

The crystal structure of CT584 from C. trachomatis is presented at 3.05 Å resolution.
Keywords: CT584, Chlamydia trachomatis, type III secretion system
Abstract
Chlamydia trachomatis is a major cause of various diseases, including blinding trachoma and pelvic inflammatory disease, and is the leading reported sexually transmitted bacterial infection worldwide. All pathogenic Chlamydiae spp. utilize a supramolecular syringe, or type III secretion system (T3SS), to inject proteins into their obligate host in order to propagate infection. Here, the structure of CT584, a T3SS-associated protein, that has been refined to a resolution of 3.05 Å is reported. The CT584 structure is a hexamer comprised of a trimer of dimers. The structure shares a high degree of similarity to the recently reported structure of an orthologous protein, Cpn0803, from Chlamydia pneumoniae, which highlights the highly conserved nature of this protein across these chlamydial species, despite different tissue tropism and disease pathology.
1. Introduction
Chlamydia trachomatis, an obligate intracellular bacterial pathogen, is the leading cause of infectious blindness and trachoma in underdeveloped countries as well as the most prevalent cause of sexually transmitted disease worldwide (Schachter, 1978 ▶; Thylefors et al., 1995 ▶; Gerbase et al., 1998 ▶). Chlamydia are phylogenetically distant organisms characterized by a high percentage of proteins of unknown function (hypothetical proteins). Type III secretion systems (T3SSs) are a common virulence determinant of pathogenic Gram-negative bacteria (Hueck, 1998 ▶). The T3SS injectisome is an energy-dependent, unidirectional supramolecular syringe that facilitates the transport of host-altering effector proteins into the cytosol during infection (Galán & Wolf-Watz, 2006 ▶). Despite the fact that Chlamydia have been demonstrated to utilize T3SS during infection (Peters et al., 2007 ▶), numerous factors essential to T3SSs have yet to be identified, including the needle-tip protein. The needle-tip protein localizes to the distal end of the T3SS apparatus, where it can sense environmental stimuli, controlling the secretion state of the injectisome (Espina et al., 2006 ▶; Olive et al., 2007 ▶; Stensrud et al., 2008 ▶; Barta et al., 2012 ▶).
Previous work by our group has demonstrated that CT584 shares biophysical characteristics with other needle-tip proteins (Markham et al., 2009 ▶), such as IpaD from Shigella flexneri (Johnson et al., 2007 ▶; Epler et al., 2012 ▶) and LcrV from Yersinia pestis (Chaudhury et al., 2013 ▶). Since then, CT584 (or its homolog in C. pneumoniae, Cpn0803) has been implicated in protein–protein interactions with T3SS core components such as the putative needle protein CdsF (Spaeth et al., 2009 ▶) and both the T3SS ATPase CdsN and C-ring CdsQ (Stone et al., 2012 ▶), and even as a T3SS chaperone, stabilizing a novel effector of unknown function CT082 (Pais et al., 2013 ▶). The 2.0 Å resolution crystal structure of Cpn0803 has recently been reported, revealing a unique hexameric assembly (Stone et al., 2012 ▶). Here, we report the refined structure of CT584 at 3.05 Å resolution, highlighting the structural similarities between CT584 and Cpn0803, despite the distinct tissue tropisms of each chlamydial species. Additionally, the interfaces involved in oligomer assembly are discussed in detail.
2. Experimental procedures
2.1. Protein expression and purification
The cloning, expression and purification of CT584 have previously been reported (Markham et al., 2009 ▶). Briefly, CT584 was PCR-amplified from C. trachomatis 434/Bu genomic DNA, restriction digested and ligated into pET-21b (Invitrogen). Following sequence confirmation, this vector was transformed into Escherichia coli BL21(DE3) cells, which were grown to an OD600 of ∼1.0 at 310 K and induced overnight at 289 K with 1 mM IPTG. Cells were harvested by centrifugation, lysed with a sonicator and clarified by high-speed centrifugation. Immobilized metal-ion affinity and gel-filtration chromatography were utilized in order to obtain >95% homogenous protein, which was dialyzed into 40 mM Tris pH 8.0, 500 mM NaCl, 5%(v/v) glycerol and concentrated to 21.9 mg ml−1.
