

Abstract
No Strain, No Gain! The first transition metal-catalyzed enantioselective α-alkylation of cyclobutanones is reported. This method employs palladium catalysis and an electron deficient PHOX type ligand to afford all–carbon α-quaternary cyclobutanones in good to excellent yields and enantioselectivities.
Keywords: Cyclobutanone, quaternary center, allylic compounds, asymmetric catalysis, palladium, allylic alkylation
The asymmetric alkylation of enolates to generate α-quaternary carbonyl compounds has become a mainstay transformation in organic synthesis.[1] In this domain, cyclobutanones have received far less attention relative to their five, six and seven membered congeners, despite the fact that these compounds and their derivatives are prevalent in important biologically active natural products[2] (Figure 1A). Additionally, cyclobutanes have been shown to serve as highly valuable synthetic intermediates for a variety of transformations.[3] The dearth of reports describing the asymmetric alkylation of cyclobutanones may be attributed to the fact that these compounds possess an estimated 26–28.6 kcal/mol of ringstrain[4] and, in turn, exhibit enhanced carbonyl electrophilicity.[5] The propensity of cyclobutanones to alleviate this strain via electrophilic ring opening is often a limiting challenge during their manipulation. Moreover, the energetic requirements for enolization of cyclobutanones are compounded by a concomitant increase in ringstrain to 31–34 kcal/mol (calculated for cyclobutene, Figure 1B)[3] as well as enforced deviation from the more favorable puckered conformation (Figure 1C).[6] In the case of α-substituted cyclobutanones, enolization is further impeded by the development of torsional strain between the putative enolate substituents (Figure 1C).[7]
Figure 1.

Figure 1A. Representative bioactive natural products possessing cyclobutanoid core structures. Figure 1B. Increased ring stain generated upon enolization. Figure 1C. Increased torsional strain upon unsaturation of 4 membered rings.
Given these data, it is not surprising that previous methods for the preparation of enantioenriched cyclobutanes have relied primarily on either [2+2] cycloaddition reactions[8] or ring expansion from various cyclopropane derivatives.[9] Recent reports from Baudoin on annulating C–H activation[10] as well as disclosures from Toste[11] and Echavarren[12] that employ gold(I) catalysis to affect cyclopropanoid rearrangements have emerged as significant new methods for the construction of cyclobutanes. Despite these advances, transformations that produce chiral cyclobutanones remain limited in scope and very few methods exist for the catalytic construction of chiral cyclobutanones from achiral starting materials.[9b], [13] In order to address these limitations and to further develop the nucleophilic chemistry of these unusually reactive compounds we report herein the first direct transition metalcatalyzed asymmetric α-alkylation of cyclobutanones to form all-carbon quaternary centers.
A longstanding interest of our research group is the asymmetric transition metal-catalyzed α-functionalization of carbonyl compounds to form all-carbon quaternary centers.[14] In the course of our studies, we have developed a series of phosphinooxazoline (PHOX) ligands with varied steric and electronic properties that exhibit a range of reactivities and selectivities. We have found that the use of electron deficient ligands (e.g. L2 and L3) often results in superior asymmetric induction where electron rich or electron neutral ligands perform poorly.[15] Examination of ligand electronic effects would, therefore, help to inform our development of a method for the asymmetric allylic alkylation of cyclobutanones.
We first established a simple and efficient reaction sequence to access allyl 1-alkyl-2-oxocyclobutanecarboxylate substrates (4) (Scheme 1). Diazotization of commercially available 1,3-cyclopentane dione (1) with para-acetamidobenzenesulfonyl azide (p-ABSA)[16] delivered the corresponding diazodiketone (2) in consistently good yields. Microwave promoted Wolff rearrangement of diketone 2 in the presense of an allylic alcohol[17] (e.g., allyl alcohol, 3), followed by alkylation with an alkyl halide (e.g., benzyl bromide) furnished the allyl 1-alkyl-2-oxocyclobutanecarboxylates in good yields over two steps. With a quick and efficient method to access the desired substrates at hand, we next examined reaction parameters to identify optimal conditions for reactivity and enantioselectivity.
Scheme 1.

Construction of allyl 1-benzyl-2-oxocyclobutanecarboxylate (4l).
