

Abstract
Properly regulated inflammation facilitates recognition and reaction to injury or infection, but inadequate or overly robust inflammation can lead to disease. Sepsis is an inflammatory disease that accounts for nearly 10% of total U.S. deaths, costing more than $17 billion. Acute inflammation in sepsis may evolve too rapidly to be modulated appropriately, and we suggest that therapies should focus not on abolishing inflammation, but rather on attenuating the positive feedback cycle of inflammation/damage/inflammation. In Gram-negative sepsis, bacterial endotoxin causes inflammation and is driven and regulated by the cytokine tumor necrosis factor-α (TNF-α), which is, in turn, negatively regulated via its endogenous inhibitor, soluble TNF-α receptor (sTNFR). We generated stably gene-modified variants of human HepG2 hepatocytes, using lentiviral constructs coding for mouse sTNFR driven by the constitutive cytomegalovirus promoter, and seeded them in a scaled-down, experimental liver bioreactor. When connected to anesthetized, cannulated rats subjected to endotoxin infusion and maintained solely by the animals’ circulation, this biohybrid device elevated circulating sTNFR, reduced the levels of TNF-α and other key inflammatory mediators, alleviated hypotension, and reduced circulating markers of organ damage. This novel class of biohybrid devices may bemodified for patient- and disease-specific application, and, thus, may represent a disruptive strategy that offers the potential for rational inflammation reprogramming.
Keywords: soluble tumor necrosis factor-alpha receptor 1, tumor necrosis factor alpha, endotoxin, sepsis, biohybrid device, multiple organ dysfunction syndrome
Introduction
Sepsis is a significant public health concern, which, by some estimates, is the 10th leading cause of death overall in the United States.1–3 Death from sepsis occurs due to inflammation-induced multiple organ dysfunction syndrome (MODS), a poorly understood syndrome of sequential and gradual loss-of-organ function.4 Current estimates put the annual cost of sepsis at nearly $17 billion,5 and this cost is expected to rise significantly due to an aging population that suffers from increasingly complex medical co-morbidities.
In Gram-negative sepsis, pro-inflammatory cytokines—primary among them being tumor necrosis factor-α (TNF-α) 6,8–15—promote immune cell activation and, therefore, feedback positively to promote further pro-production of inflammatory cytokines.15 In turn, the bioactivity of TNF-α is controlled, in part, via the cleavage of cell-surface TNF-α receptor type I, releasing soluble TNF-α receptor (sTNFR).16
Currently, there are no approved therapies for sepsis. In the present studies, we hypothesized that continuous provision of sTNFR via a biohybrid device will attenuate self-propagating acute inflammation. We report on the generation of a biohybrid device for the systemic control of experimental, endotoxin-induced acute inflammation, hypotension, and end-organ damage.7 The biohybrid device we developed is a scale-down17 of the existing, experimental bioreactors for clinical extracorporeal liver support,18 seeded with gene-modified variants of human HepG2 hepatocytes that constitutively produce mouse sTNFR (Fig. 1). This biohybrid device elevated circulating sTNFR, reduced the levels of TNF-α and other key inflammatory mediators, alleviated hypotension, and reduced the circulating markers of organ damage in rats subjected to endotoxin infusion. This novel class of biohybrid devices may be modified for patient- and disease-specific application, and, thus, may represent a disruptive strategy that offers the potential for rational inflammation reprogramming.
FIG. 1.
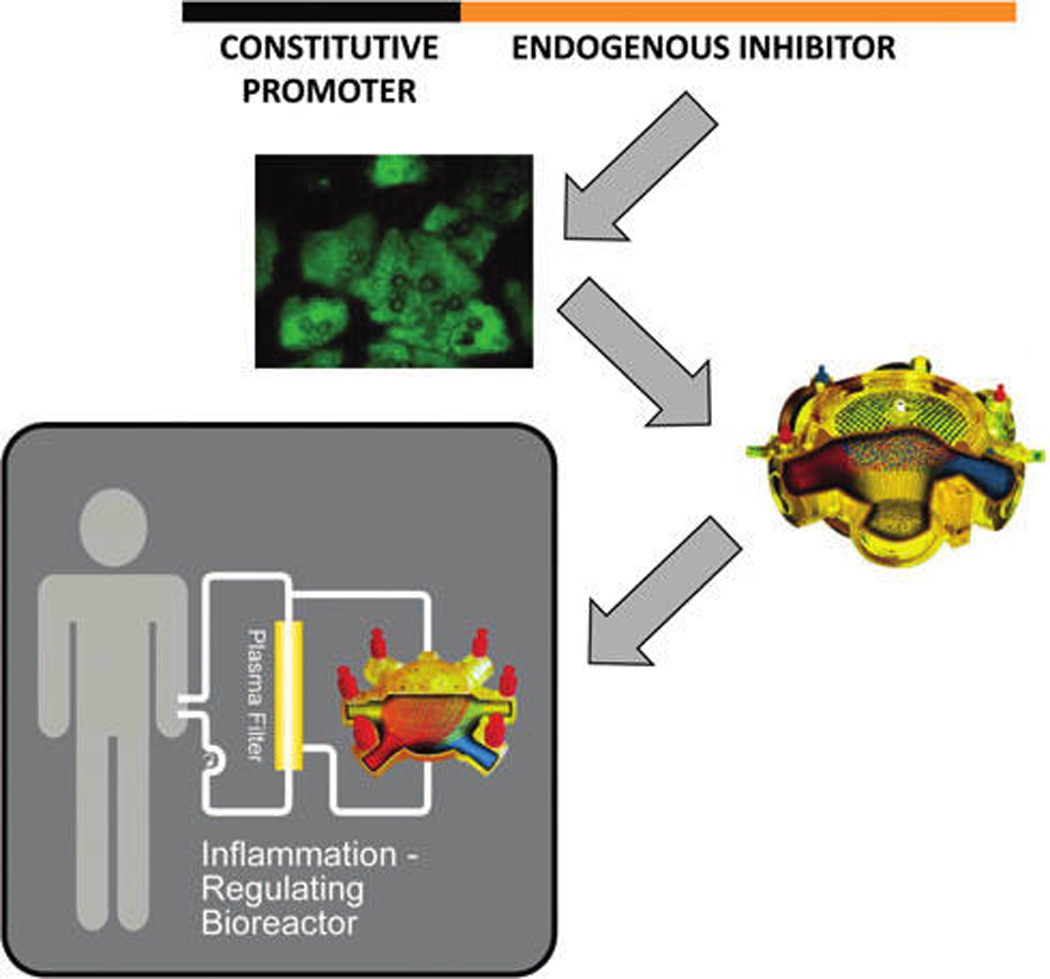
Conceptual diagram of a biohybrid device for the systemic control of acute inflammation. Lentiviral vectors consisting of constitutive promoters upstream of endogenous cytokine inhibitors are constructed using standard molecular biology techniques. The vectors are used to create stably transduced variants of hepatocyte cell lines, as these cells can resist inflammation. The cells are seeded into liver bioreactors. The resultant biohybrid devices are connected extra-corporeally to affect systemic inflammation.