2.2. Crystallization
Full-length recombinant CT584 proved recalcitrant to crystallization attempts. In situ proteolysis was utilized with purified CT584 and trace amounts of α-chymotrypsin prior to screening for crystallization in Compact Jr (Emerald BioSystems) sitting-drop vapor-diffusion plates at 293 K using equal volumes of crystallization solution and reservoir solution equilibrated against 75 µl of the latter. Prismatic crystals (Fig. 1 ▶ a) were obtained in approximately 1–2 d from Natrix screen (Hampton Research) condition No. 27 [30%(v/v) PEG 8000, 50 mM sodium cacodylate pH 6.5, 200 mM ammonium acetate, 10 mM magnesium acetate]. Samples were transferred into a fresh drop containing 75% crystallization solution and 25% PEG 400 before cooling in liquid nitrogen for data collection. Diffraction data were collected on the Advanced Photon Source IMCA-CAT beamline 17ID using a Dectris PILATUS 6M pixel-array detector (Fig. 1 ▶ b).
Figure 1.

(a) Prismatic crystals of CT584 obtained from Hampton Natrix screen condition No. 27 upon in situ proteolysis with α-chymotrypsin. Crystals were obtained in 1–2 d. (b) Representative X-ray diffraction pattern of CT584 crystals at ϕ = 90°. The crystal was exposed for 0.2 s over a 0.2° oscillation range. The edges of the detector correspond to 1.65 Å in the x axis and 2.40 Å in the y axis.
2.3. X-ray diffraction, data collection and processing
X-ray diffraction data were integrated and scaled using the XDS (Kabsch, 2010 ▶) and SCALA (Evans, 2011 ▶) packages, respectively. The Laue class was checked with POINTLESS (Evans, 2006 ▶), which indicated that most probable class was 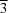 m with space group H32. The coordinates from a homologous structure, Cpn0803 (PDB entry 3q9d; Stone et al., 2012 ▶), were used in a molecular-replacement search with Phaser (McCoy et al., 2007 ▶), searching for a single molecule in the asymmetric unit. Structure refinement and manual model building were performed with PHENIX (Adams et al., 2002 ▶, 2010 ▶) and Coot (Emsley & Cowtan, 2004 ▶; Emsley et al., 2010 ▶), respectively. The Cpn0803 structure was used as a reference model during refinement. Structure validation was carried out using MolProbity (Chen et al., 2010 ▶). Additional information and refinement statistics are presented in Table 1 ▶.
m with space group H32. The coordinates from a homologous structure, Cpn0803 (PDB entry 3q9d; Stone et al., 2012 ▶), were used in a molecular-replacement search with Phaser (McCoy et al., 2007 ▶), searching for a single molecule in the asymmetric unit. Structure refinement and manual model building were performed with PHENIX (Adams et al., 2002 ▶, 2010 ▶) and Coot (Emsley & Cowtan, 2004 ▶; Emsley et al., 2010 ▶), respectively. The Cpn0803 structure was used as a reference model during refinement. Structure validation was carried out using MolProbity (Chen et al., 2010 ▶). Additional information and refinement statistics are presented in Table 1 ▶.