Our initial experiments revealed that treatment of allyl 1-benzyl-2-oxocyclobutanecarboxylate (4l) with catalytic Pd2(pmdba)3 in the presence of (S)-t-BuPHOX (L1) in THF delivered the desired α-quaternary (S)-2-allyl-2-benzylcyclobutanone (5l)[18] in 90% yield, albeit in moderate enantioselectivity (Table 1, Entry 1). The use of electron deficient ligands, resulted in considerably improved enantioinduction (Table 1, Entries 2 and 5). Although the reaction proceeds well in a number of solvents, toluene was identified as optimal for inducing asymmetry. This solvent effect is likely due to an enhanced binding between the enolate and the electrophillic sigma-allyl-Pd(II) center in the catalytic cycle, which may reinforce a tight ion pair and lead to an inner-sphere mechanism.[19] Finally, at temperatures just below ambient the reaction was found to proceed at a reasonable rate and with high enantioselectivity.
Table 1.
Reaction optimization studies.[a]
 | ||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | T[°C] | ee[%] |
| 1 | L1 | THF | 25 | 58 |
| 2 | L2 | THF | 25 | 75 |
| 3 | L2 | p-dioxane | 25 | 84 |
| 4 | L2 | benzene | 25 | 84 |
| 5 | L3 | toluene | 25 | 84 |
| 6 | L2 | toluene | 25 | 85 |
| 7 | L2 | toluene | 20 | 86 |
 | ||||
Conditions cyclobutanone 4l (1.0 equiv), Pd2(pmdba)3 (5 mol%), Ligand (L1–L3) (12.5 mol%) in stated solvent (0.033 M) at indicated temperature for 12–48 h. ee determined by chiral SFC. pmdba = 4,4’-dimethoxydibenzylidene acetone.
With these optimized conditions identified, we next explored the influence of different α-substituents (R1Table 2A) on the efficacy of the allylic alkylation process. To aid in the isolation of these highly volatile compounds, we chose β-ketoesters of higher molecular weights, bearing substitution at both the α-position and allyl fragment, (i.e. 2-phenylallyl 1-benzylyl-2-oxocyclobutanecarboxylate) as base substrates. We were pleased to find that α-alkyl substituents were well tolerated with enantiomeric excess up to 99% (Table 2A, 5a–5b). α-Benzyl substituents were found to give the respective α-quaternary cyclobutanones with uniformly excellent enantioselectivity regardless of the electronic nature of the benzyl moiety (compounds 5c–5e). In addition to alkyl- and benzyl- substituents, allyl-, TMS-protected propargyl and heteroaryl substituted 2-carboxyallyl cyclobutanones proved to be eligible substrates in the asymmetric allylic alkylation reaction providing cyclobutanones 5f–5h in high yields and enantiomeric excess.
Table 2.
Catalytic enantioselective cyclobutanone alkylation: (A) scope of α-keto substitutent tolerance; (B) functional diversity incorporated at the 2-allyl position.[a]
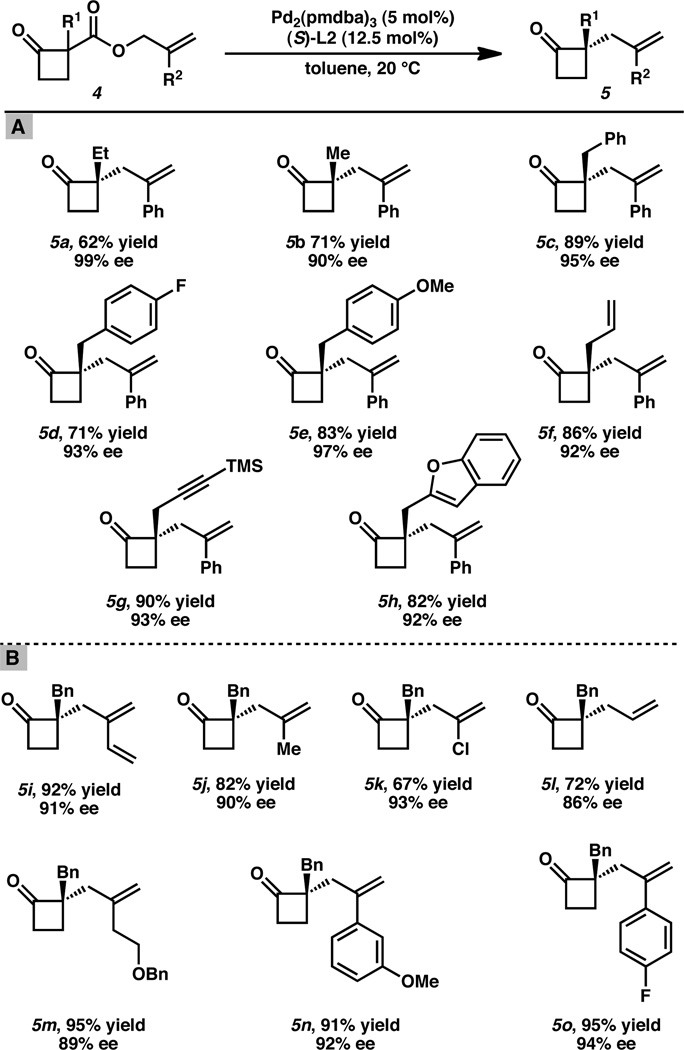 |
Conditions cyclobutanone 4 (1.0 equiv), Pd2(pmdba)3 (5 mol%), (S)-L2 (12.5 mol%) in toluene (0.033 M) at 20 °C for 12–48 h. All reported yields are for isolated products.
