

Abstract
Müller glia function as retinal stem cells in adult zebrafish. In response to loss of retinal neurons, Müller glia partially dedifferentiate, re-express neuroepithelial markers and re-enter the cell cycle. We show that the immunoglobulin superfamily adhesion molecule Alcama is a novel marker of multipotent retinal stem cells, including injury-induced Müller glia, and that each Müller glial cell divides asymmetrically only once to produce an Alcama-negative, proliferating retinal progenitor. The initial mitotic division of Müller glia involves interkinetic nuclear migration, but mitosis of retinal progenitors occurs in situ. Rapidly dividing retinal progenitors form neurogenic clusters tightly associated with Alcama/N-cadherin-labeled Müller glial radial processes. Genetic suppression of N-cadherin function interferes with basal migration of retinal progenitors and subsequent regeneration of HuC/D+ inner retinal neurons.
Keywords: Müller glia, Retinal regeneration, N-cadherin, Alcama
INTRODUCTION
In adult teleost fish, growth-related neurogenesis continues throughout the central nervous system (Kizil et al., 2012; Grandel and Brand, 2013), including in the retina, where neuroepithelial retinal progenitors in the ciliary marginal zone (CMZ; a stem cell niche) generate retinal neurons associated with ocular growth (Hitchcock et al., 2004; Raymond et al., 2006; Cerveny et al., 2012). In the teleost retina, neurogenesis continues not only in the CMZ, but also in central retina. Müller glia exhibit astrocytic properties (Bringmann et al., 2006), but in addition to physiological and structural support of the retina, they also function similar to radial glia of the embryonic mammalian cortex (Götz and Huttner, 2005; Kriegstein and Alvarez-Buylla, 2009); they divide slowly and sporadically to generate fate-restricted rod progenitors that migrate apically along the radial glial process and differentiate into rod photoreceptors (Raymond and Rivlin, 1987; Bernardos et al., 2007; Stenkamp, 2011).
Following retinal injury in teleost fish, Müller glia generate multipotent progenitors that regenerate retinal neurons (Fimbel et al., 2007; Bernardos et al., 2007; Ramachandran et al., 2010b). Gene expression profiling of mammalian Müller glia revealed unexpected similarities to retinal progenitors (Jadhav et al., 2009; Roesch et al., 2008), and evidence for intrinsic neurogenic potential of Müller glia in chick, rodent, and even human retina is growing, but we do not yet understand why the regenerative response in these vertebrates is minimal compared with the robust regenerative response in fish (Jadhav et al., 2009; Karl and Reh, 2010; Fischer and Bongini, 2010).
Although the multipotent properties of zebrafish Müller glia-derived retinal progenitors have been shown by lineage analysis (Fausett and Goldman, 2006; Bernardos, et al., 2007; Fimbel et al., 2007; Ramachandran et al., 2010a), asymmetric, self-renewing division [a hallmark of stem cells, including radial glia (Götz and Huttner, 2005)], has not been demonstrated. We also do not know whether the number of mitotic divisions of injury-induced zebrafish Müller glia is limited, as has been reported in chick and mammalian retinas (Fischer and Reh, 2003; Karl and Reh, 2010), or whether regeneration-competent Müller glia instead undergo multiple divisions to regenerate retinal neurons.
Here, we compare the early responses of Müller glia (dedifferentiation and re-entry into the cell cycle) following two distinct lesions: light damage of photoreceptors and chemical destruction of inner retinal neurons. We provide evidence for asymmetric, self-renewal divisions of injury-induced Müller glia and further show that N-cadherin-mediated adhesion is required for the formation of neurogenic clusters and basal migration of neuronal progenitors to regenerate neurons.
RESULTS
Alcama is a novel marker of multipotent retinal neuroepithelial cells
To improve our understanding of retinal stem cell properties in injury-induced zebrafish Müller glia, we further characterized the spatiotemporal changes in expression of genes associated with the transition from multipotent neuroepithelial cells to committed progenitors in the CMZ (Cayouette et al., 2006). PCNA+ proliferating progenitors in the CMZ and newly generated Müller glia (Fig. 1A, arrowheads) were immunoreactive for the retinal homeobox protein Rx1, which is associated with early eye field specification and is later expressed specifically in cone photoreceptors (Chuang et al., 1999; Raymond et al., 2006). These immature Müller glia transiently express a nuclear-targeted GFP transgene driven by the glial-specific promoter glial fibrillary acidic protein (gfap), in the zebrafish line Tg(gfap:nGFP)mi2004 (Fig. 1A) (Bernardos et al., 2007). These results show that the retinal progenitor marker Rx1 continues to be expressed in newly committed, but not fully differentiated, Müller glia.
Fig. 1.
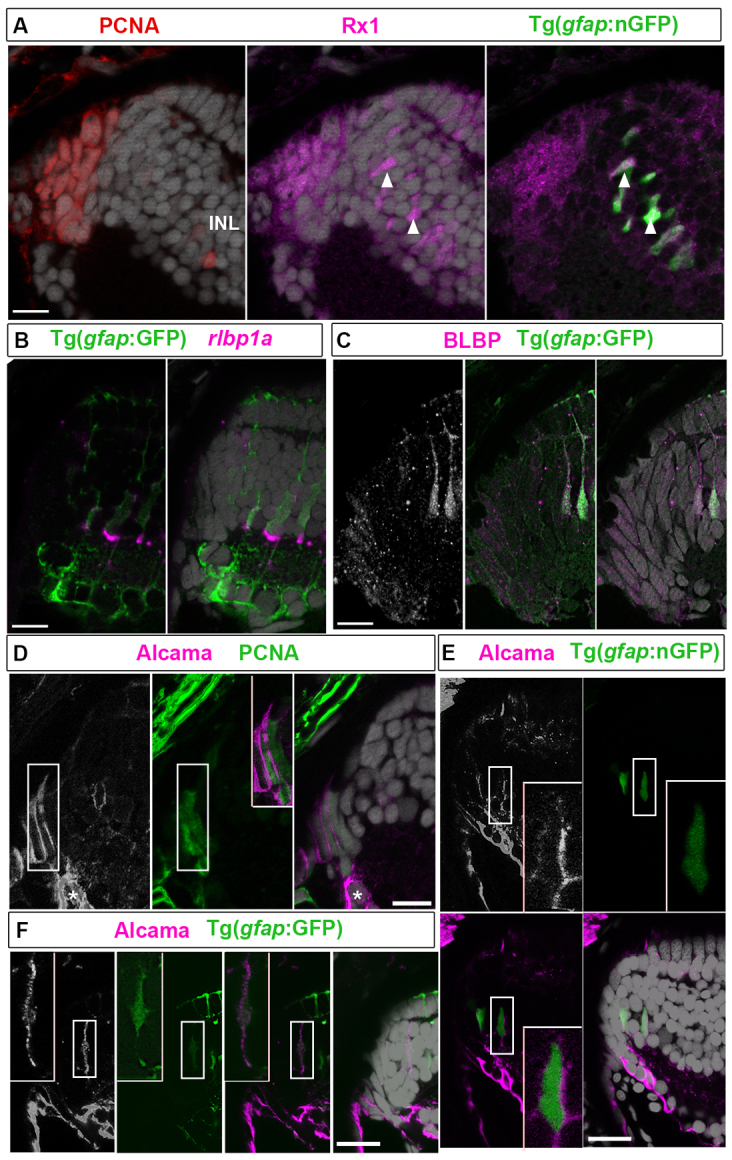
Alcama is a novel marker of multipotent retinal stem cells in adult zebrafish. (A) PCNA+ progenitors (red) in the CMZ and nGFP+ immature Müller glia (arrowheads) in the INL express Rx1 (magenta). (B) GFP+ Müller glia express rlbp1a transcripts (magenta). (C) Retinal progenitors and immature Müller glia express BLBP (white/magenta). (D) Alcama (white/magenta) in PCNA+ (green) cells in the CMZ (boxed region shown in the overlay in the middle panel). Differentiating RGCs are indicated by asterisks. (E) Alcama+ cells (white/magenta) in the INL are nGFP+ immature Müller glia (boxed regions are shown at higher magnification in the insets at bottom right). (F) Alcama (white/magenta) colocalizes with a GFP+ Müller glial process (boxed regions shown at higher magnification in the insets at the upper left). Scale bars: 10 μm.
