45–16.95) |
11.61 (5.62–20.13) |
10.00 (3.18–17.56) |
0.701 | |
| Tamponade | 7 | 7.37 (3.03–11.89) |
6.54 (4.70–13.14) |
6.71 (3.95–11.47) |
5.78 (3.79–10.48) |
5.25 (3.07–10.62) |
0.788 | |
|
| ||||||||
| Hepatic artery blood flow | Controls | 8 | 3.50 (0.71–5.27) |
3.72 (0.63–8.45) |
4.35 (0.87–7.87) |
4.31 (1.33–7.59) |
4.67 (1.95–6.03) |
0.088 |
| Peritonitis | 8 | 3.51 (1.69–11.91) |
3.47 (1.27–10.75) |
3.20 (1.01–11.13) |
5.10 (1.16–13.06) |
3.91 (1.43–11.36) |
0.111 | |
| Tamponade | 7 | 2.48 (0.95–5.48) |
3.06 (0.79–4.20) |
3.21 (1.06–4.23) |
2.45 (1.09–3.34) |
1.58 (0.56–4.45) |
0.613 | |
|
| ||||||||
| Spleen artery blood flow | Controls | 8 | 1.20 (0.46–1.74) |
1.15 (0.39–1.92) |
1.20 (0.47–2.14) |
1.19 (0.53–2.74) |
1.42 (0.52–3.14) |
0.146 |
| Peritonitis | 8 | 1.15 (0.71–1.55) |
0.73 (0.36–1.47) |
0.52 (0.22–2.04) |
0.53 (0.21–2.29) |
0.32 (0.02–2.09) |
0.076 | |
| Tamponade | 7 | 1.03 (0.73–1.79) |
0.82 (0.54–1.05) |
0.87 (0.47–1.42) |
0.67 (0.23–1.28) |
0.38 (0.23–0.74) |
<0.001 | |
All flows are in mL/kg/min. *Friedman test.
3.5. Regional and Mucosal pCO2 Gradients
In peritonitis, mixed and hepatic venous-arterial and mucosal-arterial pCO2 gradients increased, while all other regional pCO2 gradients remained unchanged (ES-Table 2). In tamponade, jejunal mucosal and all regional venous-arterial pCO2 gradients increased (ES-Table 2).
4. Discussion
Our data indicate that the whole body venous lactate efflux as an estimate of hepatic and extrahepatic organ lactate efflux is of similar magnitude in the experimental models of severe peritonitis-induced sepsis and cardiac tamponade. However, changes in arterial lactate over time were due to different mechanisms.
In peritonitis, whole body venous efflux increased early, mainly due to extrasplanchnic, extrarenal lactate production, and the liver lactate uptake was preserved. Unchanged regional pCO2 gradients and maintained regional oxygen consumption suggest that increased glycolysis rather than tissue hypoxia was the cause of increased lactate efflux. Despite a decrease in renal blood flow and an increase in local jejunal-mucosal pCO2 gradient, renal and mesenteric lactate exchange did not deteriorate, indicating that impaired perfusion in these regions was not critical either.
In contrast, in cardiac tamponade, extrasplanchnic lactate production increased, and the hepatic lactate uptake reverted to net hepatic lactate efflux. The net hepatic lactate output was associated with a lack of increase in hepatic oxygen extraction. Although hepatic arterial perfusion was preserved, portal venous blood flow and oxygen content decreased markedly. We have previously demonstrated that hepatic lactate uptake can increase acutely by several folds, if hepatic perfusion is preserved [18, 19]. The failure of hepatic lactate uptake to increase in cardiac tamponade in the present study is likely explained by the decrease of hepatic perfusion and oxygen delivery below a critical level. We found previously in an acute short-term (150 minutes) cardiac tamponade model that hepatic lactate exchange decreased when hepatic oxygen delivery decreased to approximately 20% of control values (a mean of 0.3 mL/kg/min) [15]. In the present study, the sustained reduction of hepatic oxygen delivery to approximately 30% of the control values (a mean of 0.6 mL/kg/min) was associated with an average hepatic oxygen extraction of 0.75 and resulted in net hepatic lactate production. All regional pCO2 gradients increased, but the mesenteric (and femoral) lactate gradient did not increase. Accordingly, the decreased regional blood flows did not induce severe mesenteric tissue hypoxia. Hence, the net hepatic lactate production was a result of enhanced extra-mesenteric lactate efflux and hepatic lactate production. At high arterial lactate concentrations without mesenteric ischemia, the gut and other extrahepatic tissues may even take up lactate [19, 20]. In the present study, the arterial lactate concentrations remained only slightly or moderately increased, and the extrahepatic tissues continued to release lactate.