Materials and Methods
Preparation of sTNFR-producing, gene modified cells
Multiple, stably-transduced variants HepG2 cells were generated using lentiviral vectors (Fig. 2A, B) expressing mouse sTNFR constitutively or not expressing sTNFR, as briefly described next. Lentiviral vectors were prepared using GateWay® technology (Invitrogen; see http://tools.invitrogen.com/content.cfm?pageid = 4072 for general methodological details). For the control bioreactor, we developed an HepG2 variant using the vector pLenti6-3xNFkB-sTNFR-Ires-Turbo vector (Fig. 2A). Though the pLenti6-NFKB-sTNFR-Ires-Turbo vector nominally codes for mouse sTNFR under control of the mouse NF-jB promoter/enhancer, one of the 10 stably transduced HepG2 variants generated on transduction with this lentiviral vector (clone 9) did not produce sTNFR protein, either at baseline conditions or under stimulation with mouse TNF-α (data not shown). Therefore, this cell line was used for seeding the control bioreactor. Vector pLC2PCm-GFP-sTNFR (Fig. 2B) was constructed in order to provide for the constitutive production of mouse sTNFR protein. All stably transduced HepG2 variants were obtained following antibiotic selection.
FIG. 2.
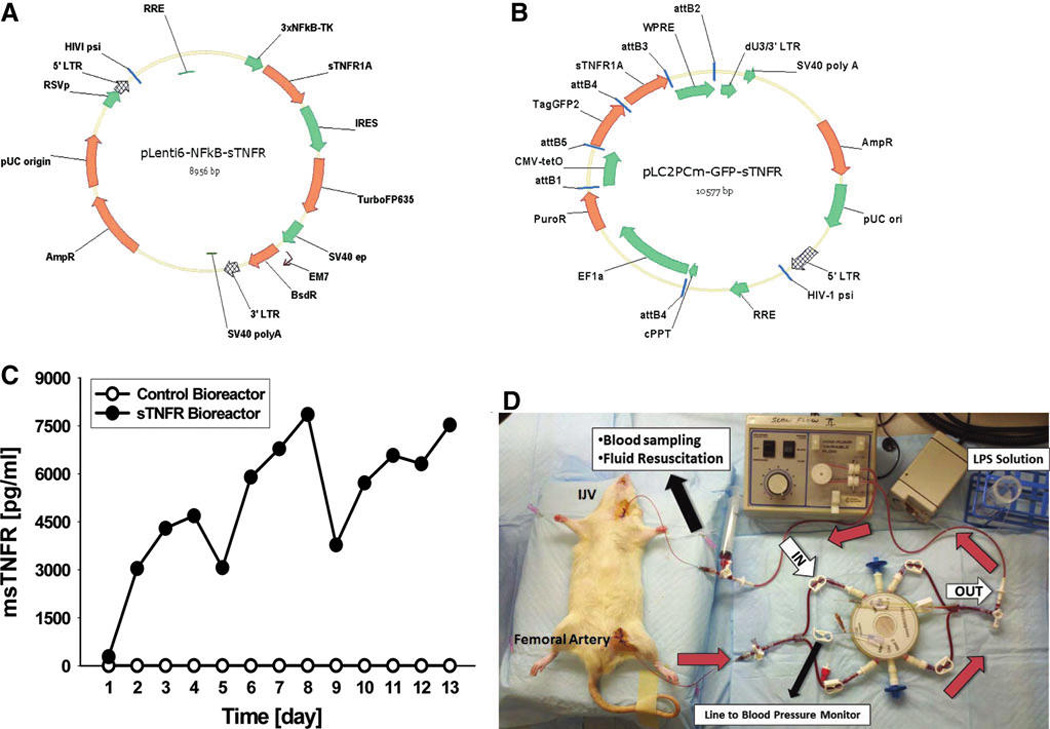
Design and implementation of a biohybrid device for systemic control of acute inflammation. Lentiviral vectors were produced as described in the Materials and Methods section, which were used as a negative control (A) or for constitutive production and delivery of mouse sTNFR (B). (C) HepG2 cells were transduced with either the mouse sTNFR gene driven by the constitutive CMV promoter (filled symbols) or with a control HepG2 variant that, though transduced with a lentivirus coding for mouse sTNFR driven by the mouse NF-jB promoter, did not produce any sTNFR protein (open symbols). These gene-modified HepG2 variants were seeded in a 0.8 mL liver bioreactor, and the cells were maintained on the bioreactor for 3-day periods, at which time the bioreactor was removed from the perfusion equipment, flushed, and connected to cannulated rats for the studies depicted in Figures 2D, 3, and 4. At the conclusion of each in vivo experiment, the bioreactors were returned to the perfusion laboratory and maintained for a further 3 days, until they were utilized for a subsequent in vivo experiment. (D) liver bioreactors seeded with the cell lines depicted in (C) were attached to anesthetized, cannulated rats undergoing endotoxemia. Black arrows indicate the various components of the experimental setup, which is detailed in the Materials and Methods section. Red arrows indicate the direction of blood flow through the bioreactor. White arrows indicate the inlet and outlet ports of the bioreactor.