Müller glia, identified by cytoplasmic expression of the GFP reporter in another transgenic line, Tg(gfap:GFP)mi2002, express the specific marker (Fleisch et al., 2008; Collery et al., 2008) retinaldehyde-binding protein 1a (rlbp1a) gene (Fig. 1B). Retinal progenitors in the CMZ and immature Müller glia also express the radial glia marker BLBP (brain lipid binding protein; also known as FABP7) (Fig. 1C) (Campbell and Götz, 2002), as we reported previously (Raymond et al., 2006).
Alcama (DM-Grasp or neurolin), which is recognized by the zebrafish monoclonal antibody zn5 (Fashena and Westerfield, 1999), is a member of the immunoglobulin superfamily implicated in axon guidance (Diekmann and Stuermer, 2009) and is expressed by differentiating retinal ganglion cells (RGCs; Fig. 1D, asterisks). We were surprised to find Alcama immunoreactivity on PCNA+ neuroepithelial cells in the most peripheral region of the CMZ (Fig. 1D, boxed region) as well as on the somata and radial processes of immature Müller glia (Fig. 1E,F, boxed regions). These results suggest that Alcama is a novel marker for multipotent neuroepithelial cells and newly generated but not yet fully differentiated Müller glia in zebrafish.
Müller glia partially dedifferentiate and express retinal progenitor markers in response to photoreceptor loss
After an acute light lesion that destroys photoreceptors in central retina of adult zebrafish, cones begin to regenerate from Müller glia-derived neuronal progenitors by 4 days post-lesion (dpl) and regeneration is largely complete by 14 dpl (Bernardos et al., 2007; Qin et al., 2009). To understand further the nature of the early response of injury-induced Müller glia, we examined temporal changes in the expression of Müller glia and progenitor markers and the mi2004 nGFP reporter.
In unlesioned retina, Müller glia, which can be recognized by their elongated, polygonal nuclei, were either unlabeled or very weakly immunoreactive for BLBP and Rx1 (Fig. 2A). Damage to cone photoreceptors was evident as early as 1 hour post-lesion (hpl) with reduced expression of Rx1, especially in ultraviolet (UV) cones (Fig. 2B). At 8 hpl, Rx1+ degenerating cone nuclei started to collapse (Fig. 2C, asterisk), and 4C4-labeled microglia were migrating into the lesion (supplementary material Fig. S1). By 4-8 hpl, Müller glia within the lesioned region began to show signs of dedifferentiation: nGFP was re-expressed in mi2004 Müller glia (Fig. 2E,F); rlbp1a transcripts were decreased by 4 hpl, and had completely disappeared by 1 dpl (Fig. 2G); and BLBP and Rx1 immunoreactivity was robust by 8 hpl (Fig. 2C).
Fig. 2.

Müller glia partially dedifferentiate and express retinal progenitor markers in response to photoreceptor lesions. (A-D) PCNA (green), BLBP (cyan) and Rx1 (white/magenta) in control (ctrl) retina. Higher magnifications of the boxed regions are shown on the right. (A) Cones are Rx1+ (asterisks, UV cone nuclei). (B) By 1 hpl, Rx1 is reduced in UV cones and BLBP is upregulated in Müller glia. (C) At 8 hpl, cones collapse (asterisk) and Müller glia express BLBP and Rx1 (boxed region). A PCNA+ microglia or rod precursor (arrowhead) can be seen in the ONL. (D) At 1 dpl, Müller glia are weakly PCNA+/BLBP+/Rx1+. (E,F) Injury-induced nGFP in lesioned area (asterisks) at 4 and 8 hpl and in immature Müller glia (arrowheads). Zpr1 (magenta), which recognizes Arrestin 3a and specifically labels red/green cones. Higher magnifications of the boxed regions are shown in insets. (C) Reduced rlbp1a transcript (white) by 4 hpl and no detectable signal at 1 dpl. Scale bars: 10 μm (A-D,G); 100 μm (E,F).
By 1 dpl, 55% of activated BLBP+ Müller glia (n=92 BLBP+ cells in four retinas) had re-entered the cell cycle as evidenced by immunoreactivity for the G1/S-phase marker PCNA (Fig. 2D). These results indicate that Müller glia respond locally and rapidly (within hours) to photoreceptor injury by dedifferentiation and re-expression of retinal progenitor markers, and re-enter the cell cycle within a day.
Injury-induced Müller glia and neurogenic progenitors are distinct populations
We previously reported apical migration of injury-induced Müller nuclei within the radial glial process towards the outer limiting membrane (OLM) and suggested that this behavior is reminiscent of interkinetic nuclear migration (IKNM) (Bernardos et al., 2007). We again used the inducible nGFP reporter to systematically examine nuclear movement of Müller glia after light lesion. By 1 dpl, Müller glial nuclei within the lesion began to migrate apically and reached the apical surface (OLM) at 2 dpl (Fig. 3A). The M-phase marker pH3 indicated that mitosis of the nGFP+ Müller glia occurs at the OLM (Fig. 3B, n=109 of 113 pH3+ cells at 2 dpl, four retinas). Nuclear membrane breakdown releases nGFP to the cytoplasm; anti-GFP was used to enhance the signal (Fig. 3B, arrows). These data indicate that injury-induced Müller glia exhibit IKNM, a distinctive property of neuroepithelial cells and radial glia (Götz and Huttner, 2005; Taverna and Huttner, 2010).
Fig. 3.

Injury-induced Müller glia and retinal progenitors are distinct cell populations. (A) By 2 dpl, Müller glial nuclei (nGFP+, green) move to the OLM (dotted line). (B) nGFP+ (anti-GFP, green)/pH3+ (magenta) mitotic Müller glia nuclei at the OLM. nGFP is diffuse (arrows). Higher magnifications of the boxed regions are shown in insets. (C) nGFP+(green)/PCNA+ (red)/Rx1+ (white/magenta) progenitors at 3 dpl. (D) Alcama (white/magenta); nGFP. (E) Alcama (white/magenta) colocalizes with GFP. Scale bars: 20 μm (A,B,D,E); 5 μm (C).
By 3-5 dpl, neurogenic clusters [elongated, tightly packed, Müller glia-derived progenitors in the inner nuclear layer (INL) (Fausett and Goldman, 2006; Bernardos et al., 2007; Thummel et al., 2008)] appeared in lesioned regions. These rapidly proliferating progenitors expressed BLBP, incorporated BrdU (data not shown), and expressed both Rx1 and PCNA (Fig. 3C; supplementary material Fig. S2A,B). Although all cells in the neurogenic clusters at 3 dpl expressed the retinal progenitor markers BLBP and Rx1, each cluster had a single, spindle-shaped, Alcama+/nGFP+ cell, with a radial process that extended to the apical and basal surfaces of the retina (Fig. 3D). Alcama immunoreactivity colocalized with the cytoplasmic, gfap:GFP, Müller glial marker in basal radial fibers extending into the inner retina (Fig. 3E). These results are consistent with the hypothesis that injury-induced Müller glia divide asymmetrically in a self-renewing division that preserves the Müller glia cell, with Alcama+ radial processes that span the retinal epithelium, and produces a daughter cell that divides repeatedly to generate neuronal progenitors.
