

Biomaterials are receiving increased attention as vaccine adjuvants owing to their chemical and compositional definition and their ability to stimulate immune responses against a variety of co-delivered antigens.[1–6] Supramolecular self-assembly can be a useful route for producing such materials-based adjuvants,[1, 2, 4–9] but it has been largely restricted to the incorporation of short peptide epitopes rather than larger protein immunogens.[1, 4–8] In this report, we developed nanofibers displaying properly folded, biologically active protein antigens. Peptides bearing p-nitrophenyl phosphonate (pNP) ligands were self-assembled into nanofibers and subsequently reacted with cutinase fusion proteins via a covalent active site-directed capture approach to afford protein-laden nanofibers. These nanofibers could be formulated to present precisely controlled amounts of protein antigen and acted as self-adjuvanting vaccines in mice. Cutinase-pNP reactions were site-selective, allowing antigens to be conjugated without disrupting their tertiary structures, making the approach broadly useful for developing protein-bearing supramolecular materials in a range of applications including immunotherapies.
Adjuvants and delivery platforms that present properly folded protein antigens are important in the development of vaccines because they allow for broad immunogenicity in outbred populations compared with peptide vaccines, and because they can include conformational epitopes.[10] Supramolecular assemblies are gaining interest in this regard, because they can be functionalized with a high density of antigens, in some cases without perturbing antigen conformation or self-assembly of the material. For example, supramolecular nanoparticle vaccines have been designed to contain both folded protein antigens and peptide antigens that mimic native epitope conformations.[7, 8, 11–13] β-sheet-rich nanofibers of peptides and peptide amphiphiles can also act as self-adjuvanting vaccines,[1, 4, 6] and they have an additional advantage of being highly modular, allowing the incorporation of multiple different molecular components with negligible compositional drift.[14,15] However, although a few instances of protein-bearing β-sheet-rich nanofibers have been reported previously,[16–18] vaccine platforms developed from these materials have employed only peptide antigens to date, which lack any intentionally designed conformation.
We developed a general approach to produce supramolecular assemblies containing properly folded proteins using green fluorescent protein (GFP) as a model antigen, and we characterized the materials’ ability to raise immune responses in mice. Proteins were attached to peptide nanofibers using the chemoselective reaction of cutinase fusion proteins with nanofiber-bound “suicide” pNP ligands (Figure 1a–b), an approach that has been used previously to conjugate proteins to solid surfaces,[19, 20] but not to construct soft materials. First, we synthesized pNP-Q11, a variant of the β-sheet fibrillizing peptide QQKFQFQFEQQ (Q11)[15, 21, 22] having a pNP ligand on its N-terminus, by reacting cysteine-terminated Q11 with maleimido-penta(ethylene glycol)-ethyl-p-nitrophenyl phosphonate, which we also synthesized (Figure 1a, detailed methods in Supplemental Information). In parallel we designed and expressed in E. coli a fusion protein containing cutinase and green fluorescent protein domains separated by a flexible linker of glycine and serine residues (cut-GFP). In phosphate-buffered saline, pNP-Q11 self-assembled into individual nanofibers and bundles of nanofibers whose morphologies were similar to previously investigated Q11 materials (Figure 1c).[21, 22] The peptides maintained this fibrillar morphology following reaction with cutinase fusion proteins (Figure 1d), which indicated that the presence of a relatively large appended protein did not perturb Q11 fibrillization.
Figure 1. Protein-bearing self-assembled peptide nanofibers.