Bioreactor seeding and determination of production of mouse sTNFR in bioreactor culture
The negative control and sTNFR-producing HepG2 variants were seeded in a scaled-down four-compartment hollow-fiber culture bioreactor17–19 (StemCell Systems, Berlin, Germany). In this class of bioreactors, cells are cultured in the interstitial spaces between the fibers. Culture medium or blood circulates from the lumens of the microfiltration fibers to the cell compartment and back to the fiber lumens, and most proteins produced by the cells seeded in the bioreactor can freely diffuse out, as the microfiltration fibers have a cutoff of 0.2 µm. Medium or blood is pumped through the two-microfiltration fiber bundles in opposing directions (counter-current flow). This complex flow pattern mimics an arterial and venous flow in the liver tissue.
The stably transfected HepG2 hepatocytes (10 million cells) were injected in the cell compartment, allowing the cells to spontaneously reassemble into tissue-like structures in a 3-D perfused cell compartment (data not shown). The volume of the bioreactor was 0.8 mL. The bioreactor was then integrated into a processor-controlled perfusion device with electronic pressure, flow, temperature, and pH regulation. The production of mouse sTNFR was assessed in a bioreactor culture (Fig. 2C) by obtaining medium samples at the indicated times, including after the removal of the bioreactor for in vivo experiments in endotoxemic rats (see next and Fig. 2 legend).
Surgical preparation, endotoxemia, bioreactor recycling, and assessment of physiology and inflammation
The study was approved by the University of Pittsburgh Institutional Animal Care and Use Committee and conforms to the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals. Adult male Sprague-Dawley rats (n = 11, 24–28 weeks old, 430–480 g body weight from Harlan Laboratories, Madison, WI) were anesthetized, and their femoral artery and the internal jugular veins (IJV) were cannulated for use in the extracorporeal circuit. The rats were then cannulated to either a bioreactor seeded with HepG2 cells producing mouse sTNFR constitutively (n = 7 rats) or to a control bioreactor (n = 4 rats) for 6 h, as indicated in Figure 2D. Blood flow through the bioreactor was maintained by a peristaltic infusion pump (Instech Laboratories, Inc., Plymouth Meeting, PA) at a rate of 1.5 mL/min.
Figure 3 depicts a schematic of the endotoxemia experiment. To induce endotoxemia, 13 µg/kg/h of Gram-negative bacterial lipopolysaccharide (LPS) solution20 (6.5 µg/mL LPS in saline, catalog # L2630; Sigma-Aldrich, St. Louis, MD) was intravenously infused (i.v.) through the IJV at a rate of 1mL/h via the peristaltic infusion pump. The LPS i.v. infusion was initiated at 0 h (immediately after establishing the extracorporeal circuit with the bioreactor connected) and continued to 4 h of the bioreactor treatment. At 4 h, the LPS infusion was discontinued while continuing treatment with the bioreactor for an additional 2 h; thus, the total bioreactor treatment time was 6 h. To monitor the mean arterial pressure (MAP), the arterial blood (from the previously cannulated femoral artery) was connected to a blood pressure analyzer (Blood Pressure Analyzer™ 400; Micro-Med, Inc., Louisville, KY). Arterial blood gases, blood electrolytes, hematocrit, hemoglobin concentration, and glucose were analyzed using an i-STAT® Handheld blood analyzer (Abbott Point of Care Inc., Princeton, NJ). Plasma cytokines/chemokines were measured using Luminex™ technology (Millipore, Billerica, MA). Plasma NO2−/NO3− was measured by the nitrate reductase method (Cayman Chemical, Ann Arbor, MI). Plasma sTNFR (as well as sTNFR collected during the bioreactor culture) using a kit from R&D Systems (Minneapolis, MN). Aspartate transaminase (AST) and alanine aminotransferase (ALT) were measured using a commercially available kit (FUJIFILM Corporation, Asaka-shi, Satama, Japan). After 6 h, the rats were euthanized by administering an overdose of pentobarbital sodium.
FIG. 3.