Injury-induced dedifferentiation is not sufficient to trigger cell cycle re-entry and regeneration
We noticed that expression of the injury-inducible nGFP reporter extended into the dorsal retina beyond the central zone in which Zpr1+ red/green double cones were ablated (supplementary material Fig. S2A), although few nGFP+ Müller glia in this region were PCNA+ (supplementary material Fig. S2B). Further examination revealed differential sensitivity of the different cone subtypes to light damage: with a double transgenic line, Tg(sws1:GFP;sws2:mCherry), in which the UV and blue cones express transgenes driven by the UV (sws1 or opn1sw1) and blue (sws2 or opn1sw2) cone opsin promoters, respectively, we found that UV cones in dorsal retina had largely been destroyed, but blue cones and red/green double cones (data not shown) were preserved (supplementary material Fig. S2C). Expression of nGFP in the dorsal retina corresponded to the region missing UV cones (supplementary material Fig. S2A,C); consistent with the near absence of proliferation, the missing UV cones in the dorsal retina failed to regenerate (supplementary material Fig. S2D). These results suggest that Müller glia respond by dedifferentiation to local loss of photoreceptors that they contact directly, but that dedifferentiation is not sufficient to promote re-entry into the cell cycle and that limited or selective loss of retinal neurons (i.e. UV cones only) is not sufficient to trigger the regenerative response in Müller glia.
Injury-induced Müller glia undergo a single asymmetric division
Previous studies of retinal regeneration have not considered whether Müller glia divide repeatedly, and presumably asymmetrically, to generate neuronal progenitors or whether glial-derived, neuronal progenitors divide symmetrically to generate neurogenic clusters, similar to a transit-amplifying population, although the former is typically assumed (Karl and Reh, 2010). Our observations that IKNM of Müller glial nuclei is observed only at the outset of the regenerative response and that a single Alcama+ radial fiber anchors each neurogenic cluster suggested that injury-induced Müller cells might divide only once, and that each neurogenic cluster is formed by a rapidly dividing progenitor produced by the initial asymmetric division of one Müller glia.
To address this question, we sequentially labeled with two thymidine analogs. We exposed fish to EdU during the initial cell cycle of Müller glia, and then labeled subsequent divisions with BrdU (Fig. 4A). If Müller glia divide only once, they will incorporate EdU but not BrdU; if they undergo multiple rounds of division, they will incorporate both EdU and BrdU. Crucial to the success of this strategy is to determine the timing of the first S phase of Müller glia, with the caveat that cell cycle progression is unlikely to be completely synchronous in the population of Müller glia. Based on detectable expression of PCNA at 1 dpl (Fig. 2D), we inferred that at least some Müller glia re-enter the cell cycle within the first day.
Fig. 4.

Injury-induced Müller glia divide asymmetrically once to generate neurogenic clusters. (A) Sequential labeling with EdU and BrdU. Two predicted alternative results are depicted: if Müller glia (MG) divide only once, they incorporate only EdU (green); if they undergo multiple divisions, they incorporate both EdU and BrdU (green+magenta=white). (B) Some nGFP+ (green) nuclei in flat-mounted retinas at 42 hpl are EdU+ (red). (C) Pair of EdU+ (red) nuclei at 42 hpl, of which only one is PCNA+ (magenta). (D) Optical z-stack projection at 72 hpl: clusters of BrdU+ nuclei (magenta) with few EdU+ nuclei (green). Right panels: single optical section of EdU+/nGFP+/BrdU- nucleus (arrows). (E) Lateral view of BrdU+ nuclei (magenta) clusters with a single EdU+ (green)/BrdU-nucleus, reconstructed from optical Z-stack. Right panel: BrdU+ clusters (magenta outline). (F) Cluster with single EdU+ cell (white/red) at 72 hpl. (G) Cluster of weakly EdU+ cells (white/red) with one strongly EdU+ cell. (H) Cluster with one PCNA-/strongly GFP+ (green) cell (arrows) and PCNA+ (magenta)/weakly GFP+ progenitors. Scale bars: 10 μm.
To determine more precisely the timing of the initial mitotic division, we compared the planimetric density of nGFP+ injury-induced Müller glia in flat-mounted mi2004 retinas (supplementary material Fig. S3A) with the number of GFP+ differentiated Müller glia in unlesioned mi2002 retinas (supplementary material Fig. S3B). The density of nGFP+ injury-induced Müller glia at 36 hpl (80±8 cells per 104 μm2, mean±s.d., n=6 retinas) was not significantly different than that of control retinas (78±16.6 cells per 104 μm2, n=6 retinas, P=0.8578; supplementary material Fig. S3D), suggesting that all Müller glia in the area of the lesion respond to the injury by upregulating the dedifferentiation marker. At 36 hpl, 66% of the nGFP+ Müller glia expressed PCNA (n=1783 nGFP+ cells in n=6 retinas; supplementary material Fig. S3A), implying that re-entry of Müller glia into the cell cycle is not synchronous and/or that not all Müller glia in this region will re-enter the cell cycle. At 42 hpl, the density of nGFP+ cells (supplementary material Fig. S3C) was significantly increased (105±17 cells per 104 μm2, n=6 retinas) compared with the density at 36 hpl (supplementary material Fig. S3D, P<0.005), indicating that some Müller glia had completed their first cell division between 36 and 42 hours post-lesion.
We therefore exposed mi2004 fish to EdU from 20 to 36 hpl to label the initial mitotic division. Previous studies of cell cycle kinetics in embryonic zebrafish retina have reported that S phase is 5.6 hours (Leung et al., 2011). To ensure that Müller glia labeled by the initial EdU exposure would complete S phase, we allowed a 6-hour gap between the end of the EdU exposure and the beginning of the BrdU exposure at 42 hpl. EdU labeling in flat-mounted retinas confirmed that the EdU label was in nGFP+ dedifferentiated Müller glia (Fig. 4B). Double labeling with PCNA at 42 hpl reveled several examples of adjacent pairs of nGFP+/EdU+ cells, in which only one of the two putative daughter cells was PCNA+ (Fig. 4C), consistent with an asymmetric division resulting in a neurogenic progenitor and a post-mitotic Müller glial cell. After a subsequent 30-hour exposure to BrdU we observed discrete clusters of BrdU+ cells and fewer EdU+ cells (Fig. 4D,E). Most of the EdU+ cells (86%, n=118 in three retinas) were negative for BrdU, and most of these EdU+/BrdU- cells (79%) were closely associated with clusters of BrdU+ cells; the BrdU+ or nGFP+ clusters were typically associated with a single, strongly labeled EdU+ cell (Fig. 4E,F). Some clusters of nGFP+ retinal progenitors were weakly EdU+ but associated with a single, strongly labeled EdU+ cell (Fig. 4G), consistent with dilution of the EdU label by subsequent divisions of the glia-derived retinal progenitors. The lack of detectable EdU in some BrdU+ or nGFP+ neurogenic clusters is consistent with asynchronous cell cycle kinetics in the population of activated Müller glia: those that entered S phase near the beginning of the 16-hour EdU exposure (Fig. 4A) would incorporate larger amounts of EdU compared with those that entered S phase nearer the end, and therefore the amount of EdU inherited by the daughter progenitor cell would vary. The robust EdU signal in a single cell in each cluster is consistent with the proposed asymmetric initial division, in which the Müller glia divides only once (thus retaining a strong EdU label) to generate a Müller glia that retains glial identity and a daughter cell progenitor that divides repeatedly, incorporating BrdU and diluting out the EdU.