Having surveyed the scope of the process with respect to various substituents at the quaternary center, we were poised to investigate the influence of different allyl substitution on the process (R2Table 2B). In accord with previous studies on the palladium-catalyzed asymmetric allylic alkylation, the catalytic system was found to be relatively inactive when terminally substituted or cyclic allyl fragments were employed.[14a, 14b] As such, we limited our survey to carboxyallyl fragments bearing substituents at the 2-allyl position. Gratifyingly, diverse substituents were found to be well tolerated (Table 2B). All α-quaternary cyclobutanones were obtained in moderate to high yield and with outstanding enantiopurity. Particularly interesting are compounds 5i5kand 5m featuring a butadiene, a vinyl chloro and a benzyl ether moiety, respectively. Each of these diverse functional groups may potentially serve as handles for various derivatization reactions (e.g., cycloaddition, annulation or transition metal-catalyzed cross-coupling).
Myriad studies have shown cyclobutanoids to be highly valuable synthetic intermediates, allowing access to enantioenriched oxazepines,[20] piperidines,[21] tetrahydropyrans,[22] α- and β-quaternary cyclopentanones,[10] benzannulated polycycles[23] as well as β-quaternary linear ketones.[2] Cyclobutanones may participate directly in a variety of robust classical transformations, such as Baeyer-Villiger oxidation and Beckmann rearrangement,[2] as well as transition metal-catalyzed ring expansion,[24] ring contraction[25] and ring-opening processes.[26] To demonstrate the utility of our asymmetric synthesis of cyclobutanones within this domain, we carried out a number of transformations on the chiral cyclobutanones generated in this study. Ring expansion by Baeyer-Villiger oxidation, treatment with trimethylsilyldiazomethane and Beckmann rearrangement all proceeded smoothly to deliver dialkyl γ-lactone 6, α-quaternary cyclopentanone 7 and dialkyl γ-lactam 8, respectively. Additionally, ring closing metathesis of diallyl substituted cyclobutanone 5f cleanly furnished quaternary [4.5]-spirocycle 9.
In summary, we have developed the first transition metalcatalyzed enantioselective α-alkylation of cyclobutanones. This method employs palladium catalysis and an electron deficient PHOX type ligand to afford α-quaternary cyclobutanones in good to excellent yields and enantioselectivities. A wide variety of substitutents are tolerated at both the α-keto and 2-allyl positions. The mild nature of our method is reflected in its compatibiilty with otherwise highly electrophilic cyclobutanones. We have further demonstrated the utility of chiral cyclobutanones as synthetic building blocks to access a variety of enantioenriched derivative compounds including dialkyl γ-lactams, dialkyl γ-lactones, α-quaternary cyclopentanones and quaternary [4.5]-spirocycles. We believe that this novel synthetic method will enable the expeditious synthesis of complex bioactive natural products and pharmaceutical components by providing unique access to previously unknown and inaccessible enantioenriched α-quaternary cyclobutanones. Efforts toward this end are currently underway in our laboratory.