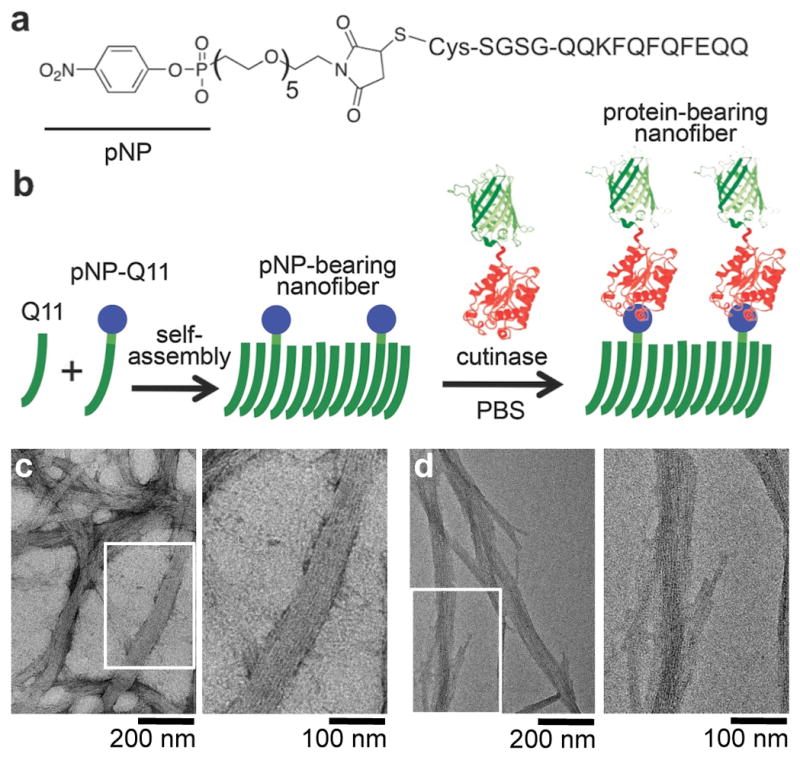
a) pNP-Q11. b) Schematic of the non-covalent assembly of Q11 and pNP-Q11 into nanofibers, and the subsequent covalent capture of cutinase-GFP by pNP-bearing Q11 nanofibers. c-d) TEM of pNP-Q11 nanofibers before (c) and after (d) conjugation with cut-GFP.
One of the advantages of supramolecular systems is that the relative amounts of different functional components in the final material can often be controlled simply by mixing specific combinations of precursor molecules and inducing self-assembly.[23–25] The phosphonate-cutinase system also lent itself to this modularity, as the amount of antigen coupled to the peptide nanofibers could be controlled by specifying the amount of pNP-Q11 co-assembled with non-functionalized Q11 (Figure 2). Protein conjugation was assessed both directly by measuring GFP fluorescence on sedimented nanofibers, and indirectly using a colorimetric assay for residual unreacted cutinase following conjugation.[26] GFP fluorescence additionally served as an indication of proper protein folding. Self-assembled Q11 peptide nanofibers bearing increasing amounts of co-assembled pNP-Q11 bound predictably increasing amounts of cut-GFP, whether measured by the fluorescence of bound GFP (Figure 2a) or by residual cutinase activity (Figure 2b, c). Q11 fibrils lacking pNP bound negligible amounts of cut-GFP non-specifically, whereas pNP-bearing fibrils incubated with a molar equivalent of cut-GFP bound the protein with about 80% efficiency (Figure 2a). A 3-fold molar excess of cut-GFP led to nearly complete reaction of the pNP ligand (not shown). In this way, the amount of protein displayed on the fibrils could be controlled with precision in a simple, straightforward manner, by dosing pNP-Q11 into Q11 nanofibers and reacting them with a slight molar excess of cut-GFP. Importantly, the pNP-cutinase conjugation proceeded to the same extent whether cut-GFP was added to freshly dissolved pNP-Q11 or to peptide that had been allowed to assemble into more mature peptide fibrils over the course of 24 h (Figure 2a), indicating that the assembly process did not adversely affect the availability of the ligand. The precision of the reaction was also reflected in the amount of active cut-GFP that remained after conjugation. Nanofibers bearing increasing amounts of pNP-Q11 were added to cutinase solutions and incubated overnight, after which the cutinase activity was measured using p-nitrophenyl butyrate (pNB) at a concentration below the Km for cutinase-pNB (Supplemental Figure S4).[26] The progress of these reactions showed diminishing initial velocities (v0) with increasing pNP content on the nanofibers, indicating progressively diminishing amounts of active residual cutinase (Figure 2b), presumably because the balance was conjugated to the nanofibers. By calculating the ratio of v0 values for the various pNP-containing samples to those containing only Q11, it was observed that almost all of the available pNP ligands were reacted when there was a molar excess of cut-GFP (Figure 2c). When there were equimolar concentrations of the ligand and protein (5 μM of both), about 80% of the ligands were bound with protein. This observed decrease in reaction efficiency at a high pNP-Q11 concentration suggested that we may be approaching the steric limit for GFP conjugation onto Q11 nanofibers, despite a pNP:Q11 ratio of 1:200 within these materials. These results corresponded closely with the direct measures of GFP fluorescence discussed above and in Figure 2a, illustrating that the conjugation reaction likely proceeded through the predicted mechanism, that the GFP domain retained its proper folding, and that collectively the strategy provided predictable control over protein loading on the peptide fibrils.