Table 1. Crystallographic data for CT584 refined to 3.05 Å resolution.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Unit-cell parameters (Å, °) | a = b = 112.24, c = 82.07, α = β = 90, γ = 120 |
| Space group | H32 |
| Resolution (Å) | 62.71–3.05 (3.21–3.05) |
| Wavelength (Å) | 1.0000 |
| Temperature (K) | 100 |
| Observed reflections | 38926 |
| Unique reflections | 3900 |
| 〈I/σ(I)〉 | 20.3 (3.6) |
| Completeness (%) | 100 (100) |
| Multiplicity | 10.0 (10.5) |
| R merge † (%) | 9.3 (71.3) |
| R meas ‡ (%) | 9.8 (74.9) |
| R p.i.m. § (%) | 3.1 (23.0) |
| Refinement | |
| Resolution (Å) | 32.40–3.05 |
| Reflections (working/test) | 3717/178 |
| R work/R free ¶ (%) | 22.5/25.3 |
| No. of non-H atoms (protein) | 1243 |
| Model quality | |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.074 |
| Average B factor (Å2) | |
| All atoms | 75.4 |
| Coordinate error based on maximum likelihood (Å) | 0.36 |
| Ramachandran plot | |
| Favored (%) | 97.9 |
| Allowed (%) | 2.1 |
| PDB code | 4mlk |
R
merge = 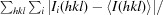
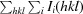 , where Ii(hkl) is the intensity measured for the ith reflection and 〈I(hkl)〉 is the average intensity of all reflections with indices hkl.
, where Ii(hkl) is the intensity measured for the ith reflection and 〈I(hkl)〉 is the average intensity of all reflections with indices hkl.
R meas is the redundancy-independent (multiplicity-weighted) R merge (Evans, 2006 ▶).
R p.i.m. is the precision-indicating (multiplicity-weighted) R merge (Weiss, 2001 ▶).
R factor = 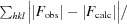
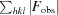 ; R
free is calculated in an identical manner using a randomly selected 5% of reflections that were not included in the refinement.
; R
free is calculated in an identical manner using a randomly selected 5% of reflections that were not included in the refinement.
2.4. Multiple sequence alignments and figure modeling
Multiple sequence alignments were carried out using ClustalW (Thompson et al., 1994 ▶) and were aligned with secondary-structure elements using ESPript (Gouet et al., 1999 ▶). Three-dimensional structures were superimposed using the local–global alignment method (LGA; Zemla, 2003 ▶). Representations of all structures were generated using PyMOL (DeLano, 2002 ▶).
3. Results and discussion
The final model of CT584 was refined to 3.05 Å resolution in space group H32 with a single polypeptide in the asymmetric unit (Fig. 2 ▶ a). Strong model–map correlation was observed for residues Thr14–Thr180 (Fig. 2 ▶ b). However, the following solvent-exposed residues within loop regions of CT584 could not be modeled owing to poor electron density: Gln47–Gly48, Leu66–Gln73 and Ala110–Arg114. In addition, potentially owing to the use of limited proteolysis in order to crystallize CT548, residues at both the amino-terminus (Met1–Asn13) and carboxy-terminus (Lys181–Val183) could not be modeled due to poor electron density. Overall, the structure of CT584 is highly similar to the recently determined structure of Cpn0803 (PDB entry 3q9d; Stone et al., 2012 ▶), an ortholog of CT584 from C. pneumoniae, with an r.m.s.d. of 0.55 Å over 152/175 Cα residues (Fig. 2 ▶ c) as determined using the LGA server (Zemla, 2003 ▶). Given the high level of sequence identity between these two proteins (88.8% of the aligned region), the observed structural similarity is not surprising. In contrast, the structure of CT584 aligns very poorly with that of LcrV from Y. pestis (PDB entry 4jbu; Chaudhury et al., 2013 ▶), with an r.m.s.d. of 3.13 Å over just 47/152 Cα residues, suggesting that CT584 is either not a bona fide T3SS needle-tip protein or that this function has been retained in the absence of structural conservation.
Figure 2.

Crystal structure of CT584 (residues Thr14–Thr180) refined to 3.05 Å resolution. (a) A single polypeptide is found in the asymmetric unit. The cartoon ribbon format is colored according to secondary structure, with α-helices in orange and β-strands in cyan. Loop regions that were unmodeled owing to poor electron density are represented by dashed lines and the solid arrow highlights the π-bulge. (b) OMIT F o − F c weighted electron-density map (contoured at 3.0σ, omitting residues 95–100 and 123–127), calculated using PHENIX (Adams et al., 2002 ▶, 2010 ▶), represented as an orange cage around the region highlighted (dashed lines) in (a). (c) Superposition of of CT584 (cyan) and Cpn0803 (purple). The calculated r.m.s.d. was 0.53 Å over 152/175 Cα atoms within 5.0 Å. The alignment is rotated 180° about the vertical axis with respect to (a). Cpn0803 terminal residues are labeled.