Supplementary Material
Footnotes
The authors wish to thank NIH-NIGMS (R01GM080269-01), Deutsche Forschungsgemeinschaft, DFG postdoctoral fellowship to C.E., Amgen, Abbott, Boehringer Ingelheim and Caltech for financial support. Materia, Inc. is gratefully acknowledged for the donation of catalysts. The Varian 400 MR instrument used in this study was purchased via an NIH award to Caltech (NIH RR027690). Dr. Mona Shahgholi and Naseem Torian are acknowledged for highresolution mass spectrometry assistance. Mr. Jonny Gordon is acknowledged for insightful discusions. Mr. Robert Craig is gratefully acknowledged for providing compound L3 and for insightful discussion.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For general reviews see: Cain D. In: Carbon-Carbon Bond Formation, Vol. 5. Augustine RL, editor. Vol. 1. New York: Marcel Dekker Inc.; 1979. pp. 85–250. Seebach D. Angew. Chem., Int. Ed. Engl. 1988;27:1624–1654. Hughes in DL. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 2. Berlin: Springer; 1999. pp. 1273–1294.
- 2.a) Belluš D, Ernst B. Angew. Chem. Int. Ed. Engl. 1988;27:797–827. [Google Scholar]; b) Sergeiko A, Poroikov VV, Hanus LO, Dembitsky VM. Open Med. Chem. J. 2008;2:26–37. doi: 10.2174/1874104500802010026. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dembitsky VM. J Nat. Med. 2008;62:1–33. doi: 10.1007/s11418-007-0166-3. [DOI] [PubMed] [Google Scholar]; d) Yoshikawa K, Kaneko A, Matsumoto Y, Hama H, Arihara S. J. Nat. Prod. 2006;69:1267–1270. doi: 10.1021/np068006a. [DOI] [PubMed] [Google Scholar]; e) Marrero J, Rodríguez AD, Baran P, Raptis RG, Sánchez JA, Ortega-Barria E, Capson TL. Org. Lett. 2004;6:1661–1664. doi: 10.1021/ol049495d. [DOI] [PubMed] [Google Scholar]; f) Li Y-S, Matsunaga K, Ishibashi M, Ohizumi Y. J. Org. Chem. 2001;66:2165–2167. doi: 10.1021/jo001460d. [DOI] [PubMed] [Google Scholar]; g) Tanaka N, Okasaka M, Ishimaru Y, Takaishi Y, Sato M, Okamoto M, Oshikawa T, Ahmed SU, Consentino LM, Lee KH. Org. Lett. 2005;7:2997–2999. doi: 10.1021/ol050960w. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Jiang B, Qiao J, Yang Y, Lu Y. J. Cell. Biochem. 2012;113:2560–2566. doi: 10.1002/jcb.22173. [DOI] [PubMed] [Google Scholar]
- 3.a) Seiser T, Saget T, Tran DN, Cramer N. Angew. Chem. 2011;123:7884–7896. doi: 10.1002/anie.201101053. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:7740–7752. doi: 10.1002/anie.201101053. [DOI] [PubMed] [Google Scholar]; b) Fu N-Y, Chan S-H, Wong HNC. In: Patai Series: the Chemistry of Functional Groups. Rappoport Z, Liebman JF, Patai S, editors. Chichester, UK: John Wiley & Sons, Ltd; 2005. pp. 357–440. [Google Scholar]
- 4.Bauzá A, Quiñonero D, Deyà PM, Frontera A. Chem. Phys. Lett. 2012;536:165–169. [Google Scholar]
- 5.Wiberg KB. In: Patai Series: the Chemistry of Functional Groups. Rappoport Z, Liebman JF, Patai S, editors. John Wiley & Sons, Ltd: Chichester, UK; 2005. pp. 1–15. [Google Scholar]
- 6.Bauld NL, Cessac J, Holloway RL. J. Am. Chem. Soc. 1977;99:8140–8144. [Google Scholar]
- 7.Berg in U. In: Patai Series: the Chemistry of Functional Groups. Rappoport Z, Liebman JF, Patai S, editors. Chichester, UK: John Wiley & Sons, Ltd; 2005. pp. 83–131. [Google Scholar]