Also consistent with the proposal that Müller glia divide once is the heterogeneity of cells in the neurogenic clusters at 72 hpl; elongated, PCNA+ dividing retinal progenitors, weakly labeled with GFP, were typically associated with a single, PCNA-negative, strongly GFP+ cell with an angular shape characteristic of Müller glia cell bodies (Fig. 4H). Strong activation of the gfap:EGFP reporter indicates Müller glial identity, and a weaker signal is interpreted as perseverance of the GFP protein in proliferating, glial-derived neuronal progenitors. Also consistent with self-renewing divisions is that the planimetric density of GFP+ radial fibers of Müller glia, counted at the level of the inner plexiform layer in retinal flat-mounts (e.g. supplementary material Fig. S3B), was not changed during or after regeneration was complete (number of cells per 104 μm2: control, 83±10; 2 dpl, 90±11, 7 dpl, 77±5; 14 dpl, 78±8; P=0.33, three retinas per sample). These data are consistent with the hypothesis that injury-induced Müller glia typically have one self-renewing division to produce a rapidly dividing progenitor that generates a neurogenic cluster.
Müller glia partially dedifferentiate and express retinal progenitor markers in response to retinal ganglion cell loss, but with a delay
Regeneration of retinal neurons in adult zebrafish is not limited to photoreceptors (Raymond and Hitchcock, 2000); inner retinal neurons destroyed by ouabain also regenerate (Fimbel et al., 2007), but whether Müller glia undergo IKNM and divide asymmetrically in this paradigm is not known. To test these questions, we injected ouabain intraocularly, and at 1 day post-injection (dpi) the neuronal marker HuC/D, which labels amacrine cells (AC) in the inner nuclear layer (INL) and RGCs, was no longer detectable in the ganglion cell layer (GCL) and was reduced in the INL (Fig. 5A,B). At 2 dpi, basal processes of Müller glia in the inner retina had collapsed, as revealed by the GFP reporter and Müller glial marker GS (glutamine synthetase) (supplementary material Fig. S4A,B). By contrast, photoreceptors in the outer retina were largely intact, as revealed by normal lamination of the outer nuclear layer (ONL), Rx1 immunostaining in cones (Fig. 5C), and intact apical Müller glial processes (supplementary material Fig. S4A,B). At 2 dpi, PCNA+/4C4+ microglial cells were abundant in the inner retina (Fig. 5C; supplementary material Fig. S4C). These results confirm that an intraocular injection of ouabain at this dose preferentially destroys RGCs and disrupts the basal processes of Müller glia.
Fig. 5.

Induction of neuroepithelial markers in Müller glia is delayed after ouabain. (A) Control (ctrl) HuC/D+ neurons. (B) HuC/D immunoreactivity is absent in the GCL and reduced in the INL at 1 dpi. (C) PCNA (green), BLBP (cyan) and Rx1 (magenta) staining at 2 dpi. Right panel: BLBP+/Rx1+ Müller glia. Scattered PCNA+/BLBP-/Rx1- cells in the INL and GCL are microglia. (D) At 3 dpi, nGFP+/PCNA+ (red)/Rx1+ (white/magenta) Müller glial nuclei are apically displaced. (E) At 3 dpi, pH3+ nuclei (magenta) are apical in the INL but not at the OLM (dotted line). (F) Alcama+ (white/magenta) Müller glia radial processes at 5 dpi. (G) At 5 dpi, nGFP+/HuC/D+ (red) regenerated neurons (higher magnification of boxed area shown in inset). Scale bars: 20 μm (A-G); 2 μm (inset in C).
We next asked whether the Müller glia response to loss of inner retinal neurons is similar to photoreceptor loss. At 1 dpi, the expression of rlbp1a was no longer detectable in Müller glia (supplementary material Fig. S4D,E), and immunoreactivity for Rx1 and BLBP increased by 2 dpi (Fig. 5C; compare with controls shown in Fig. 2A). Re-entry into the cell cycle was delayed compared with photoreceptor lesions. At 2 days after intense light exposure, over half of activated Müller glia were PCNA+ (supplementary material Fig. S3A), but at 2 days after ouabain injection, only 4% of activated BLBP+ Müller glia were PCNA+ (n=73 BLBP+ cells in four retinas). By 3 dpi (Fig. 5D), 77% of activated BLBP+ Müller glia became PCNA+ (n=117 BLBP+ cells in four retinas). These results suggest that Müller glia partially dedifferentiate (as shown by loss of rlbp1a, upregulation of BLBP and Rx1, and expression of the inducible nGFP reporter) and then re-enter the cell cycle in response to both types of lesion, but the time course of the response is slower after loss of inner neurons compared with loss of photoreceptors.
We next asked whether Müller glial nuclei undergo IKNM after loss of RGCs. In the undamaged retina, Müller glial nuclei are located in the inner half of the INL (supplementary material Fig. S4A), but at 3 dpi nGFP+/Rx1+/PCNA+ nuclei were scattered throughout the INL (Fig. 5D), and pH3+ mitotic nuclei were at the outer boundary of the INL (Fig. 5E). These results indicate that Müller glia nuclei migrate apically after inner retinal damage, but in the absence of photoreceptor loss, they fail to penetrate the ONL to reach the apical surface (OLM).
At 3 dpi, weak Alcama immunoreactivity was observed in some nGFP+ Müller glial processes at the outer boundary of the INL (supplementary material Fig. S4F). At 5 dpi, nGFP+ progenitors formed neurogenic clusters similar to those associated with photoreceptor regeneration (Fig. 5F,G). Again, we typically found one Alcama+ cell with radial processes associated with each neurogenic cluster (Fig. 5F), consistent with an initial asymmetric division of Müller glia. Most progenitors in the neurogenic clusters at 5 dpi were weakly immunoreactive for Rx1 (Fig. 5G), and lineage tracing with the nGFP reporter confirmed that regenerated HuC/D+ neurons are derived from Müller glia (Fig. 5G). These data show that the Müller glia-derived neurogenic clusters contain multipotent progenitors competent to regenerate photoreceptors and other retinal neurons.
N-cadherin is necessary for the formation of neurogenic clusters in the inner retina
The model suggested by data presented thus far is that loss of retinal neurons induces Müller glia to partially dedifferentiate and re-enter the cell cycle with IKNM; the Müller cell retains its radial processes and divides once asymmetrically to produce a progenitor cell, which migrates basally and proliferates rapidly to generate a cluster of neuronal progenitors that remain associated with the Alcama+ radial Müller glial process. The tightly packed clusters of progenitors surrounding each radial Müller glia is suggestive of differential cell adhesion, and we previously reported injury-induced upregulation of cdh2 transcript and N-cadherin (Cadherin 2) protein (Liu et al., 2002; Raymond et al., 2006) and N-cadherin distribution along the radial processes of Müller glia and progenitors in the neurogenic cluster (Fig. 6A,B). We therefore asked whether N-cadherin-mediated homophilic adhesion is responsible for the tight clustering of neuronal progenitors.
Fig. 6.

Reduction of N-cadherin function interferes with formation of neurogenic clusters after light lesions. (A) N-cadherin (white/magenta) at OLM (between arrowheads) in unlesioned (ctrl) retina. (B) At 3 dpl, N-cadherin (white/magenta) in Müller glia and progenitors (green). (C) RT-PCR products of cdh2 digested with BsmBI. (D,E) N-cadherin (white/magenta) in the ONL at 3 dpl in cdh2+/m117; mi2002 (D) and cdh2+/m117; mi2004 (E). (F) Neurogenic clusters (PCNA+; green) in sib retinas at 3 dpl. (G) PCNA+ (green)/BLBP+ (red)/Rx1+ (white/magenta) progenitors in cdh2+/m117 hets at 3 dpl. (H) Counts of PCNA+ progenitors in the INL and ONL at 3 dpl. *P<0.005, **P<0.0005. (I) Nuclei of nGFP+/Alcama+ (white/magenta) Müller glia in the INL and nGFP+/Alcama- progenitors in the ONL at 3 dpl in cdh2+/m117; mi2004. (J) pH3+ (magenta) nuclei at 3 and 5 dpl in sibs. (K) pH3+ (magenta) nuclei at 3 and 5 dpl in cdh2+/m117 hets. (L) Fraction of pH3+ cells in INL and ONL after lesion. *P<0.0001. Box plots: median, 25th and 75th percentiles; whiskers show maximum and minimum data points. Scale bars: 20 μm.