Figure 2. Covalent capture of cut-GFP by pNP-bearing Q11 nanofibers.

a) Conjugation of cut-GFP to nanofibers bearing different amounts of pNP-Q11, which were reacted with cut-GFP either immediately upon hydration or after extended assembly in water for 24 h. Immobilized cut-GFP was measured directly by GFP fluorescence on sedimented and resuspended nanofibers. Inset: Fluorescent micrograph of fibril aggregates of the formulation indicated. b) Reaction progress curves illustrating the residual activity of cutinase after overnight reaction with Q11 nanofibers bearing different amounts of pNP-Q11, as measured by p-nitrophenyl butyrate hydrolysis. (c) Initial velocities (v0) derived from (b) were used to calculate the % of active cutinase remaining after conjugation (5 μM cut-GFP, 0–5 μM pNP-Q11). All data show means ± s.d., n=3. pNP amounts are reported as the concentration of the enantiomerically active species (half of the total pNP racemate).
In mice, Q11 nanofibers bearing cut-GFP (Q11-cut-GFP) acted as self-adjuvanting vaccines, eliciting robust and durable anti-GFP antibody responses following subcutaneous delivery. C57BL/6 mice immunized with 9.3 μg cut-GFP conjugated to Q11 fibrils or emulsified in CFA raised significant anti-cut-GFP antibodies over 16 weeks, without boosting (Figure 3a). On the other hand, mice immunized with the same amount of cut-GFP in PBS did not raise detectable anti-cut-GFP antibodies, and an intermediate reaction was observed for mice immunized with the same amount of cut-GFP mixed with but not conjugated to Q11 fibers. One possible explanation for the weak anti-GFP responses following immunization with Q11 + cut-GFP could be the presence of a short-lived depot of co-localized protein and nanofibers in the subcutaneous space that can elicit immune responses against the delivered antigen due to the adjuvant properties of the nanofibers. Reducing the dose to 1.86 μg Q11-cut-GFP also resulted in similar antibody titers, and these titers were strongly boostable with a second dose of half this protein amount. Conversely, 1.86 μg of soluble cut-GFP mixed with Q11 fibers failed to raise a detectable primary response at all (Figure 3b) and were accordingly not boosted. The antibodies raised against Q11-cut-GFP or cut-GFP in CFA were reactive towards both the cutinase and GFP domains, whereas antibodies raised against soluble cut-GFP mixed with Q11 fibers were more reactive towards the cutinase domain (Figure 3c and Supplemental Figure S5) illustrating that peptide nanofiber conjugation not only increased the overall immunogenicity of the protein but also promoted the immunogenicity of the model antigen (GFP) relative to the cutinase domain.
Figure 3. Antibody responses to GFP-bearing peptide nanofibers.