Despite the well-maintained hepatic perfusion in peritonitis, the liver lactate uptake did not increase. We have previously shown that hepatic lactate increases acutely during hyperlactatemia induced by lactate infusion [19] and in response to increased splanchnic lactate production during acute mesenteric ischemia [18], providing that the hepatic perfusion is preserved. The lack of increase in hepatic lactate uptake in peritonitis may be related to either the relatively modest increase in portal venous lactate, to sepsis-induced impairment of hepatic function unrelated to tissue perfusion, or to a combination of the two mechanisms. Regardless of the mechanism(s), the increased whole body lactate efflux in peritonitis was mainly due to increased extrasplanchnic lactate production. The lack of increase in regional pCO2-gradients suggests that the increased lactate efflux was not related to impaired regional perfusion. The etiology of hyperlactatemia in clinical sepsis without shock is multifactorial, but it is rarely associated with an increase in lactate to pyruvate ratio [21]. Accordingly, increased aerobic lactate production due to increased glycolysis and hypermetabolism or inhibition of pyruvate dehydrogenase activity and hepatic dysfunction may contribute. Indeed, it has been shown that stress-induced aerobic glycolysis in skeletal muscle occurs in many forms of shock including human and experimental septic shock and also hemorrhagic shock [22, 23]. Under these circumstances, hyperlactatemia (and increased pyruvate concentrations) is linked at least in part to epinephrine induced β 2-stimulation of sarcolemmal Na+-K+-ATPase [24, 25].
In most patients with severe sepsis, the hepatosplanchnic region as a whole does not produce lactate [26]. This was also the case in peritonitis in the present study, where the hepatic-venous-arterial lactate gradient remained negative throughout the experiment.
Our study has important limitations. It has been shown in rabbits that fasting prevents or attenuates endotoxin-induced systemic and hepatic blood flow reduction, metabolic acidosis, and hyperglycemia which are observed in fed animals [27]. We cannot therefore exclude that feeding our animals until sepsis induction would have revealed a different metabolic (and maybe hemodynamic) response. Small doses of heparin infused to maintain intravenous catheter patency in our study may theoretically have interfered with the inflammatory profile in peritonitis animals [28]. In cardiac tamponade, the increased central venous pressures related to the model may have reduced the hepatic blood flow more than in cardiogenic shock caused by other mechanisms. This may have contributed to the net hepatic lactate production. The peritonitis model was fluid resuscitated throughout the study. In clinical sepsis, a variable delay in starting fluid resuscitation may modify both the etiology of lactate production and the lactate kinetics.
In conclusion, we found that venous lactate efflux from the systemic circulation was of a similar magnitude in peritonitis and in severe cardiogenic shock. In peritonitis, this was the consequence of an early increase in peripheral, extrasplanchnic, and extrarenal lactate production without signs of tissue hypoxia but impaired capability to increase hepatic lactate uptake. In cardiogenic shock, the increased venous lactate efflux was the result of increased net lactate release from both the periphery and from the liver.
Supplementary Material
The Supplementary Material contains 4 figures, indicating mesenteric (ES-Figure 1) and renal (ES-Figure 2) lactate exchanges, and hepatic (ES-Figure 3) and whole body vein lactate efflux (ES-Figure 4), as well as 2 tables indicating regional oxygen transport (ES-Table 1) and regional pCO2 gradients (ES-Table 2).
Conflict of Interests
The Department of Intensive Care Medicine has, or has had in the past, research contracts with Orion Pharma, Abbott Nutrition International, B. Braun Medical AG, CSEM SA, Edwards Lifesciences Services GmbH, Kenta Biotech Ltd., Maquet Critical Care AB, and Omnicare Clinical Research AG and research and development/consulting contracts with Orion Pharma, Edwards Lifesciences SA, Maquet Critical Care AB, and Néstle. The money is/was paid into a departmental fund; no author receives/received any personal financial gain. The department has received unrestricted educational grants from the following organizations for organizing a quarterly postgraduate educational symposium, the Berner Forum for Intensive Care: Fresenius Kabi, GSK, MSD, Lilly, Baxter, Astellas, AstraZeneca, B. Braun, CSL Behring, Maquet, Novartis, Covidien, Mycomed, and RobaPharma.