To investigate this, we used a semi-dominant mutant allele of N-cadherin, cdh2m117, which disrupts homophilic adhesion (Malicki et al., 2003; Harrington et al., 2007). The homozygous mutant is embryonic lethal, so we examined retinal regeneration in heterozygous cdh2+/m117; RT-PCR confirmed that both alleles are expressed in the adult heterozygote (het) retinas (Fig. 6C). We found no differences in N-cadherin localization (data not shown), morphology and number of Müller glia (data not shown) and morphology and distribution of cone photoreceptors between wild-type siblings (sib) (supplementary material Fig. S5A,C) and cdh2+/m117 hets (supplementary material Fig. S5B,E).
We then subjected adult cdh2+/m117 hets to light damage. At 3 dpl, N-cadherin distribution was altered; the strongest labeling was in the ONL (Fig. 6D,E) compared with a more widely distributed labeling, including Müller glial basal processes, in wild-type (Fig. 6B). To determine whether Müller glial responses to injury were impaired in cdh2+/m117 hets, we examined gene expression, nuclear migration and formation of neurogenic clusters. We found no differences in BLBP and Rx1 expression between cdh2+/m117 hets and wild-type sibs, and Müller glial nuclei underwent IKNM (data not shown). However, at 3 dpl neurogenic clusters were not as elongated in cdh2+/m117 hets and did not extend into the INL (Fig. 6E compared with Fig. 3D), and PCNA+/BLBP+/Rx1+ progenitors instead accumulated in the ONL (compare Fig. 6F,G). In wild-type sib retinas at 3 dpl, the number of PCNA+ progenitors in the INL was greater than in the ONL (Fig. 6H, ONL: n=164 cells; INL: n=481 cells in four retinas; P<0.005), whereas the reverse was true in the cdh2+/m117 retinas (Fig. 6H, ONL: n=600 cells; INL: n=192 cells in four retinas; P<0.0005). At 3 dpl, the nuclei of Alcama+ Müller glia were correctly positioned in the INL (Fig. 6I), suggesting that neuronal progenitors, produced by the initial division of Müller glia at the apical surface, continue to divide and accumulate in the ONL (Fig. 6E,G), whereas Müller glial nuclei return to the INL.
The laminar distribution of pH3+ mitotic nuclei is consistent with this interpretation. In cdh2+/m117 hets, >90% of pH3+ cells were in the ONL at 3 and 5 dpl whereas in wild-type sibs, by 5 dpl 45% pH3+ nuclei were in the INL (Fig. 6J-L; n=133 pH3+ cells in four retinas, P<0.0001). These results suggest that when N-cadherin function is reduced, progenitors produced by the initial asymmetric division of Müller glia failed to migrate to the INL. We conclude that N-cadherin-mediated adhesion regulates basal migration of progenitors along the radial Müller glial processes, and this adhesive interaction is necessary to form neurogenic clusters in the inner retina.
To examine whether the migration defect in cdh2+/m117 disrupts photoreceptor regeneration, we again used the double transgenic opsin reporter line for UV and blue cones and Zpr1 for red and green cones. At 14 dpl, all four types of cones had regenerated in both wild-type sibs [supplementary material Fig. S5D; number of cells/104 μm2: 97±15 (UV), 52±8 (blue), 158±9 (double cones), n=3 retinas] and cdh2+/m117 hets [supplementary material Fig. S5F; number of cells/104 μm2: 80±12 (UV), 66±3 (blue), 162±14 (double cones), n=3 retinas]. These results indicate that the defect in progenitor migration and formation of neurogenic clusters in the INL in N-cadherin mutants does not impede photoreceptor regeneration.
N-cadherin-mediated adhesion in neurogenic clusters is necessary for retinal ganglion cell regeneration
To regenerate inner retinal neurons, such as HuC/D+ AC and RGC, progenitors must migrate basally to the GCL. Because basal migration of the progenitors into the INL is impeded in cdh2+/m117 hets, we asked whether loss of N-cadherin function would prevent their regeneration. To test this, we injected ouabain intraocularly into cdh2+/m117;mi2002, cdh2+/m117;mi2004 hets, and wild-type sibs to destroy neurons in the inner retina.
In wild-type sibs, N-cadherin protein was upregulated in the collapsed GFP+ Müller glial basal processes at 3 dpi (Fig. 7A), and in the spindle-shaped nGFP+ neurogenic clusters at 5 dpi (Fig. 7B). In hets at 3 dpi, N-cadherin protein was also upregulated in Müller glia (Fig. 7C), but at 5 dpi, the shape and positioning of neurogenic clusters was abnormal; nGFP+/Rx1+ progenitors formed irregular, rounded clumps (Fig. 7D,E) not elongated clusters as in sibs (Fig. 7B). Proliferation of Müller glia and retinal progenitors was not affected in the hets, although individual PCNA+ cells were more rounded and less spindle-shaped than in sibs (supplementary material Fig. S6A,B). The number of PCNA+ cells was the same in sibs and hets at 3, 5 or 7 dpi (Fig. 7F), but retinal progenitors failed to correctly specify and differentiate. Although some nGFP+/HuC/D+ regenerated neurons were observed in cdh2+/m117; mi2004 hets at 5 dpi (Fig. 7E), at 14 dpi HuC/D+ neurons in the hets (Fig. 7H,I) were significantly reduced (n=1299 HuC/D+ cells in four retinas) compared with sibs (Fig. 7G,I; n=2590 HuC/D+ cells in four retinas, P<0.0001). These results suggest that N-cadherin-dependent cell-cell adhesion is necessary for the formation of neurogenic clusters and subsequent cell fate specification to allow regeneration of HuC/D+ inner retinal neurons.
Fig. 7.

Regeneration of retinal ganglion cells is reduced in cdh2+/m117 heterozygotes. (A,C) At 3 dpi, GFP+/N-cadherin+ (white/magenta) basal Müller glia processes collapse in siblings (A) and hets (C). (B,D) N-cadherin+ (white/magenta) neurogenic clusters in sib retinas (B, arrows) and abnormal clusters in hets (D) at 5 dpi. (E) Regenerated nGFP+/Rx1-/HuC/D+ (red) inner retinal neuron (boxed; enlarged in inset) in het at 5 dpi. (F) Counts of PCNA+ cells in sib and cdh2+/m117. (G) HuC/D+ (magenta) neurons are destroyed at 1 dpi; some regenerate by 14 dpi. (H) Regenerated HuC/D+ neurons in hets. (I) Counts of HuC/D+ neurons. *P<0.0001. Box plots: median, 25th and 75th percentiles; whiskers show maximum and minimum data points. Scale bars: 20 μm.
DISCUSSION
Our results show that loss of retinal neurons in adult zebrafish triggers two separable responses in Müller glia: partial dedifferentiation followed by re-entry into the cell cycle (Fig. 8A,C). Shortly after destruction of photoreceptors, injury-induced dedifferentiated Müller glial nuclei migrate apically and undergo a single, asymmetric division at the OLM. This self-renewing division allows the Müller cell to retain its glial identity and its radial process and to produce a single daughter cell (a neuronal progenitor) that divides rapidly and migrates basally into the INL to generate neurons for retinal repair.
Fig. 8.