The structure of CT584 (Fig. 2 ▶ a) consists of a four α-helix bundle at the N-terminus (α1–α2–α3–α4; residues Phe15–Ala27, Thr33–Cys45, Gln50–Asn62 and Gln75–His96, respectively), followed by a three-stranded antiparallel β-sheet (β1–β2–β3; residues Asn102–Leu109, Val116–Arg123 and Gly126–Thr133, respectively) and finally two α-helices arranged in a kinked antiparallel manner at the C-terminus (α5-α6; residues Val135–Lys154 and Pro156–Leu179, respectively). This kink bends α6 by nearly 82° (as measured through the Cα atoms of Glu163, Glu164 and Glu167; Fig. 2 ▶ a) and forms a dimerization interface with the twofold crystallographic symmetry mate. Given the absence of Pro residues within this region of α6, it appears that electrostatic interactions between Asp168 and His140/Arg143 (detailed below) stabilize this noncanonical kink (Barlow & Thornton, 1988 ▶). The overall topology of CT584 is α1–α2–α3–α4–β1–β2–β3–α5–α6.
Despite the presence of a single polypeptide in the asymmetric unit, the most likely oligomeric state of CT584, as predicted using the protein-interface server PISA (Krissinel & Henrick, 2007 ▶), was that of a dimer and a hexamer. The dimeric state of CT584 (Fig. 3 ▶ a) is formed with a polypeptide related by a crystallographic twofold operator (−x, −x + y, −z + 1). The C-terminal two-helix bundle of each polypeptide intercalates with the other, burying 2837.9 Å2 of each monomer, or nearly 25% of the available surface area contributed by each polypeptide. A network of electrostatic interactions and hydrogen bonds stabilizes this interface through a set of paired side chains from each twofold crystallographic symmetry mate (Fig. 3 ▶ b). Additional hydrophobic interactions are observed throughout the antiparallel β-sheet and α-helix bundle that further stabilize the dimeric state of CT584. Within the intercalated two-helix bundles, side chain–side chain interactions occur between each polypeptide. Asp168 of α6 forms salt bridges with the guanidinium group of Arg143 and the imidazole group of His140 as well as a hydrogen bond to the sulfhydryl group of Cys139, all of which are located on α5 of the neighboring symmetry mate. A hydrogen-bonding network, primarily backbone–backbone interactions between the side chain of Arg123 and Glu99 O, between Met132 O and Ile101 N and between Met132 N and Leu130 O, between antiparallel β-sheets further stabilizes the dimer interface. In the 47 available sequences of CT584 orthologs (Supplementary Fig. S11), all of the aforementioned amino acids are invariantly conserved, with the exception of those involving backbone interactions and Asp168, which is also an acidic residue (Glu) in C. pneumoniae strains. While this level of conservation supports the biophysical importance of dimerization to protein function, there is an extensive level of overall sequence conservation (>80% identity) among all Chlamydiaceae orthologs (Supplementary Fig. S1). Also of note, the sequence of the encoded CT584 protein is highly conserved but is found only within the Chlamydiaceae family and is not encoded by other species within the Chlamydiae phylum (e.g. Parachlamydiaceae). Associated species of this family predominately infect mammals and birds, suggesting a more specific biological role within these hosts.
Figure 3.

Dimerization of CT584. (a) The dimer interface as predicted by PISA is formed by a CT584 polypeptide in the asymmetric unit (orange) with a polypeptide related by a crystallographic twofold operator (cyan). (b) Amino-acid side chains relevant to dimer formation from each polypeptide are shown in ball-and-stick representation. Side chains are colored according to bonding interaction (red dashed line), with salt bridges in magenta and hydrogen bonds in lime. The backbone coloring is as in (a). The interface is highlighted in (a) (dashed lines) and is rotated 90° counterclockwise.