- 8.a) Pete J-P. Advances in Photochemistry. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 1996. [Google Scholar]; b) Hehn JP, Mueller C, Bach T. In: Handbook of Synthetic Photochemistry. Albini A, Fagnoni M, editors. Weinheim: Wiley-VCH; 2010. pp. 171–215. [Google Scholar]; c) Hoffmann N. In: Handbook of Synthetic Photochemistry. Albini A, Fagnoni M, editors. Weinheim: Wiley-VCH; 2010. pp. 137–169. [Google Scholar]; d) Lee-Ruff E. In: Methods of Organic Chemistry. De Meijere A, editor. Vol. E17e. Stuttgart: Georg Thieme Verlag; 1997. pp. 179–187. [Google Scholar]
- 9.a) Fitjer L. De Meijere A. Methods of Organic Chemistry. Vol. E17e. Stuttgart: Georg Thieme Verlag; 1997. pp. 251–316. [Google Scholar]; b) Lee-Ruff E. In: Patai Series: the Chemistry of Functional Groups. Rappoport Z, Liebman JF, Patai S, editors. Chichester, UK: John Wiley & Sons, Ltd; 2005. pp. 281–355. [Google Scholar]
- 10.Chaumontet M, Piccardi R, Audic N, Hitce J, Peglion J-L, Clot E, Baudoin O. J. Am. Chem. Soc. 2008;130:15157–15166. doi: 10.1021/ja805598s. [DOI] [PubMed] [Google Scholar]
- 11.a) Markham JP, Staben ST, Toste FD. J. Am. Chem. Soc. 2005;127:9708–9709. doi: 10.1021/ja052831g. [DOI] [PubMed] [Google Scholar]; b) Kleinbeck F, Toste FD. J. Am. Chem. Soc. 2009;131:9178–9179. doi: 10.1021/ja904055z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.López-Carrillo V, Echavarren AM. J. Am. Chem. Soc. 2010;132:9292–9294. doi: 10.1021/ja104177w. [DOI] [PubMed] [Google Scholar]
- 13.a) Lee-Ruff E, Mladenova G. Chem. Rev. 2003;103:1449–1484. doi: 10.1021/cr010013a. [DOI] [PubMed] [Google Scholar]; b) Trost BM, Yasukata T. J. Am. Chem. Soc. 2001;123:7162–7165. doi: 10.1021/ja010504c. [DOI] [PubMed] [Google Scholar]; c) Nemoto H, Ishibashi H, Nagamochi M, Fukumoto K. J. Org. Chem. 1992;57:1707–1712. [Google Scholar]
-
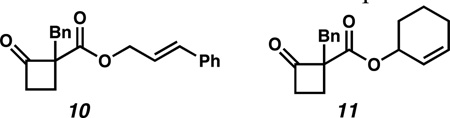
14.a) Cursory studies on substrates bearing terminally substituted and cyclic allyl fragments were conducted. Subjecting compounds 10 and 11 (below) to the reaction conditions described in Scheme 2 resulted in minimal conversion to the desired products (<5% and <1%, respectively, as determined by 1H NMR of the crude reaction mixtures) and these substrate classes were not pursued further.
 Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x.
Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem. 2005;117:7084–7087. doi: 10.1002/anie.200502018.
Angew. Chem. Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018.
Seto M, Roizen JL, Stoltz BM. Angew. Chem. 2008;120:6979–6982. doi: 10.1002/anie.200801424.
Angew. Chem. Int. Ed. 2008;47:6873–6876. doi: 10.1002/anie.200801424.
Streuff J, White DE, Virgil SC, Stoltz BM. Nature Chem. 2010;2:192–196. doi: 10.1038/nchem.518.
Behenna DC, Liu Y, Yurino T, Kim J, White DE, Virgil SC, Stoltz BM. Nature Chem. 2012;4:130–133. doi: 10.1038/nchem.1222.
Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x.
Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem. 2005;117:7084–7087. doi: 10.1002/anie.200502018.
Angew. Chem. Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018.
Seto M, Roizen JL, Stoltz BM. Angew. Chem. 2008;120:6979–6982. doi: 10.1002/anie.200801424.
Angew. Chem. Int. Ed. 2008;47:6873–6876. doi: 10.1002/anie.200801424.
Streuff J, White DE, Virgil SC, Stoltz BM. Nature Chem. 2010;2:192–196. doi: 10.1038/nchem.518.
Behenna DC, Liu Y, Yurino T, Kim J, White DE, Virgil SC, Stoltz BM. Nature Chem. 2012;4:130–133. doi: 10.1038/nchem.1222.