Cell adhesion in photoreceptor and retinal ganglion cell regeneration. (A,B) Time course of responses after light lesions in sibs (A) and cdh2+/m117 hets (B). (C,D) Time course of response after intraocular injection of ouabain in sibs (C) and cdh2+/m117 hets (D). Green, Müller glia; red outline, N-cadherin; cyan outline, both N-cadherin and Alcama. See text for details.
That Müller glia divide only once is somewhat unexpected. Most studies have assumed that the neurogenic clusters in the regenerating zebrafish retina were produced from multiple divisions of injury-induced Müller glia, and the key to successful retina regeneration was thought to rely on Müller glial dedifferentiation into a cycling population of progenitor cells (Karl and Reh, 2010; Ramachandran et al., 2010b; Ramachandran et al., 2011; Wan et al., 2012). By contrast, Thummel and colleagues (Thummel et al., 2010) suggested that continued division of Müller glia is not responsible for generating neurogenic clusters; they showed that morpholino knockdown of Pax6b did not affect the initial Müller glial division, but inhibited the early divisions of neuronal progenitors, resulting in the absence of neurogenic clusters and failure of cone regeneration. The implications of a single, self-renewing division of Müller glia are several. First, it explains how the damaged retina maintains its highly patterned, laminated cytoarchitecture, which is known to depend on the integrity of Müller glia (Bringmann et al., 2006), while repair is underway. If instead Müller glia dedifferentiated completely and were transformed into neuroepithelial cells, then defects in lamination would result, as observed when Müller glia are eliminated by the gliotoxin DL-α-aminoadipic acid (Rich et al., 1995). Second, it explains why the planimetric density of Müller glia is unchanged after regeneration is completed (this study) (Lenkowski et al., 2013). Third, it provides an explanation for why retinal injury in fish does not provoke reactive gliosis and formation of a ‘glial scar’ characteristic of retinal lesions in mammals (Bringmann et al., 2006; Karl and Reh, 2010). This insight has implications for potential therapeutic strategies to enhance the neurogenic potential of Müller glia in the damaged human retina. Finally, these observations support the suggestion (Raymond et al., 2006) that Müller glia and their associated neurogenic clusters resemble stem cell niches in the adult mammalian brain, with their slowly cycling radial glial stem cells and rapidly dividing neuronal progenitors (Alvarez-Buylla and Lim, 2004; Johansson et al., 2010; Grandel and Brand, 2013).
After completing the injury-induced cell cycle, Müller glia express the adhesion molecule Alcama, which we also show is a novel marker of multipotent retinal stem cells in the CMZ. Alcama is expressed in mesenchymal, hematopoietic, neuronal and stromal stem cell populations in mammals (Wai Wong et al., 2012). The absence of Alcama in the proliferating cells of the neurogenic cluster provides additional evidence that retinal progenitors responsible for regeneration are distinct from Müller glia, per se. Although we have no functional data on the role of Alcama in retinal regeneration, its distribution along the radial Müller glial processes, which guide migration of rod precursors in the uninjured, growing fish retina (Raymond and Rivlin, 1987), suggests that Alcama-dependent adhesive interactions may mediate migration of progenitors in the neurogenic clusters. Interestingly, mammalian ALCAM also colocalizes with VE- and N-cadherin at lateral membrane domains in endothelial cell junctions (Ofori-Acquah et al., 2008); it is a marker for metastasis in human tumor cells, recruits N-cadherin and β-catenin to adherens junctions, and promotes motility in uveal melanoma cell lines (Jannie et al., 2012).
The importance of cell-cell adhesion and directed cell migration in retinal regeneration has received scant attention, and instead the focus has been on examining transcriptional networks and secreted signals that promote Müller-dependent retinal neurogenesis (Karl and Reh, 2010; Fischer and Bongini, 2010; Thummel et al., 2010; Ramachandran et al., 2010b; Ramachandran et al., 2011; Wan et al., 2012; Nelson et al., 2012; Lenkowski et al., 2013). We showed previously that cells in the neurogenic clusters strongly upregulate both N-cadherin and Notch components (Raymond et al., 2006), and recent studies demonstrate a functional role for Notch signaling along with secreted heparin-binding epidermal growth factor (HB-EGF) and Wnt/β-catenin, which cooperatively modulate a regulatory network activated during retinal regeneration targeting the neurogenic transcription factors Pax6b and Ascl1a (Ramachandran et al., 2011; Wan et al., 2012). Here, we provide direct evidence for the involvement of N-cadherin in basal migration of neuronal progenitors along radial Müller glial fibers. Spindle-shaped clusters of N-cadherin+ retinal progenitors surround N-cadherin+/Alcama+ radial processes of Müller glia to form ‘regeneration niches’ (Fig. 8A,C). When N-cadherin function is reduced, following light damage the retinal progenitors produced at the apical surface by Müller glia fail to migrate into the INL to form neurogenic clusters, and instead accumulate in the ONL (Fig. 8B). Despite the abnormal spatial distribution of progenitors, cone photoreceptors regenerate in cdh2+/m117 hets, possibility because extrinsic, laminar-specific photoreceptor determinants are present in the ONL. However, regeneration of HuC/D+ inner retinal neurons is partially blocked in cdh2+/m117 hets; progenitors proliferate but they fail to migrate basally and differentiate (Fig. 8D). Interestingly, regulation of N-cadherin expression also controls migration and laminar position of neurons in the developing cortical plate in the embryonic mammalian brain (Solecki, 2012).
Further evidence implicating cell adhesion in Müller glia-dependent regeneration is that photoreceptor loss triggers a more rapid response in Müller glia compared with loss of inner retinal neurons. Because the injury mechanism is different in the two lesion paradigms, the time course of neuronal damage and degeneration is not necessarily the same, and a major difference is that apical processes of Müller glia form adherens junctions with photoreceptors at the OLM (Gosens et al., 2008). Damage to photoreceptors disrupts the adherens junctions, which are important regulators of differentiation and proliferation in radial glia (Götz and Huttner, 2005). In particular, β-catenin links the intracellular domain of cadherins at the adherens junctions, and separately functions as the intracellular mediator of canonical Wnt signaling; β-catenin-mediated signal transduction regulates radial glia proliferation in the developing mouse cortex (Johansson et al., 2010) as well as progenitors in the regenerating retina (Wan et al., 2012; Meyers et al., 2012).
Dedifferentiation of Müller glia is not sufficient to trigger re-entry into the cell cycle and retinal regeneration. We observed that in the dorsal retina, in which only UV cones were destroyed by intense light, Müller glia responded by partial dedifferentiation, but they did not proliferate and UV cones never regenerated. In the damaged mammalian retina, reactive Müller glia respond by non-proliferative hypertrophy or proliferative gliosis (Karl and Reh, 2010). Several previous studies have suggested that a minimum level of retinal cell loss is necessary to provoke regeneration in fish retina. Selective loss of dopaminergic neurons in goldfish does not induce regeneration (Braisted and Raymond, 1992), and depletion of a subset of rod photoreceptors does not induce Müller glia-dependent regeneration in zebrafish (Montgomery et al., 2010).
Our data refine and expand the model for retinal regeneration in zebrafish. When injury-induced Müller glia re-enter the cell cycle they undergo a single, self-renewing division and re-express a novel retinal stem cell marker, Alcama. Müller glia contribute to retinal regeneration as both stem cells and niche cells: they generate neuronal progenitors and provide an N-cadherin-dependent migratory pathway to support the basal migration of progenitors necessary for regeneration of inner neurons.
MATERIALS AND METHODS
Animals and retinal lesions
Zebrafish (Danio rerio) lines Tg(gfap:GFP)mi2002 (Bernardos and Raymond, 2006), Tg(gfap:nGFP)mi2004 (Bernardos et al., 2007), Tg(sws1:GFP;sws2:mCherry) and cdh2+/m117 (Malicki et al., 2003) were maintained at 28°C in a 14 hour/10 hour light/dark cycle according to standard husbandry protocols. The University of Michigan Committee on the Use and Care of Animals approved all experimental procedures.