a) Primary anti-cut-GFP serum Ig responses following immunization with 9.3 μg cut-GFP delivered either in PBS, conjugated to 0.5 mM Q11 via 5 μM co-assembled pNP-Q11 (Q11-cut-GFP), mixed with 0.5 mM Q11 (cut-GFP + Q11), or emulsified in CFA. b) Anti-cut-GFP serum Ig titers following immunization with 1.86 μg cut-GFP either mixed with 0.1 mM Q11 or conjugated to 0.1 mM Q11 via 1 μM co-assembled pNP-Q11. Arrow represents a booster of one-half the primary dose delivered to the Q11-cut-GFP group at week 16. c) Relative reactivity of serum Ig towards GFP or cutinase domains, and (d-e) anti-GFP Ig isotype profiles from mice shown in (a), at 2 weeks and 5 weeks. f) Primary anti-cut-GFP serum Ig responses in C3H/HeJ mice following immunization with 9.3 μg cut-GFP delivered with 0.5 mM Q11 (Q11 + cut-GFP) or conjugated to 0.5 mM Q11 via 5 μM co-assembled pNP-Q11 (Q11-cut-GFP). g) Isotype profile for C3H/HeJ mice shown in (f), at week 5. N = 5 for all serum Ig titer plots, N = 4 for all reactivity and Ig isotype profile plots. * p<0.05 (compared to cut-GFP + Q11 group in a, b, c, f; compared to Q11-cut-GFP group (panel d) at the respective time point in e), # p<0.05 (compared to PBS group in A), by ANOVA with Tukey post-hoc testing.
Primary responses to Q11-cut-GFP were skewed towards TH2-type responses, indicated by the predominance of GFP-reactive IgG1 in sera as early as two weeks following immunization, and an increasing predominance of IgG1 at five weeks (Figure 3d). In contrast, cut-GFP emulsified in CFA elicited a broader spectrum of GFP-reactive immunoglobulin (Ig) isotypes, which became particularly apparent by week 5 (Figure 3e). Unconjugated cut-GFP mixed with Q11 fibers lacked a discernible anti-GFP isotype profile (Figure 3e), which was consistent with the previous observation that much of the antibody response was directed towards the cutinase domain, not the GFP domain, for this formulation.
Given the bacterial source of cut-GFP, we undertook methods to greatly minimize the endotoxin content in the nanofiber-adjuvanted materials, and we used endotoxin-insensitive mice to rule out any role for endotoxin in the materials’ efficacy. During purification of cutinase fusion proteins, endotoxin was removed using Triton X-114 cloud-point precipitation (see methods),[27] ultimately producing materials for which the endotoxin content was measured to be below 1 EU/mL immediately preceding immunizations. This endotoxin level is consistent with guidelines for the preclinical testing of protein therapeutics.[28] The TH2-type skewing of immune responses raised by Q11-cut-GFP (Figure 3d) also suggested that any residual endotoxin was not unduly influencing responses to nanofiber-conjugated proteins, as any Toll-like receptor-4 (TLR-4) activation would be expected to skew the immune response towards a TH1-type response.[29] To further rule out the role of any residual endotoxin, we immunized C3H/HeJ mice with Q11-cut-GFP or soluble cut-GFP mixed with Q11. This strain has a mutation in the locus of the Toll-like receptor-4 (TLR-4) gene that renders them non-responsive to endotoxin.[30] High anti-cut-GFP titers were observed in C3H/HeJ mice following immunization with 9.3 μg Q11-cut-GFP (Figure 3f), which were comparable to serum titers from C57BL/6 mice immunized with the same dose (Figure 3a). Like the responses raised in C57BL/6, antibodies raised against Q11-cut-GFP in C3H/HeJ mice were skewed towards IgG1 (Figure 3g). In addition to being a measure for endotoxin insensitivity, the study in C3H/H3J mice also demonstrated that Q11-cut-GFP was immunogenic in strains with different haplotypes (C57BL/6 mice are H-2b; C3H/HeJ mice are H-2k). This result emphasizes one of the key advantages of protein-displaying nanofibers over previously developed peptide-displaying nanofibers, as full proteins contain a greater diversity of possible epitopes and are more likely to be reactive in a broad, outbred population containing multiple haplotypes.