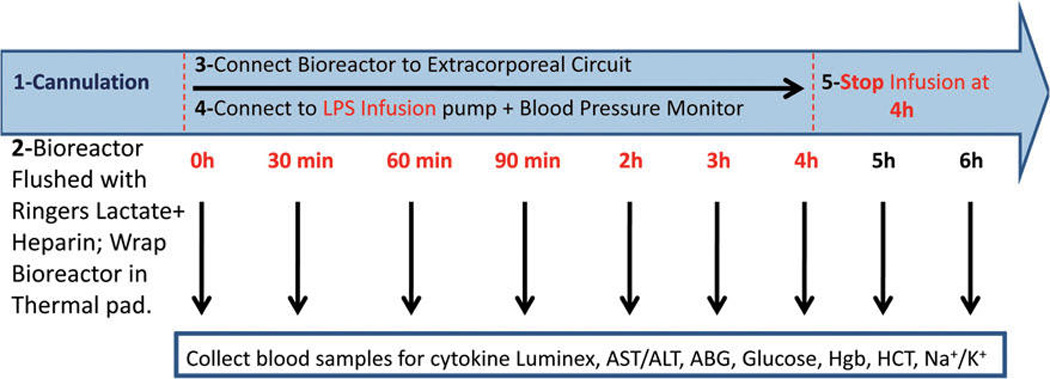
Schematic of endotoxemia study. Rats were anesthetized and cannulated. Intravenous infusion of LPS was initiated immediately after establishing the extracorporeal circuit with the bioreactor connected, and continued to 4 h of bioreactor treatment. At 4 h, the LPS infusion was discontinued while continuing treatment with the bioreactor for an additional 2 h; thus, the total bioreactor treatment time was 6 h. Blood samples were collected at the indicated times and assayed as described. Mean arterial pressure was monitored throughout the experiment. See Materials and Methods section for experimental details. LPS, Gram-negative bacterial lipopolysaccharide; AST, Aspartate transaminase; ALT, alanine aminotransferase; ABG, arterial blood gases; HCT, hematocrit; Hgb, hemoglobin.
The bioreactor was then disconnected from the rat and flushed with HEPES buffer solution and 1% antibiotic-antimycotic solution to clear blood from the inlet and outlet portals of the medium compartments. The bioreactor was then reintegrated to the perfusion device (see above) for 2–3 days before starting the next experiment. While the bioreactor was connected to the perfusion device, daily samples were taken from the culture media for the measurement of sTNFR, pH, lactate dehydrogenase, lactate, PaO2, PaCO2, HCO3, and glucose. After 3 days of maintaining the bioreactor with the perfusion device, another extracorporeal circuit was established, and the bioreactor was connected to another rat using the same experimental procedure just mentioned.
Statistical analyses
All data are expressed as mean ± SEM. Statistical analysis was performed by Two-Way analysis of variance (ANOVA) followed by the Tukey post hoc test using SigmaStat software (Systat Software, San Jose, CA) and S-Plus software (Statistical Sciences, Seattle, WA), with p < 0.05 considered significant.
Results
Generation of stably gene-modified variants of HepG2 cells
We hypothesized that continuous provision of sTNFR would ameliorate inflammation, pathophysiology, and organ damage in the setting of endotoxin-induced acute inflammation. We reasoned that the gene-modified cells would be ideal in order to synthesize and infuse sTNFR continuously into the systemic circulation. We further reasoned that hepatocyte-like cells would be ideal for withstanding an acute inflammatory milieu. Finally, we hypothesized that the existing liver bioreactor designs would optimize the growth and metabolism of such cells (Fig. 1). In order to facilitate future clinical use, we sought to use a human cell line as the basis for this biohybrid device, and, accordingly, we used the human HepG2 cell line. In order to facilitate preclinical studies in rodents, we generated variants of the human HepG2 cell line that were stably modified using lentiviral vectors to produce mouse sTNFR. One variant was produced in which mouse sTNFR was produced constitutively under the control of the CMV promoter, as determined in standard cell culture (data not shown). Coincidentally, one such variant produced no sTNFR in standard cell culture (data not shown), and this served as a negative control. Neither HepG2 variant produced any detectable human sTNFR (which could be distinguished from mouse sTNFR; data not shown).
Production of sTNFR in bioreactor culture
We next seeded two separate four-compartment, hollow-fiber membrane liver bioreactors with the negative-control and sTNFR-producing HepG2 cells, respectively, and assessed their production of mouse sTNFR. As seen in Figure 2C, the HepG2 cells that produce sTNFR constitutively were capable of long-term production of mouse sTNFR. This is especially remarkable, given that every fourth day the bioreactor was removed from the perfusion equipment and cannulated to an endotoxemic rat (see next). The robustness of the HepG2 cells in the bioreactor was further demonstrated by the fact that the in vitro perfusion and the in vivo endotoxemia studies (Fig. 2D; see next) were carried out in different buildings, requiring 15–30 min of transport time. In contrast with the sTNFR-producing bioreactor, the control bioreactor produced no detectable mouse sTNFR over the same time period (Fig. 2C).
Effects of sTNFR bioreactor on circulating levels of inflammatory mediators and markers of organ damage
Having demonstrated that we could observe the differential production of sTNFR in a bioreactor culture, we tested the hypothesis that the constitutive production and infusion of mouse sTNFR would ameliorate inflammation and patho-physiology in endotoxemic rats. We cannulated rats and established an extracorporeal circuit that included either the sTNFR-producing bioreactor or the negative control bioreactor (Fig. 2D), and carried out the experimental procedure depicted schematically in Figure 3. We initially sought to determine the plasma sTNFR concentration between the sTNFR bioreactor group and the control group. It should be noted that the ELISA kit used for this measurement cannot distinguish between endogenous rat sTNFR and the infused mouse sTNFR; thus, the levels of sTNFR observed in the control bioreactor group likely represent rat sTNFR, while the levels present in the sTNFR bioreactor group consist of a combination of both rat and mouse sTNFR. The concentration of sTNFR was very significantly higher in the sTNFR bioreactor group at all time points when compared with the control group (Fig. 4A). Based on two-way ANOVA, we discerned a highly significant treatment linear-time interaction effect, but no time effect ( p < 0.001). Accordingly, we hypothesize that once the levels of infused mouse sTNFR reached a plateau, they remained elevated due to equilibration with the rats’ own circulating sTNFR, and that the sTNFR infusion did not cause the additional production of rat sTNFR.