Brief exposures (30 minutes) to intense light destroyed photoreceptors as described previously (Bernardos et al., 2007). To destroy inner retinal neurons, 0.04 mM ouabain in 0.9% sodium chloride was injected intraocularly to achieve an estimated intraocular concentration of 2 μM, as described previously (Fimbel et al., 2007; Raymond et al., 1988).
Histology
Immunocytochemistry on retinal cryosections and flat mounts was as previously described (Barthel and Raymond, 1990; Raymond et al., 2006; Bernardos et al., 2007). Nuclei were stained with Hoechst and slides mounted in Prolong Gold (Invitrogen). Antibodies are listed in supplementary material Table S1. A guinea pig polyclonal antibody against zebrafish Rx1 was generated by Open Biosystems with the following peptides: aa 63-83 (KVGGESLGEPGKLDQRVQPYGH) and aa 109-126 (GDLGDLRKAIESKSKSPD). Zebrafish rlbp1a (Collery et al., 2008) was used for in situ hybridization (Raymond et al., 2006).
To label proliferating cells, fish were placed in aquarium system water with the following thymidine analogs for up to 2 days: 1 mM 5-bromo-2′-deoxyuridine (BrdU; Sigma) buffered with 10 mM HEPES (pH 7.2), or 125 μM or 250 μM 5-ethynyl-2′-deoxyuridine (EdU; Invitrogen).
Isolated retinas were fixed (Barthel and Raymond, 1990), rinsed, treated with 2 N HCl in PBS with 0.5% Triton X-100 for 45 minutes and rinsed with 3% bovine serum albumin (BSA) in PBS/1% Tween/1% Triton X-100. To detect EdU, retinas were incubated with Click-iT reaction mixture (Invitrogen) for 45 minutes, rinsed with PBS/1% Tween/1% Triton X-100, blocked for 2 hours in 10% normal goat serum/1% Triton X-100/1% Tween 20/1% DMSO, and incubated in primary and secondary antibodies overnight at room temperature. BrdU detection was as previously described (Bernardos et al., 2007).
Microscopy and imaging
Immunofluorescence images were collected on a Zeiss AxioImager with ApoTome and AxioCam (Carl Zeiss Microimaging) or a Leica SP5 laser scanning confocal microscope (Leica Microsystems). Contrast of the entire image was adjusted with Adobe Photoshop (Adobe Systems). Using ImageJ (National Institutes of Health), the number of PCNA+ and HuC/D+ nuclei at 100 μm or 400 μm dorsal to the optic disc, respectively, was counted in radial retinal cryosections. All pH3+ nuclei were counted in radial retinal cryosections containing the optic disc in three to four sections per retina from four fish. GFP+ Müller glia and regenerated cone photoreceptors were counted in the temporal region of flat-mounted retina (six or three retinas from three fish, respectively). All BLBP+/PCNA+ Müller glia were counted in two radial cryosections per retina from four fish. Statistical significance was determined by ANOVA with Tukey’s post-hoc test with JMP statistics software (SAS Institute).
RT-PCR
The cdh2m117 allele has a T-to-G point mutation introducing a BsmBI site (Malicki et al., 2003). Total RNA was isolated (TRIzol RNA Isolation Regent; Invitrogen) and reverse transcribed (Superscript II Reverse Transcriptase; Invitrogen). PCR products amplified with cadherin-2 specific primers (5′-ccgagggacaagttctgctc-3′: 5′-tactgaggcaccttctcacg-3) were digested with BsmBI; three biological replicates, two retinas per sample.
Supplementary Material
Acknowledgments
We thank Dilip Pawar for fish husbandry; Shoji Kawamura for zebrafish opsin transgenic lines and plasmid constructs; and Breandan Kennedy for the rlbp1a plasmid. The cdh2m117 mutant line was obtained from the Zebrafish International Resource Center.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
P.A.R. and M.N. designed the experiments and wrote the paper; M.N. performed the experiments with assistance from L.K.B.
Funding
Supported by the National Institutes of Health [R01EY004318 to P.A.R.]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.090738/-/DC1
References
- Alvarez-Buylla A., Lim D. A. (2004). For the long run: maintaining germinal niches in the adult brain. Neuron 41, 683–686 [DOI] [PubMed] [Google Scholar]
- Barthel L. K., Raymond P. A. (1990). Improved method for obtaining 3-microns cryosections for immunocytochemistry. J. Histochem. Cytochem. 38, 1383–1388 [DOI] [PubMed] [Google Scholar]
- Bernardos R. L., Raymond P. A. (2006). GFAP transgenic zebrafish. Gene Expr. Patterns 6, 1007–1013 [DOI] [PubMed] [Google Scholar]
- Bernardos R. L., Barthel L. K., Meyers J. R., Raymond P. A. (2007). Late-stage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. J. Neurosci. 27, 7028–7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braisted J. E., Raymond P. A. (1992). Regeneration of dopaminergic neurons in goldfish retina. Development 114, 913–919 [DOI] [PubMed] [Google Scholar]
- Bringmann A., Pannicke T., Grosche J., Francke M., Wiedemann P., Skatchkov S. N., Osborne N. N., Reichenbach A. (2006). Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 25, 397–424 [DOI] [PubMed] [Google Scholar]
- Campbell K., Götz M. (2002). Radial glia: multi-purpose cells for vertebrate brain development. Trends Neurosci. 25, 235–238 [DOI] [PubMed] [Google Scholar]
- Cayouette M., Poggi L., Harris W. A. (2006). Lineage in the vertebrate retina. Trends Neurosci. 29, 563–570 [DOI] [PubMed] [Google Scholar]
- Cerveny K. L., Varga M., Wilson S. W. (2012). Continued growth and circuit building in the anamniote visual system. Dev. Neurobiol. 72, 328–345 [DOI] [PubMed] [Google Scholar]
- Chuang J. C., Mathers P. H., Raymond P. A. (1999). Expression of three Rx homeobox genes in embryonic and adult zebrafish. Mech. Dev. 84, 195–198 [DOI] [PubMed] [Google Scholar]
- Collery R., McLoughlin S., Vendrell V., Finnegan J., Crabb J. W., Saari J. C., Kennedy B. N. (2008). Duplication and divergence of zebrafish CRALBP genes uncovers novel role for RPE- and Muller-CRALBP in cone vision. Invest. Ophthalmol. Vis. Sci. 49, 3812–3820 [DOI] [PubMed] [Google Scholar]
- Diekmann H., Stuermer C. A. (2009). Zebrafish neurolin-a and -b, orthologs of ALCAM, are involved in retinal ganglion cell differentiation and retinal axon pathfinding. J. Comp. Neurol. 513, 38–50 [DOI] [PubMed] [Google Scholar]
- Fashena D., Westerfield M. (1999). Secondary motoneuron axons localize DM-GRASP on their fasciculated segments. J. Comp. Neurol. 406, 415–424 [DOI] [PubMed] [Google Scholar]
- Fausett B. V., Goldman D. (2006). A role for alpha1 tubulin-expressing Müller glia in regeneration of the injured zebrafish retina. J. Neurosci. 26, 6303–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimbel S. M., Montgomery J. E., Burket C. T., Hyde D. R. (2007). Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. J. Neurosci. 27, 1712–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Bongini R. (2010). Turning Müller glia into neural progenitors in the retina. Mol. Neurobiol. 42, 199–209 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., Reh T. A. (2003). Potential of Müller glia to become neurogenic retinal progenitor cells. Glia 43, 70–76 [DOI] [PubMed] [Google Scholar]
- Fleisch V. C., Schonthaler H. B., von Lintig J., Neuhauss S. C. (2008). Subfunctionalization of a retinoid-binding protein provides evidence for two parallel visual cycles in the cone-dominant zebrafish retina. J. Neurosci. 28, 8208–8216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosens I., den Hollander A. I., Cremers F. P., Roepman R. (2008). Composition and function of the Crumbs protein complex in the mammalian retina. Exp. Eye Res. 86, 713–726 [DOI] [PubMed] [Google Scholar]