The CT584 hexamer is composed of a trimer of dimers, with each dimeric assembly related by the crystallographic threefold operators (−y, x − y, z) and (−x + y, −x, z). While this trimeric dimer interface only buries ∼5% of the available polypeptide surface area (625.1 Å2) at each of the six symmetrical sites, numerous hydrogen bonds and a single salt bridge stabilize hexameric CT584 (Fig. 4 ▶ b). These interactions occur between each antiparallel β-sheet and α2, with Arg36 forming electrostatic interactions with Asp107 as well as hydrogen bonding to Thr105. Further interactions within this region include Asn102–Glu37, Glu128–Thr33 (backbone and side chain) and Glu128–Tyr32 backbone bonding. Finally, a single hydrogen bond between Lys24 of α1 and the backbone O atom of Val82 stabilizes each four-α-helix bundle to its symmetry mate. This final interaction occurs at a kink in α4 introduced by a π-bulge (Fig. 2 ▶ a), which results from the insertion of an extra amino acid into an α-helix and is commonly found at functional sites within proteins (Weaver, 2000 ▶). As seen for the CT584 dimer interface, all of these amino acids are invariantly conserved (Supplementary Fig. S1), with the exception of Val82 (Ala or Thr in other chlamydial orthologs). This suggests that the structural nature of this α-helix (i.e. the presence of a π-bulge and the resulting kinked α-helix) is more important than the type of side chain that is present. Given the high degree of structural similarity between the CT584 hexamer and the Cpn0803 hexamer (Supplementary Fig. S2; Stone et al., 2012 ▶), the biological unit of CT584 appears to exist in a hexameric state (Fig. 4 ▶ a) rather than only as a dimer, which is supported by previous size-exclusion chromatography of CT584 (Markham et al., 2009 ▶).
Figure 4.

Oligomerization of CT584. (a) The hexamer interface as predicted by PISA is formed by six polypeptides related by a crystallographic twofold operator (dimer; Fig. 3 ▶) and by two crystallographic threefold operators of that dimer forming a trimer of dimers (hexamer). Each dimer pair is independently colored. (b) Amino-acid side chains relevant to dimer trimerization (hexamer) from each polypeptide dimer are shown in ball-and-stick representation. Side chains are colored according to bonding interactions (red dashed lines), with salt bridges in magenta and hydrogen bonds in cyan. The backbone coloring is as in (a). The interface is highlighted in (a) (solid arrow and dashed lines) and is rotated 90° clockwise about the vertical axis. (c) Proposed steps in the oligomerization of CT584. Each polypeptide is shown as a surface representation and colored as in (a).
CT584 would seem to be most stable in dimeric and hexameric assemblies (Fig. 4 ▶ c) based upon the lowest energy ΔG values as judged by PISA. Thus, it appears likely that CT584 assembles as a trimer of dimers rather than a dimer of trimers, which is consistent with the reported oligomeric assembly of Cpn0803 (Stone et al., 2012 ▶). Unfortunately, the structure of CT584 provides relatively little information with regard to insight into its biological role or functional annotation within Chlamydia. Monomeric, dimeric and hexameric forms of CT584 were used to search for structurally related motifs within proteins deposited in the Protein Data Bank using the DALI server (Holm & Rosenström, 2010 ▶). The only statistically significant hit (Z-score > 8.0) was Cpn0803, the previously mentioned C. pneumoniae ortholog. As previously highlighted, numerous reports have demonstrated protein–protein interactions between CT584 and T3SS-related proteins, including both apparatus components and putative effectors. While structural homology does not support a role as either T3SS machinery or as a chaperone, a hypothesis confluent with these previous reports is that CT584 is a secreted effector protein. If CT584 is secreted through the type III secretion system, folding and proper assembly of the oligomeric structure will likely depend on local concentration and interactions with host cell partners. Future studies to determine whether CT584 is secreted in a T3SS-dependent manner and what oligomeric state is adopted following secretion will be very informative in regard to the potential biological function and relevance of the reported hexameric structure.