FIG. 4.
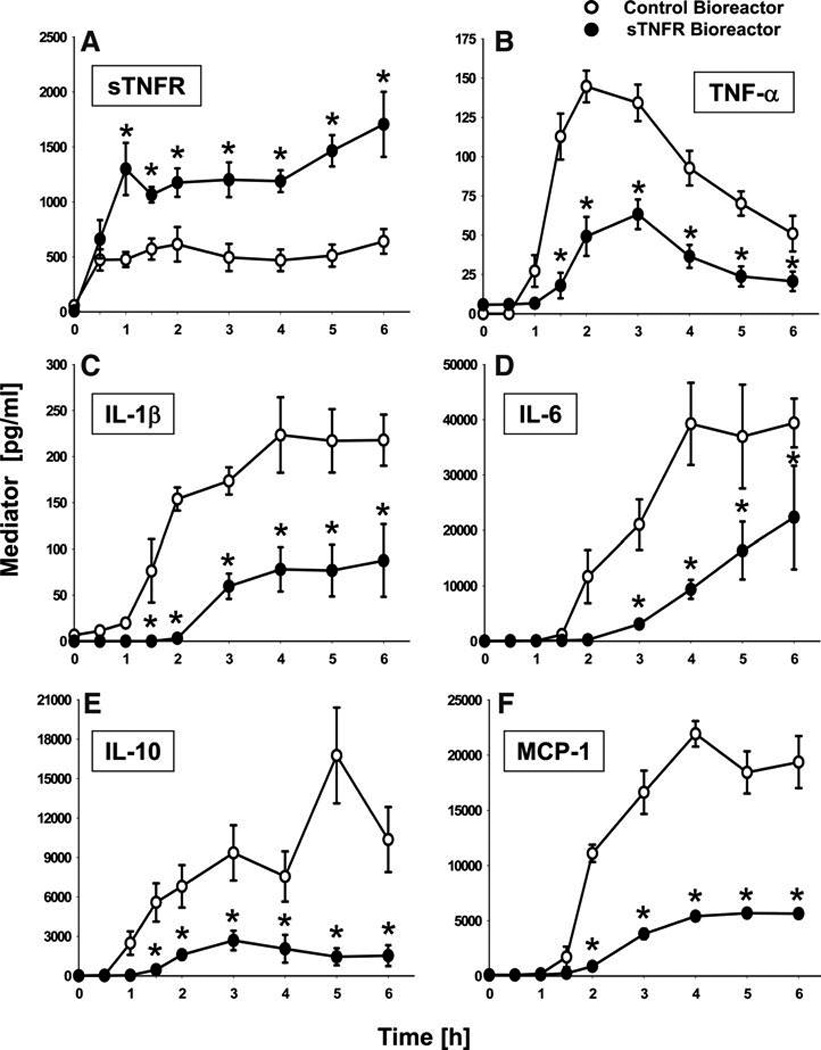
Elevated sTNFR and suppression of endotoxin-induced systemic inflammation by a biohybrid device that produces sTNFR. Rats were subjected to endotoxemia as depicted in Figure 3, in the presence of 0.8 mL liver bioreactors seeded with either HepG2 cells expressing mouse sTNFR constitutively (filled symbols; n = 7 rats) or with a control HepG2 variant that did not produce any sTNFR (open symbols; n = 4 rats). A single sTNFR-producing and a single control bioreactor was used to treat all the animals in each experimental group, respectively. In between each in vivo study, the cells were maintained on the bioreactor for 3-day periods, at which time the bioreactor was removed from the perfusion equipment, flushed, and connected to the subsequent cannulated rat. (A) plasma sTNFR concentrations as a function of time and treatment. (B) plasma TNF-α concentration as a function of time and treatment. (C) plasma IL-1β concentration as a function of time and treatment. (D) plasma IL-6 concentration as a function of time and treatment. (E) plasma IL-10 concentration as a function of time and treatment. (F) plasma MCP-1 concentration as a function of time and treatment. *statistically significant differences from the control bioreactor (p < 0.05 by 2-way ANOVA followed by Holm–Sidak post hoc test). sTNFR, soluble tumor necrosis factor-alpha receptor I; TNF-α, tumor necrosis factor alpha; IL, interleukin; ANOVA, analysis of variance.
We then tested the hypothesis that the sTNFR bioreactor would alter the systemic balance of inflammatory mediators in the setting of endotoxemia. Accordingly, we examined the effect of the sTNFR bioreactor on circulating cytokines. The differences in plasma cytokines for animals treated with the sTNFR bioreactor (n = 7) versus The control bioreactor (n = 4) are shown in Figure 4B–F. Postcannulation baseline values (i.e., 0 h after establishing the extracorporeal circuit along with the i.v. LPS infusion) for all cytokines were not significantly different between the two experimental groups. However, the concentrations of TNF-α (Fig. 4B), interleukin (IL)-1β (Fig. 4C), IL-6 (Fig. 4D), IL-10 (Fig. 4E), and MCP-1 (Fig. 4F) were significantly lower in the sTNFR bioreactor group after treatment as compared with the control bioreactor group. Based on two-way ANOVA, we discerned significant (p < 0.001) treatment-time interactions in the dynamics of these inflammatory mediators, suggesting that continuous treatment with sTNFR affected the synthesis and/or degradation of these mediators. In contrast, the levels of circulating interferon-γ, IL-12 p70, IL-18, and IL-1α were unchanged over time in both sTNFR bioreactor and control bioreactor animals (data not shown).