- Götz M., Huttner W. B. (2005). The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777–788 [DOI] [PubMed] [Google Scholar]
- Grandel H., Brand M. (2013). Comparative aspects of adult neural stem cell activity in vertebrates. Dev. Genes Evol. 223, 131–147 [DOI] [PubMed] [Google Scholar]
- Harrington M. J., Hong E., Fasanmi O., Brewster R. (2007). Cadherin-mediated adhesion regulates posterior body formation. BMC Dev. Biol. 7, 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock P., Ochocinska M., Sieh A., Otteson D. (2004). Persistent and injury-induced neurogenesis in the vertebrate retina. Prog. Retin. Eye Res. 23, 183–194 [DOI] [PubMed] [Google Scholar]
- Jadhav A. P., Roesch K., Cepko C. L. (2009). Development and neurogenic potential of Müller glial cells in the vertebrate retina. Prog. Retin. Eye Res. 28, 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannie K. M., Stipp C. S., Weiner J. A. (2012). ALCAM regulates motility, invasiveness, and adherens junction formation in uveal melanoma cells. PLoS ONE 7, e39330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson P. A., Cappello S., Götz M. (2010). Stem cells niches during development - lessons from the cerebral cortex. Curr. Opin. Neurobiol. 20, 400–407 [DOI] [PubMed] [Google Scholar]
- Karl M. O., Reh T. A. (2010). Regenerative medicine for retinal diseases: activating endogenous repair mechanisms. Trends Mol. Med. 16, 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizil C., Kaslin J., Kroehne V., Brand M. (2012). Adult neurogenesis and brain regeneration in zebrafish. Dev. Neurobiol. 72, 429–461 [DOI] [PubMed] [Google Scholar]
- Kriegstein A., Alvarez-Buylla A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski J. R., Qin Z., Sifuentes C. J., Thummel R., Soto C. M., Moens C. B., Raymond P. A. (2013). Retinal regeneration in adult zebrafish requires regulation of TGFβ signaling. Glia 61, 1687–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung L., Klopper A. V., Grill S. W., Harris W. A., Norden C. (2011). Apical migration of nuclei during G2 is a prerequisite for all nuclear motion in zebrafish neuroepithelia. Development 138, 5003–5013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Londraville R. L., Azodi E., Babb S. G., Chiappini-Williamson C., Marrs J. A., Raymond P. A. (2002). Up-regulation of cadherin-2 and cadherin-4 in regenerating visual structures of adult zebrafish. Exp. Neurol. 177, 396–406 [DOI] [PubMed] [Google Scholar]
- Malicki J., Jo H., Pujic Z. (2003). Zebrafish N-cadherin, encoded by the glass onion locus, plays an essential role in retinal patterning. Dev. Biol. 259, 95–108 [DOI] [PubMed] [Google Scholar]
- Meyers J. R., Hu L., Moses A., Kaboli K., Papandrea A., Raymond P. A. (2012). β-catenin/Wnt signaling controls progenitor fate in the developing and regenerating zebrafish retina. Neural Dev. 7, 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery J. E., Parsons M. J., Hyde D. R. (2010). A novel model of retinal ablation demonstrates that the extent of rod cell death regulates the origin of the regenerated zebrafish rod photoreceptors. J. Comp. Neurol. 518, 800–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. M., Gorsuch R. A., Bailey T. J., Ackerman K. M., Kassen S. C., Hyde D. R. (2012). Stat3 defines three populations of Müller glia and is required for initiating maximal müller glia proliferation in the regenerating zebrafish retina. J. Comp. Neurol. 520, 4294–4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofori-Acquah S. F., King J., Voelkel N., Schaphorst K. L., Stevens T. (2008). Heterogeneity of barrier function in the lung reflects diversity in endothelial cell junctions. Microvasc. Res. 75, 391–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z., Barthel L. K., Raymond P. A. (2009). Genetic evidence for shared mechanisms of epimorphic regeneration in zebrafish. Proc. Natl. Acad. Sci. USA 106, 9310–9315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Reifler A., Parent J. M., Goldman D. (2010a). Conditional gene expression and lineage tracing of tuba1a expressing cells during zebrafish development and retina regeneration. J. Comp. Neurol. 518, 4196–4212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Fausett B. V., Goldman D. (2010b). Ascl1a regulates Müller glia dedifferentiation and retinal regeneration through a Lin-28-dependent, let-7 microRNA signalling pathway. Nat. Cell Biol. 12, 1101–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Zhao X. F., Goldman D. (2011). Ascl1a/Dkk/β-catenin signaling pathway is necessary and glycogen synthase kinase-3β inhibition is sufficient for zebrafish retina regeneration. Proc. Natl. Acad. Sci. USA 108, 15858–15863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond P. A., Hitchcock P. F. (2000). How the neural retina regenerates. Results Probl. Cell Differ. 31, 197–218 [DOI] [PubMed] [Google Scholar]
- Raymond P. A., Rivlin P. K. (1987). Germinal cells in the goldfish retina that produce rod photoreceptors. Dev. Biol. 122, 120–138 [DOI] [PubMed] [Google Scholar]
- Raymond P. A., Reifler M. J., Rivlin P. K. (1988). Regeneration of goldfish retina: rod precursors are a likely source of regenerated cells. J. Neurobiol. 19, 431–463 [DOI] [PubMed] [Google Scholar]
- Raymond P. A., Barthel L. K., Bernardos R. L., Perkowski J. J. (2006). Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev. Biol. 6, 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich K. A., Figueroa S. L., Zhan Y., Blanks J. C. (1995). Effects of Müller cell disruption on mouse photoreceptor cell development. Exp. Eye Res. 61, 235–248 [DOI] [PubMed] [Google Scholar]
- Roesch K., Jadhav A. P., Trimarchi J. M., Stadler M. B., Roska B., Sun B. B., Cepko C. L. (2008). The transcriptome of retinal Müller glial cells. J. Comp. Neurol. 509, 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki D. J. (2012). Sticky situations: recent advances in control of cell adhesion during neuronal migration. Curr. Opin. Neurobiol. 22, 791–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenkamp D. L. (2011). The rod photoreceptor lineage of teleost fish. Prog. Retin. Eye Res. 30, 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna E., Huttner W. B. (2010). Neural progenitor nuclei IN motion. Neuron 67, 906–914 [DOI] [PubMed] [Google Scholar]
- Thummel R., Kassen S. C., Enright J. M., Nelson C. M., Montgomery J. E., Hyde D. R. (2008). Characterization of Müller glia and neuronal progenitors during adult zebrafish retinal regeneration. Exp. Eye Res. 87, 433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R., Enright J. M., Kassen S. C., Montgomery J. E., Bailey T. J., Hyde D. R. (2010). Pax6a and Pax6b are required at different points in neuronal progenitor cell proliferation during zebrafish photoreceptor regeneration. Exp. Eye Res. 90, 572–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai Wong C., Dye D. E., Coombe D. R. (2012). The role of immunoglobulin superfamily cell adhesion molecules in cancer metastasis. Int. J. Cell Biol. 2012, 340296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J., Ramachandran R., Goldman D. (2012). HB-EGF is necessary and sufficient for Müller glia dedifferentiation and retina regeneration. Dev. Cell 22, 334–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.