In summary, we have demonstrated that Q11 nanofibers presenting pNP ligands can covalently capture a cutinase fusion protein without disrupting fibrillization. The amount of cutinase fusion conjugated onto these assemblies can be precisely controlled by varying the concentration of pNP-Q11 co-assembled within the material. We have previously demonstrated that precisely controlling the concentration of peptide ligands incorporated into Q11 assemblies can enable the use of statistical methods to optimize the biological responses elicited by these materials.[14] Importantly, cutinase-pNP capture may allow these approaches to be extended to protein-functionalized non-covalent assemblies. In addition, co-assembly of pNP-Q11 and Q11 variants terminated with ligands that selectively capture other enzymes (e.g. SNAP-tag[31]) may enable the future development of materials functionalized with multiple different protein ligands, in which the stoichiometry of proteins within the material could be precisely controlled and optimized. Q11 fibers bearing GFP elicited robust anti-GFP antibodies, which demonstrated that supramolecular assemblies can act as self-adjuvanting vaccines for whole-protein antigens. Eliciting anti-GFP antibodies following immunization with Q11-cut-GFP did not depend on endotoxin-mediated activation of TLR-4, which suggested that the adjuvanticity arises from the supramolecular assembly itself, analogous to supramolecular assemblies bearing peptide antigens.[5] Additionally, although it is conceivable that protein antigens could be more susceptible to proteolysis compared with previously studied peptide antigens, this possibility does not seem to have precluded the ability of the protein-bearing materials to also raise strong antibody responses. Q11 nanofibers carrying GFP strongly skewed anti-GFP humoral immune responses to a TH2-type, which is similar to alum adjuvants in current clinical use.[32] These observations suggest that using Q11-based materials as an adjuvant for whole-protein antigens may be advantageous in the development of anti-bacterial or extracellular antigen vaccines, which benefit from enhanced antibody and TH2-type responses to elicit protection. Notably, these responses were achieved without addition of any immunomodulatory agents, such as cytokines or TLR ligands. However, in light of the modular nature of non-covalent assemblies, it may also be possible to engineer Q11 variants with TLR agonists or pathogen-associated molecular patterns that can be co-assembled with phos-Q11 to develop materials that elicit mixed TH1/TH2, TH0, or TH1-type adaptive immune responses. In total, cutinase-pNP capture is likely to enable the design of non-covalent assemblies with defined and tunable protein ligand composition for diverse biomedical and biotechnological applications, including self-adjuvanting prophylactic vaccines against clinically relevant pathogens, as well as therapeutic vaccines for cancer or autoimmune disorders.
Experimental
Peptides and proteins
The peptides Q11 (QQKFQFQFEQQ) and Cys-SGSG-Q11 were synthesized using standard Fmoc solid-phase peptide synthesis, as previously reported [21, 22]. pNP-Q11 was produced by first synthesizing ethyl (4-nitrophenyl) (17-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)-3,6,9,12,15-pentaoxaheptadecyl) phosphonate, and then conjugating it to Cys-SGSG-Q11 (see Supporting Information for synthesis details) [expected m/z [M+Na]+ = 2515.04; observed m/z [M+Na]+ = 2515.3]. Cut-GFP was expressed in E. coli using previously reported methods for cutinase fusion proteins (See Supporting Information for expression details and sequences) [19]. Endotoxin was removed using an established protocol [27] by adding Triton X-114 to proteins at a 1:10 (v/v) ratio at 4 °C, maintaining them on ice for 20 min, and then heating them to 37°C for 10 min. Endotoxin-loaded Triton X-114 micelles were then removed by centrifugation at 5000xg, and the process was repeated twice. Endotoxin levels in all final immunization formulations were below 1 EU/mL, measured immediately prior to injection using the limulus amoebocyte lysate chromogenic end-point assay (Lonza).
Preparation and characterization of supramolecular assemblies
Stock solutions of Q11 and pNP-Q11 (10 m and 1 mM, respectively) were prepared in deionized water. Aqueous peptides were mixed to give the desired molar ratio, vortexed and sonicated. For experiments characterizing cut-GFP conjugation onto nanofibers, aqueous peptides were diluted 10-fold in phosphate buffered saline (PBS) containing cut-GFP (final [cut-GFP] = 4.5 or 5 μM, as indicated in Figure 2), and incubated overnight at room temperature. For immunization experiments, aqueous solutions containing Q11 and pNP-Q11 (9.9 mM and 0.1 mM, respectively) were diluted 10-fold in PBS containing cut-GFP (5 μM), and incubated overnight at 4 °C. Protein conjugation efficiency was not significantly influenced by incubation temperature. For all experiments, buffered solutions containing protein and peptides were incubated on a Barnstead/Thermolyne LabQuake rotator to ensure even mixing. Protein-bearing nanofibers were collected and unreacted protein was removed by three rounds of sedimentation at 12000xg and resuspension in PBS. TEM was conducted as previously reported [21, 22], and nanofiber-conjugated protein was measured via GFP fluorescence (395 nm excitation, 503 nm emission, [GFP] calculated from standard curves of un-conjugated cut-GFP). Residual cutinase activity was measured as previously described, using p-nitrophenyl butyrate as a substrate [19, 26]. For immunization experiments, nanofibers bearing cut-GFP were diluted 2-fold or 10-fold in sterile PBS to achieve the desired antigen dose (9.3 μg or 1.86 μg, as indicated in Figure 3). Additional detailed methods for preparation and characterization of nanofibers are provided in the Supplemental Information.