We next tested the hypothesis that the sTNFR bioreactor, having reduced the intensity of systemic inflammation, would likewise ameliorate the pathophysiology associated with endotoxemia. Circulating levels of the organ damage marker ALT (but not AST) were significantly lower in the sTNFR bioreactor group versus the control bioreactor group at 1, 3, and 6 h after treatment (Fig. 5A). Additionally, the hypotension characteristic of endotoxemia7 was partially, but significantly, reversed by treatment with the sTNFR bioreactor versus the control bioreactor (Fig. 5B). Since nitric oxide (NO) is thought to be the main vasodilator in sepsis and endotoxemia,21,22 we hypothesized that the reduced hypotension was due to decreased NO production (presumably produced enzymatically by the inducible isoform of NO synthase22,23). In support of this hypothesis, the circulating concentrations of the stable NO reaction products NO2−/NO3− at 4, 5, and 6 h of treatment were significantly lower in the sTNFR bioreactor group when compared with the control bioreactor group (Fig. 5C).
FIG. 5.
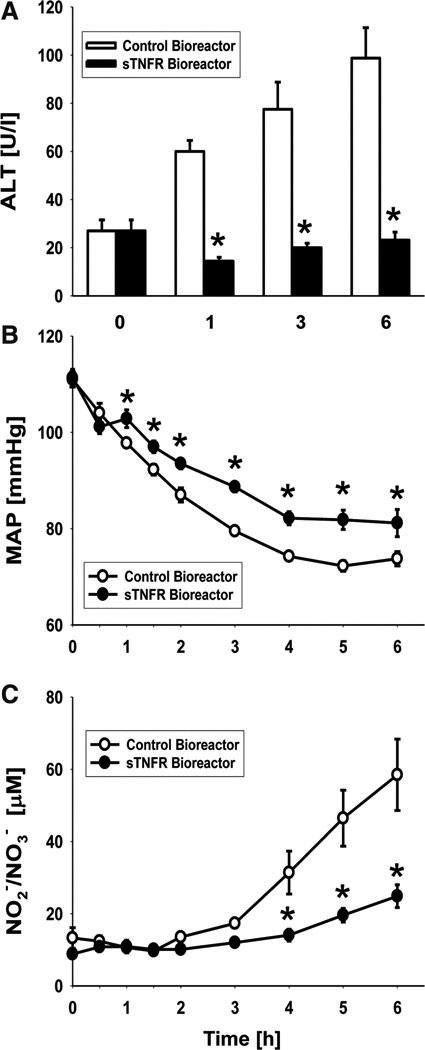
Amelioration of endotoxin-induced pathophysiology by a biohybrid device that produces sTNFR. Various indices of pathophysiology were assessed in the rats described in Figure 4. (A) plasma ALT concentrations as a function of time and treatment. (B) MAP as a function of time and treatment. (C) plasma NO2−/NO3− concentration as a function of time and treatment. *statistically significant differences from the control bioreactor (p < 0.05 by 2-way ANOVA followed by Holm–Sidak post hoc test). MAP, mean arterial pressure.
Discussion
A decade of work on systems approaches to inflammation in our group has led us to suggest that acute inflammation goes awry when the positive feedback loop of inflammation/tissue damage/inflammation fails to resolve under the influence of anti-inflammatory/pro-healing mediators. We suggest that the acute inflammatory response, which forms the core of the detrimental effects of inflammation, may simply evolve too rapidly to be modulated appropriately, given the time necessary for the proper diagnosis and administration of therapy. A central question, then, is as follows: How do we harness the beneficial effects of inflammation while simultaneously not allowing inflammation to exceed a threshold that becomes self-sustaining? We suggest that the current approach to anti-inflammatory therapeutics—extinguishing inflammation to the greatest degree possible—is misguided. Rather, the therapeutic goal should not be to abolish inflammation, but rather to attenuate the feed-forward behavior of inflammation and thereby allow the inflammatory response to resolve itself. To accomplish this goal, we should allow the body to re-equilibrate its inflammatory response through a repeated, incremental reduction of pro-inflammatory drivers. We hypothesized that the most efficient mechanism for doing so involves using gene-modified cells that are active in an extracorporeal support device and temporarily in exchange with the blood circulation via a bioreactor.
Herein, we report on a novel class of cell-based (biohybrid) devices for such extracorporeal treatment of inflammatory diseases. We demonstrate that in a paradigm of sterile acute inflammation induced by Gram-negative bacterial endotoxins—in which inflammation is driven by the cytokine TNF-α—the constitutive delivery of its endogenous inhibitor, sTNFR,16 results in reduced inflammation and pathophysiology. We show that after an initial ramp-up time of approximately 60 min, maximal levels of circulating sTNFR could be achieved. These elevated levels of sTNFR resulted in reduced circulating TNF-α, supporting the hypothesis that sTNFR bound TNF-α and also possibly reduced the auto-inducing effect of TNF-α. The levels of other pro-inflammator mediators, including IL-1β, MCP-1, and IL-6, were also reduced, presumably as a consequence of reduced circulating TNF-α. Interestingly, IL-10 was also reduced, suggesting that the provision of sTNFR acted to reduce overall inflammation, rather than inducing anti-inflammatory cytokines. Though we did not assess a larger panel of biomarkers, we would hypothesize that other circulating markers of the inflammatory and acute phase responses, such as soluble CD14, are also reduced in animals treated with the sTNFR bioreactor. Presumably as a consequence of reduced systemic inflammation, multiple indices of organ function were improved by treatment with the sTNFR-producing biohybrid device versus the control, with no discernible adverse effects. This latter finding is especially remarkable, as this device contained human cells that produce a mouse protein and was used to treat rats.