Acknowledgment
This study was supported by Grant 3200BO/102268 from the Swiss National Fund.
References
- 1.Bakker J, Gris P, Coffernils M, Kahn RJ, Vincent J-L. Serial blood lactate levels can predict the development of multiple organ failure following septic shock. American Journal of Surgery. 1996;171(2):221–226. doi: 10.1016/S0002-9610(97)89552-9. [DOI] [PubMed] [Google Scholar]
- 2.Trzeciak S, Dellinger RP, Chansky ME, et al. Serum lactate as a predictor of mortality in patients with infection. Intensive Care Medicine. 2007;33(6):970–977. doi: 10.1007/s00134-007-0563-9. [DOI] [PubMed] [Google Scholar]
- 3.Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Critical Care Medicine. 2009;37(5):1670–1677. doi: 10.1097/CCM.0b013e31819fcf68. [DOI] [PubMed] [Google Scholar]
- 4.Chioléro RL, Revelly J-P, Leverve X, et al. Effects of cardiogenic shock on lactate and glucose metabolism after heart surgery. Critical Care Medicine. 2000;28(12):3784–3791. doi: 10.1097/00003246-200012000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Meregalli A, Oliveira RP, Friedman G. Occult hypoperfusion is associated with increased mortality in hemodynamically stable, high-risk, surgical patients. Critical Care. 2004;8(2):R60–R65. doi: 10.1186/cc2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S-W, Hong Y-S, Park D-W, et al. Lactic acidosis not hyperlactatemia as a predictor of inhospital mortality in septic emergency patients. Emergency Medicine Journal. 2008;25(10):659–665. doi: 10.1136/emj.2007.055558. [DOI] [PubMed] [Google Scholar]
- 7.Maciel AT, Park M. Differences in acid-base behavior between intensive care unit survivors and nonsurvivors using both a physicochemical and a standard base excess approach: a prospective, observational study. Journal of Critical Care. 2009;24(4):477–483. doi: 10.1016/j.jcrc.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Chrusch C, Bands C, Bose D, et al. Impaired hepatic extraction and increased splanchnic production contribute to lactic acidosis in canine sepsis. American Journal of Respiratory and Critical Care Medicine. 2000;161(2):517–526. doi: 10.1164/ajrccm.161.2.9902403. [DOI] [PubMed] [Google Scholar]
- 9.Michaeli B, Martinez A, Revelly JP. Effects of endotoxin on lactate metabolism in humans. Critical Care. 2012;16, article R139 doi: 10.1186/cc11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giovannini I, Chiarla C, Boldrini G. The relationship between oxygen extraction and venous pH in sepsis. Shock. 1997;8(5):373–377. doi: 10.1097/00024382-199711000-00010. [DOI] [PubMed] [Google Scholar]
- 11.Curtis SE, Cain SM. Regional and systemic oxygen delivery/uptake relations and lactate flux in hyperdynamic, endotoxin-treated dogs. American Review of Respiratory Disease. 1992;145(2):348–354. doi: 10.1164/ajrccm/145.2_Pt_1.348. [DOI] [PubMed] [Google Scholar]
- 12.Gore DC, Jahoor F, Hibbert JM, DeMaria EJ. Lactic acidosis during sepsis is related to increased pyruvate production, not deficits in tissue oxygen availability. Annals of Surgery. 1996;224(1):97–102. doi: 10.1097/00000658-199607000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruokonen E, Takala J, Kari A, Saxen H, Mertsola J, Hansen EJ. Regional blood flow and oxygen transport in septic shock. Critical Care Medicine. 1993;21(9):1296–1303. doi: 10.1097/00003246-199309000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Sair M, Etherington PJ, Winlove CP, Evans TW. Tissue oxygenation and perfusion in patients with systemic sepsis. Critical Care Medicine. 2001;29(7):1343–1349. doi: 10.1097/00003246-200107000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Jakob SM, Tenhunen JJ, Laitinen S, Heino A, Alhava E, Takala J. Effects of systemic arterial hypoperfusion on splanchnic hemodynamics and hepatic arterial buffer response in pigs. American Journal of Physiology: Gastrointestinal and Liver Physiology. 2001;280(5):G819–G827. doi: 10.1152/ajpgi.2001.280.5.G819. [DOI] [PubMed] [Google Scholar]