Immunizations and ELISA
Immunizations were prepared containing Q11-cut-GFP in PBS, soluble cut-GFP, soluble cut-GFP mixed with Q11 fibers, or cut-GFP emulsified 1:1 in complete Freund’s adjuvant. Female C57BL/6 mice (6–8 weeks old, Taconic Farms, IN) or female C3H/HeJ mice (8–10 weeks old, Jackson Labs, ME) were each given two 50 μL subcutaneous injections near the shoulder blades, together containing the amounts of cut-GFP and Q11 indicated in the text for each experiment, similar to previously reported methods [6]. Some groups received booster immunizations of one half the primary dose at week 16. Blood was collected weekly via the submandibular vein. Serum antibody titers were measured as previously reported, using ELISA [6]. Detailed ELISA methods are provided in the Supplemental Information. For Ig isotyping, serum diluted 1:103 was applied, followed by goat anti-mouse IgG1, IgG2a/2c, IgG2b, IgG3, and IgM (Sigma). Institutional guidelines for the care and use of laboratory animals were strictly followed under a protocol approved by the University of Chicago’s Institutional Animal Care and Use Committee.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (NIBIB, 1R01EB009701; NCI, U54 CA151880; NIAID, 1F32AI096769) and the National Science Foundation (CHE-0802286). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Biomedical Imaging and BioEngineering, the National Institute of Allergy and Infectious Disease, the National Cancer Institute, or the National Institutes of Health.
Footnotes
Supporting Information is available online from Wiley InterScience or from the author.
References
- 1.Black M, Trent A, Kostenko Y, Lee JS, Olive C, Tirrell M. Adv Mater. 2012;24(28):3845–9. doi: 10.1002/adma.201200209. [DOI] [PubMed] [Google Scholar]
- 2.Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ. Nat Mater. 2011;10(3):243–51. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, Hubbell JA. Nat Biotechnol. 2007;25(10):1159–64. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 4.Rudra JS, Mishra S, Chong AS, Mitchell RA, Nardin EH, Nussenzweig V, Collier JH. Biomaterials. 2012;33(27):6476–84. doi: 10.1016/j.biomaterials.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudra JS, Sun T, Bird KC, Daniels MD, Gasiorowski JZ, Chong AS, Collier JH. ACS Nano. 2012;6(2):1557–64. doi: 10.1021/nn204530r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudra JS, Tian YF, Jung JP, Collier JH. Proc Natl Acad Sci U S A. 2010;107(2):622–7. doi: 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pimentel TA, Yan Z, Jeffers SA, Holmes KV, Hodges RS, Burkhard P. Chem Biol Drug Des. 2009;73(1):53–61. doi: 10.1111/j.1747-0285.2008.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wahome N, Pfeiffer T, Ambiel I, Yang Y, Keppler OT, Bosch V, Burkhard P. Chem Biol Drug Des. 2012;80(3):349–57. doi: 10.1111/j.1747-0285.2012.01423.x. [DOI] [PubMed] [Google Scholar]
- 9.Moon JJ, Suh H, Polhemus ME, Ockenhouse CF, Yadava A, Irvine DJ. PLoS One. 2012;7(2):e31472. doi: 10.1371/journal.pone.0031472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tainer JA, Getzoff ED, Alexander H, Houghten RA, Olson AJ, Lerner RA, Hendrickson WA. Nature. 1984;312(5990):127–34. doi: 10.1038/312127a0. [DOI] [PubMed] [Google Scholar]