We suggest that this approachmay have utility in sepsis and trauma, in which TNF-α is a central driver of pathophysiology. More broadly, we describe a completely newplatform technology that has the potential to impact all inflammatory diseases. Inflammation can be thought of as acute (in settings such as sepsis, trauma, and wound healing) or chronic (in diseases such as psoriasis, rheumatoid arthritis, ulcerative colitis, Crohn’s disease, etc.). Earlier, chronic and acute inflammatory processes were thought to be driven by different causes, through the activities of different cells and inflammatory mediators, and to result in quite different ultimate outcomes. However, a more modern view suggests that these processes are interlinked. Thus, chronic inflammation may be thought of as a constant re-starting of acute inflammation.24 Since TNF-α is a key mediator in many of these inflammatory diseases, the provision of sTNFR via a device such as the one described herein could attenuate the forward-feedback loop that results in self-sustaining, chronic inflammation. However, more broadly, this may serve as a new platform for the continuous synthesis and delivery of therapeutic biological agents in vivo.
Disruptive Science and Technology
The concept of a biohybrid device for the systemic control of acute inflammation is a demonstration of disruptive science and technology, for several reasons. This device has the potential to benefit society, given the complete lack of Food and Drug Administration-approved therapies for sepsis and related disorders. This is the first report of a device—in essence an auxiliary organ—that can reprogram the inflammatory response at the whole-organism level. This biohybrid device is a platform technology for a nearly infinite array of applications, given the myriad ways that cells can be modified genetically, and the fact that multiple, differently modified cells can be seeded in the device, thereby tailoring its characteristics in numerous, subtle ways. A few examples include the possibility to have the biological therapeutic be produced in response to stimuli (reflecting either the patient’s own inflammatory response or in response to the administration of drugs), rather than constitutively; the ability to visualize fluorescence or luminescence in proportion to the amount of biological therapeutic produced (or, in the case of a biologically adaptive variant, the amount of stimulus); the ability to deliver different biological agents depending on the stimulus; and the ability to code for antisense or siRNA in order to knock down host or pathogen genes. This degree of flexibility suggests that this class of devices could create new markets or alter existing business models; for example, this device could simultaneously combine diagnosis and therapy. Given the potential for this type of disruption of existing business models and modes of thinking about therapies for acute inflammation, it is distinctly possible that this class of device could engender resistance in both academia and industry.
Barriers to be Overcome
The present article comprises a first-generation approach toward device-based inflammation control, one that is effective in an animal model in which there are no adverse effects of overly suppressed inflammation. In true (or experimental) sepsis, as well as posttrauma/hemorrhage, TNF-α is necessary in order to combat infection and to drive tissue healing. Furthermore, the acute inflammatory response is a complex system, in both structure and behavior,25 and yet the approach presented herein is fairly reductionist: inhibition of a known and central pro-inflammatory mediator. Ultimately, therefore, this type of device should produce sTNFR, or any other therapeutic molecule, in a manner that adapts to an individual’s inflammatory dynamics.
How can this be accomplished? Mathematical modeling has emerged as a viable means of predicting the behavior of complex systems such as the acute inflammatory response.26–28 In addition, mathematical modeling has been used as a tool for the rational design of synthetic biology strategies.29,30 Therefore, we suggest that mathematical modeling will likely be required both to tailor the operating characteristics of the biohybrid device and to predict the effects of the device on the disease. An adaptive variant of this type of device holds the promise of solving the current need for a personalized (yet standardized) inflammation-modulating therapy. Such devices could be standardized, as, for a given disease, a single bioreactor would be used, thereby reducing regulatory hurdles. Such devices could be personalized, as a given patient’s individual production of inflammatory mediators would be counteracted in a precise fashion and only as required (and, ideally, as described and predicted through mechanstic mathematical modeling).
Our initial studies reported herein demonstrate the feasibility of the components required to create such a device (capability of engineering molecular circuits and the use of gene-modified cells to modulate inflammation in vivo), and, thus, represent a key step toward the concept of a self-adaptive, biohybrid device for the control of systemic inflammation. In order to proceed toward clinical use, we will need to generate GMP-grade HepG2 (or other) cell lines expressing the human versions of the gene constructs, and test them in a device applied to small animals, large animals (possibly nonhuman primates), and studies in humans culminating in Phase III clinical trials. Regulatory challenges include the fact that such a biohybrid, adaptive device currently has no known predicates, but may compete with the nonadaptive administration of sTNFR or other TNF-neutralizing therapies, neither of which, it should be noted, is currently being used in sepsis or related diseases. Reimbursement may also be a barrier for this class of device, given the lack of predicate devices.
Acknowledgments
This work was funded by the Department of Defense grants and Advancing Regenerative Medicine 2, X81XWH-07-1-0415, and W81XWH-08-2-0032; and by the NIH grant P50-GM-53789.
Footnotes
Author Disclosure Statement
Maxim Mikheev, Jörg Gerlach, and Yoram Vodovotz are co-inventors on U.S. Patent application: “Self-Regulating Device for Modulating Inflammation” (U.S. Patent Application No. 13/121,013).