Supplementary Material
PDB reference: CT584, 4mlk
Supplementary material file. DOI: 10.1107/S1744309113027371/sw5067sup1.pdf
Acknowledgments
Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman–Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. MLB, LMH, KEK and PSH were supported in part by National Institutes of Health grants AI079083 and RR17708.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: SW5067).
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Barlow, D. J. & Thornton, J. M. (1988). J. Mol. Biol. 201, 601–619. [DOI] [PubMed]
- Barta, M. L., Guragain, M., Adam, P., Dickenson, N. E., Patil, M., Geisbrecht, B. V., Picking, W. L. & Picking, W. D. (2012). Proteins, 80, 935–945. [DOI] [PMC free article] [PubMed]
- Chaudhury, S., Battaile, K. P., Lovell, S., Plano, G. V. & De Guzman, R. N. (2013). Acta Cryst. F69, 477–481. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Epler, C. R., Dickenson, N. E., Bullitt, E. & Picking, W. L. (2012). J. Mol. Biol. 420, 29–39. [DOI] [PMC free article] [PubMed]
- Espina, M., Olive, A. J., Kenjale, R., Moore, D. S., Ausar, S. F., Kaminski, R. W., Oaks, E. V., Middaugh, C. R., Picking, W. D. & Picking, W. L. (2006). Infect. Immun. 74, 4391–4400. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Galán, J. E. & Wolf-Watz, H. (2006). Nature (London), 444, 567–573. [DOI] [PubMed]
- Gerbase, A. C., Rowley, J. T. & Mertens, T. E. (1998). Lancet, 351, Suppl. 3, 2–4. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Hueck, C. J. (1998). Microbiol. Mol. Biol. Rev. 62, 379–433. [DOI] [PMC free article] [PubMed]
- Johnson, S., Roversi, P., Espina, M., Olive, A., Deane, J. E., Birket, S., Field, T., Picking, W. D., Blocker, A. J., Galyov, E. E., Picking, W. L. & Lea, S. M. (2007). J. Biol. Chem. 282, 4035–4044. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Markham, A. P., Jaafar, Z. A., Kemege, K. E., Middaugh, C. R. & Hefty, P. S. (2009). Biochemistry, 48, 10353–10361. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Olive, A. J., Kenjale, R., Espina, M., Moore, D. S., Picking, W. L. & Picking, W. D. (2007). Infect. Immun. 75, 2626–2629. [DOI] [PMC free article] [PubMed]
- Pais, S. V., Milho, C., Almeida, F. & Mota, L. J. (2013). PLoS One, 8, e56292. [DOI] [PMC free article] [PubMed]
- Peters, J., Wilson, D. P., Myers, G., Timms, P. & Bavoil, P. M. (2007). Trends Microbiol. 15, 241–251. [DOI] [PubMed]
- Schachter, J. (1978). N. Engl. J. Med. 298, 490–495. [DOI] [PubMed]
- Spaeth, K. E., Chen, Y.-S. & Valdivia, R. H. (2009). PLoS Pathog. 5, e1000579. [DOI] [PMC free article] [PubMed]
- Stensrud, K. F., Adam, P. R., La Mar, C. D., Olive, A. J., Lushington, G. H., Sudharsan, R., Shelton, N. L., Givens, R. S., Picking, W. L. & Picking, W. D. (2008). J. Biol. Chem. 283, 18646–18654. [DOI] [PMC free article] [PubMed]
- Stone, C. B., Sugiman-Marangos, S., Bulir, D. C., Clayden, R. C., Leighton, T. L., Slootstra, J. W., Junop, M. S. & Mahony, J. B. (2012). PLoS One, 7, e30220. [DOI] [PMC free article] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Thylefors, B., Négrel, A. D., Pararajasegaram, R. & Dadzie, K. Y. (1995). Bull. World Health Organ. 73, 115–121. [PMC free article] [PubMed]
- Weaver, T. M. (2000). Protein Sci. 9, 201–206. [DOI] [PMC free article] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Zemla, A. (2003). Nucleic Acids Res. 31, 3370–3374. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: CT584, 4mlk
Supplementary material file. DOI: 10.1107/S1744309113027371/sw5067sup1.pdf