- 11.Richert LE, Servid AE, Harmsen AL, Rynda-Apple A, Han S, Wiley JA, Douglas T, Harmsen AG. Vaccine. 2012;30(24):3653–65. doi: 10.1016/j.vaccine.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venter PA, Dirksen A, Thomas D, Manchester M, Dawson PE, Schneemann A. Biomacromolecules. 2011;12(6):2293–301. doi: 10.1021/bm200369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riedel T, Ghasparian A, Moehle K, Rusert P, Trkola A, Robinson JA. Chembiochem. 2011;12(18):2829–36. doi: 10.1002/cbic.201100586. [DOI] [PubMed] [Google Scholar]
- 14.Jung JP, Moyano JV, Collier JH. Integr Biol (Camb) 2011;3(3):185–96. doi: 10.1039/c0ib00112k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung JP, Nagaraj AK, Fox EK, Rudra JS, Devgun JM, Collier JH. Biomaterials. 2009;30(12):2400–10. doi: 10.1016/j.biomaterials.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baldwin AJ, Bader R, Christodoulou J, MacPhee CE, Dobson CM, Barker PD. J Am Chem Soc. 2006;128(7):2162–3. doi: 10.1021/ja0565673. [DOI] [PubMed] [Google Scholar]
- 17.Leng Y, Wei HP, Zhang ZP, Zhou YF, Deng JY, Cui ZQ, Men D, You XY, Yu ZN, Luo M, Zhang XE. Angew Chem Int Ed Engl. 2010;49(40):7243–6. doi: 10.1002/anie.201002452. [DOI] [PubMed] [Google Scholar]
- 18.Baxa U, Speransky V, Steven AC, Wickner RB. Proc Natl Acad Sci U S A. 2002;99(8):5253–60. doi: 10.1073/pnas.082097899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodneland CD, Lee YS, Min DH, Mrksich M. Proc Natl Acad Sci U S A. 2002;99(8):5048–52. doi: 10.1073/pnas.072685299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy WL, Mercurius KO, Koide S, Mrksich M. Langmuir. 2004;20(4):1026–30. doi: 10.1021/la035733m. [DOI] [PubMed] [Google Scholar]
- 21.Collier JH, Messersmith PB. Bioconjug Chem. 2003;14(4):748–55. doi: 10.1021/bc034017t. [DOI] [PubMed] [Google Scholar]
- 22.Jung JP, Jones JL, Cronier SA, Collier JH. Biomaterials. 2008;29(13):2143–51. doi: 10.1016/j.biomaterials.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collier JH. Soft Matter. 2008;4(12):2310–2315. doi: 10.1039/b805563g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collier JH, Rudra JS, Gasiorowski JZ, Jung JP. Chem Soc Rev. 2010;39(9):3413–24. doi: 10.1039/b914337h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matson JB, Stupp SI. Chem Commun (Camb) 2012;48(1):26–33. doi: 10.1039/c1cc15551b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolattukudy PE, Purdy RE, Maiti IB. In: Methods in Enzymology. John ML, editor. Vol. 71. Academic Press; Waltham, MA: 1981. pp. 652–664. [Google Scholar]
- 27.Aida Y, Pabst MJ. J Immunol Methods. 1990;132(2):191–5. doi: 10.1016/0022-1759(90)90029-u. [DOI] [PubMed] [Google Scholar]
- 28.Malyala P, Singh M. J Pharm Sci. 2008;97(6):2041–4. doi: 10.1002/jps.21152. [DOI] [PubMed] [Google Scholar]
- 29.Re F, Strominger JL. J Biol Chem. 2001;276(40):37692–9. doi: 10.1074/jbc.M105927200. [DOI] [PubMed] [Google Scholar]
- 30.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Science. 1998;282(5396):2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 31.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. Nat Biotechnol. 2003;21(1):86–9. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 32.Grun JL, Maurer PH. Cell Immunol. 1989;121(1):134–45. doi: 10.1016/0008-8749(89)90011-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.