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Vincent JL, Sakr Y, Sprung CL, et al. Sepsis in European intensive care units: results of the SOAP study. Crit Care Med. 2006;34:344–353. doi: 10.1097/01.ccm.0000194725.48928.3a. [DOI] [PubMed] [Google Scholar]
- 4.Moore FA, Moore EE, Sauaia A. In: Postinjury multiple-organ failure in Trauma. Mattox KL, Feliciano DV, Moore EE, editors. New York, NY: McGraw-Hill; 1999. pp. 1427–1459. [Google Scholar]
- 5.Angus DC, Wax RS. The epidemiology of sepsis: An update. Crit Care Med. 2001;29(Suppl 7):S109–S116. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 6.Beutler B. Science review: Key inflammatory and stress pathways in critical illness - the central role of the Toll-like receptors. Crit Care. 2003;7:39–46. doi: 10.1186/cc1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker SJ, Watkins PE. Experimental models of gram-negative sepsis. Br J Surg. 2001;88:22–30. doi: 10.1046/j.1365-2168.2001.01632.x. [DOI] [PubMed] [Google Scholar]
- 8.Bellingan G. Inflammatory cell activation in sepsis. Br Med Bull. 1999;55:12–29. doi: 10.1258/0007142991902277. [DOI] [PubMed] [Google Scholar]
- 9.Jones AL, Selby P. Tumour necrosis factor: clinical relevance. Cancer Surv. 1989;8:817–836. [PubMed] [Google Scholar]
- 10.Cavaillon JM. Cytokines and macrophages. Biomed Pharmacother. 1994;48:445–453. doi: 10.1016/0753-3322(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 11.Kox WJ, Volk T, Kox SN, et al. Immunomodulatory therapies in sepsis. Intensive Care Med. 2000;26(Suppl 1):S124–S128. doi: 10.1007/s001340051129. [DOI] [PubMed] [Google Scholar]
- 12.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 13.Pinsky MR. Sepsis: A pro- and anti-inflammatory disequilibrium syndrome. Contrib Nephrol. 2001;132:354–366. doi: 10.1159/000060100. [DOI] [PubMed] [Google Scholar]
- 14.Baugh JA, Bucala R. Mechanisms for modulating TNF alpha in immune and inflammatory disease. Curr Opin Drug Discov Devel. 2001;4:635–650. [PubMed] [Google Scholar]
- 15.Chen G, Goeddel DV. TNF-R1 signaling: A beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Botran R, Crespo FA, Sun X. Soluble cytokine receptors in biological therapy. Expert Opin Biol Ther. 2002;2:585–605. doi: 10.1517/14712598.2.6.585. [DOI] [PubMed] [Google Scholar]
- 17.Zeilinger K, Schreiter T, Darnell M, et al. Scaling down of a clinical three-dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies. Tissue Eng Part C. Methods. 2011;17:549–556. doi: 10.1089/ten.TEC.2010.0580. [DOI] [PubMed] [Google Scholar]
- 18.Schmelzer E, Mutig K, Schrade P, et al. Effect of human patient plasma ex vivo treatment on gene expression and progenitor cell activation of primary human liver cells in multi-compartment 3D perfusion bioreactors for extra-corporeal liver support. Biotechnol Bioeng. 2009;103:817–827. doi: 10.1002/bit.22283. [DOI] [PubMed] [Google Scholar]
- 19.Gerlach JC. Bioreactors for extracorporeal liver support. Cell Transplant. 2006;15(Suppl 1):S91–S103. doi: 10.3727/000000006783982296. [DOI] [PubMed] [Google Scholar]
- 20.Ivanov AI. Does the formation of lipopolysaccharide tolerance require intact vagal innervation of the liver. Auton Neurosci. 2000;85:111–118. doi: 10.1016/S1566-0702(00)00229-0. [DOI] [PubMed] [Google Scholar]
- 21.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 22.Zamora R, Vodovotz Y, Billiar TR. Inducible nitric oxide synthase and inflammatory diseases. Mol Med. 2000;6:347–373. [PMC free article] [PubMed] [Google Scholar]
- 23.Nathan C, Xie Q-W. Nitric oxide synthases: Roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 24.Vodovotz Y. Roy S, Sen C. Chronic Inflammation: Nutritional & Therapeutic Interventions. Florence, KY: Taylor & Francis; 2012. At the Interface between acute and chronic inflammation: Insights from computational Modeling. [Google Scholar]
- 25.Oda K, Kitano H. A comprehensive map of the toll-like receptor signaling network. Mol Syst Biol. 2006;2:15–34. doi: 10.1038/msb4100057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vodovotz Y, Csete M, Bartels J, et al. Translational systems biology of inflammation. PLoS Comput Biol. 2008;4:1–6. doi: 10.1371/journal.pcbi.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vodovotz Y. Translational systems biology of inflammation and healing. Wound Repair Regen. 2010;18:3–7. doi: 10.1111/j.1524-475X.2009.00566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foteinou PT, Yang E, Androulakis IP. Networks, biology and systems engineering: A case study in inflammation. Comput Chem Eng. 2009;33:2028–2041. doi: 10.1016/j.compchemeng.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arkin AP, Fletcher DA. Fast, cheap and somewhat in control. Genome Biol. 2006;7:114. doi: 10.1186/gb-2006-7-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber W, Fussenegger M. Emerging biomedical applications of synthetic biology. Nature Rev Genet. 2012;13:21–35. doi: 10.1038/nrg3094. [DOI] [PMC free article] [PubMed] [Google Scholar]