- 15.McDougal NT, Virgil SC, Stoltz BM. Synlett. 2010:1712–1716. doi: 10.1055/s-0030-1258094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baum JS, Shook DA, Davies HML, Smith HD. Synth. Commun. 1987;14:1709–1716. [Google Scholar]
- 17.Presset M, Coquerel Y, Rodriguez J. J. Org. Chem. 2009;74:415–418. doi: 10.1021/jo8021567. [DOI] [PubMed] [Google Scholar]
-
18.
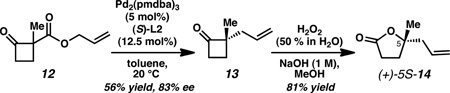
Derivatization of allylic alkylation product 2-allyl-2-methylcyclobutanone 13 to known compound (5S)-5-allyl-5-methyldihydrofuran-2(3H)-one 14 (below) allowed for comparison of optical rotation data and, by chemical correlation, confirmed (5S) absolute configuration (see supporting information). The absolute stereo-configuration of all other products generated in this study were assigned by analogy. These data are consistent with the absolute configurations observed in previous studies on 5-, 6- and 7- membered rings (see ref. 14).
- 19.a) Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Angew. Chem. Int. Ed. 2009;48:6840–6843. doi: 10.1002/anie.200902575. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Behenna DC, Mohr JT, Sherden NH, Marinescu SC, Harned AM, Tani K, Seto M, Ma S, Novák Z, Krout MR, McFadden RM, Roizen JL, Enquist JA, Jr., White DE, Levine SR, Petrova KV, Iwashita A, Virgil SC, Stoltz BM. Chem.—Eur. J. 2011;17:14199–14223. doi: 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Keith JA, Behenna DC, Sherden N, Mohr JT, Ma S, Marinescu SC, Nielsen RJ, Oxgaard J, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2012;134:19050–19060. doi: 10.1021/ja306860n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stevens SJ, Bérubé A, Wood JL. Tetrahedron. 2011;67:6479–6481. doi: 10.1016/j.tet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moustafa MMAR, Pagenkopf BL. Org. Lett. 2010;12:4732–4735. doi: 10.1021/ol102062t. [DOI] [PubMed] [Google Scholar]
- 22.Moustafa MMAR, Stevens AC, Machin BP, Pagenkopf BL. Org. Lett. 2010;12:4736–4738. doi: 10.1021/ol102063f. [DOI] [PubMed] [Google Scholar]
- 23.a) Nishimura T, Ohe K, Uemura S. J. Am. Chem. Soc. 1999;121:2645–2646. [Google Scholar]; b) Matsuda T, Makino M, Murakami M. Angew. Chem. 2005;117:4684–4687. doi: 10.1002/anie.200500799. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:4608–4611. doi: 10.1002/anie.200500799. [DOI] [PubMed] [Google Scholar]; c) Matsuda T, Shigeno M, Murakami M. J. Am. Chem. Soc. 2007;129:12086–12087. doi: 10.1021/ja075141g. [DOI] [PubMed] [Google Scholar]; d) Shigeno M, Yamamoto T, Murakami M. Chem.–Eur. J. 2009;15:12929–12931. doi: 10.1002/chem.200902593. [DOI] [PubMed] [Google Scholar]
- 24.O’Brien JM, Kingsbury JS. J. Org. Chem. 2011;76:1662–1672. doi: 10.1021/jo102257k. [DOI] [PubMed] [Google Scholar]
- 25.Murakami M, Amii H, Shigeto K, Ito Y. J. Am. Chem. Soc. 1996;118:8285–8290. [Google Scholar]
- 26.a) Matsuda T, Makino M, Murakami M. Bull. Chem. Soc. Jpn. 2005;78:1528–1533. [Google Scholar]; b) Matsuda T, Makino M, Murakami M. Org. Lett. 2004;6:1257–1259. doi: 10.1021/ol049833a. [DOI] [PubMed] [Google Scholar]
Scheme 2.

Derivation of enantioenriched α-quaternary cyclobutanones. Conditions: i) 5cH2O2 (55 wt% in H2O), 1 M NaOH, MeOH, 23 °C, 80% yield. ii) 5g, TMSCHN2, BF3·Et2O, Et2O, HCl (aq.), DCM, 69% yield, two steps. iii) 5o, HONH2·HCl, pyridine, EtOH; p-TsCl, Et3N, DMAP, DCM, 22% yield, two steps. iv) 5f, Grubbs-Hoveyda G2, PhH, 97% yield.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.