- 16.Brandt S, Regueira T, Bracht H, et al. Effect of fluid resuscitation on mortality and organ function in experimental sepsis models. Critical Care. 2009;13(6, article R186) doi: 10.1186/cc8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regueira T, Djafarzadeh S, Brandt S, et al. Oxygen transport and mitochondrial function in porcine septic shock, cardiogenic shock, and hypoxaemia. Acta Anaesthesiologica Scandinavica. 2012;56:846–859. doi: 10.1111/j.1399-6576.2012.02706.x. [DOI] [PubMed] [Google Scholar]
- 18.Jakob SM, Merasto-Minkkinen M, Tenhunen JJ, Heino A, Alhava E, Takala J. Prevention of systemic hyperlactatemia during splanchnic ischemia. Shock. 2000;14(2):123–127. doi: 10.1097/00024382-200014020-00008. [DOI] [PubMed] [Google Scholar]
- 19.Barthelmes D, Jakob SM, Laitinen S, Rahikainen S, Ahonen H, Takala J. Effect of site of lactate infusion on regional lactate exchange in pigs. British Journal of Anaesthesia. 2010;105(5):627–634. doi: 10.1093/bja/aeq214. [DOI] [PubMed] [Google Scholar]
- 20.Naylor JM, Kronfeld DS, Freeman DE, Richardson D. Hepatic and extrahepatic lactate metabolism in sheep: effects of lactate loading and pH. The American Journal of Physiology. 1984;247(6):E747–E755. doi: 10.1152/ajpendo.1984.247.6.E747. [DOI] [PubMed] [Google Scholar]
- 21.Suistomaa M, Ruokonen E, Kari A, Takala J. Time-pattern of lactate and lactate to pyruvate ratio in the first 24 hours of intensive care emergency admissions. Shock. 2000;14(1):8–12. doi: 10.1097/00024382-200014010-00002. [DOI] [PubMed] [Google Scholar]
- 22.Levy B, Gibot S, Franck P, Cravoisy A, Bollaert P-E. Relation between muscle Na+K+ATPase activity and raised lactate concentrations in septic shock: a prospective study. The Lancet. 2005;365:871–875. doi: 10.1016/S0140-6736(05)71045-X. [DOI] [PubMed] [Google Scholar]
- 23.Levy B, Desebbe O, Montemont C, Gibot S. Increased aerobic glycolysis through β2 stimulation is a common mechanism involved in lactate formation during shock states. Shock. 2008;30(4):417–421. doi: 10.1097/SHK.0b013e318167378f. [DOI] [PubMed] [Google Scholar]
- 24.James JH, Fang C-H, Schrantz SJ, Hasselgren P-O, Paul RJ, Fischer JE. Linkage of aerobic glycolysis to sodium-potassium transport in rat skeletal muscle: implications for increased muscle lactate production in sepsis. Journal of Clinical Investigation. 1996;98(10):2388–2397. doi: 10.1172/JCI119052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bundgaard H, Kjeldsen K, Suarez Krabbe K, et al. Endotoxemia stimulates skeletal muscle Na+ K+ ATPase and raises blood lactate under aerobic conditions in humans. American Journal of Physiology: Heart and Circulatory Physiology. 2003;284(3):H1028–H1034. doi: 10.1152/ajpheart.00639.2002. [DOI] [PubMed] [Google Scholar]
- 26.De Backer D, Creteur J, Silva E, Vincent J-L. The hepatosplanchnic area is not a common source of lactate in patients with severe sepsis. Critical Care Medicine. 2001;29(2):256–261. doi: 10.1097/00003246-200102000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Losser M-R, Bernard C, Beaudeux J-L, Pison C, Payen D. Glucose modulates hemodynamic, metabolic, and inflammatory responses to lipopolysaccharide in rabbits. Journal of Applied Physiology. 1997;83(5):1566–1574. doi: 10.1152/jappl.1997.83.5.1566. [DOI] [PubMed] [Google Scholar]
- 28.Ding R, Zhao D, Guo R, Zhang Z, Ma X. Treatment with unfractionated heparin attenuates coagulation and inflammation in endotoxemic mice. Thrombosis Research. 2011;128(6):e160–e165. doi: 10.1016/j.thromres.2011.07.044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplementary Material contains 4 figures, indicating mesenteric (ES-Figure 1) and renal (ES-Figure 2) lactate exchanges, and hepatic (ES-Figure 3) and whole body vein lactate efflux (ES-Figure 4), as well as 2 tables indicating regional oxygen transport (ES-Table 1) and regional pCO2 gradients (ES-Table 2).