

The tertiary structure of invertebrate-type lysozyme from the common orient clam was determined and compared with that of V. philippinarum lysozyme.
Keywords: i-type lysozyme, tertiary structure, invertebrates, Meretrix lusoria
Abstract
To evaluate the structure–function relationships of invertebrate lysozymes, a new invertebrate-type (i-type) lysozyme was isolated from the common orient clam (Meretrix lusoria) and the tertiary structure of this enzyme was determined. Comparison of the tertiary structure of this enzyme with those of chicken and Venerupi philippinarum lysozymes revealed that the location of the side chain of the second catalytic residue, an aspartic acid, and the N-acetylglucosamine trimer bound at subsites A–C were different. Furthermore, the amino acid electrostatically interacting with Asp30 in V. philippinarum lysozyme, Lys108, was substituted by Gly in M. lusoria lysozyme and no other possible amino acid that could contribute to this interaction was found in M. lusoria lysozyme. It therefore seems that the substitutions of the amino acids at the interface of the V. philippinarum lysozyme dimer are likely to change the oligomeric state of the M. lusoria lysozyme.
1. Introduction
Lysozyme (EC 3.2.1.17) is a widely distributed glycosidase classified into chicken (c), goose (g), bacterial and phage types. C-type lysozyme, which is one of the best characterized glycosidases, catalyzes the hydrolysis of the β-1,4-glycosidic bonds of heteropolymers of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid in bacterial cell walls and of the homopolymer of GlcNAc, chitin (Jollès & Jollès, 1975 ▶). The tertiary structure of this enzyme was the first to be analyzed by X-ray crystallography (Blake et al., 1965 ▶; Kelly et al., 1979 ▶; Pincus & Scheraga, 1981 ▶) and has been extensively studied. C-type lysozyme catalyzes not only the hydrolysis of sugar chains but also a transglycosylation reaction that requires an acceptor oligomer instead of a water molecule during hydrolysis (Smith-Gill et al., 1984 ▶; Pollock & Sharon, 1970 ▶; Inoue et al., 1992 ▶; Fukamizo et al., 1989 ▶). The c-type lysozyme hen egg-white lysozyme (HEL) contains six substrate-binding sites, subsites A–F (Pincus et al., 1977 ▶; Kuhara et al., 1982 ▶). The interaction of the HEL molecule and (GlcNAc)3 that is important in substrate binding and determining the cleavage specificity of HEL was revealed by the X-ray structure of the HEL–(GlcNAc)3 complex.
The additional classification invertebrate-type (i-type) lysozyme was reported by Ito, Yoshikawa et al. (1999 ▶). An invertebrate lysozyme was first reported in 1975 (Jollès & Jollès, 1975 ▶) and a complete structure was first determined for lysozyme from the marine bivalve Venerupis philippinarum (VPL) composed of 123 amino acids (Ito, Yoshikawa et al., 1999 ▶; Takeshita et al., 2003 ▶). The crystal structure of the VPL–(GlcNAc)3 complex at 1.6 Å resolution and mutational analyses demonstrated that Glu18 and Asp30 are candidate catalytic residues in VPL (Goto et al., 2007 ▶). The lytic activity of bivalve lysozymes was found to be sensitive to the ionic strength of the assay solution (Goto et al., 2007 ▶; Olsen et al., 2003 ▶). Analysis of the tertiary structure of VPL revealed a dimer formed by electrostatic interactions between the catalytic residues in one molecule and positive residues at the C-terminus of helix 6 in the other molecule. These findings show that the lysozyme activity of VPL is modulated by its quaternary structure. However, the molecular mechanism of i-type lysozymes is not as well characterized as that of c-type lysozymes.
In this study, we report the X-ray crystallographic structure of an i-type lysozyme from the common orient clam Meretrix lusoria (MLL) complexed with the substrate analogue (GlcNAc)3. We compared the structure of MLL with those of HEL and VPL. The results revealed that MLL has a largely similar structure to VPL, but that the location of the side chain of the second catalytic residue is different and amino-acid substitutions at the dimer-formation interface of VPL may be responsible for the lack of salt-dependent activation of MLL (Kuwano et al., 2013 ▶).
2. Materials and methods
2.1. Reagents and equipment
Ion-exchange chromatography was performed on CM-Toyopearl 650M (Tosoh). C4 reverse-phase high-performance liquid chromatography (RP-HPLC) was performed on a Hi-Pore RP304 reverse-phase column (4.6 × 250 mm; Bio-Rad). C18 RP-HPLC was performed on a YMC-Pack ODS-A column (120 Å, 5 µm, 4.6 × 250 mm; Yamamura Chemical). Crystallizations were performed using Crystal Screen and Crystal Screen 2 (Hampton Research) and the Wizard Cryo 1 and 2 (Emerald BioSystems) sparse-matrix crystallization kits.
Common orient clams (M. lusoria) of shell length 40–60 mm growing in the Ariake Sea were purchased in Kumamoto, Japan and stored at 253 K until use in purification.
2.2. Purification of lysozyme
One of the M. lusoria lysozyme isozymes (MLL-B) was purified according to the method described by Kuwano et al. (2013 ▶). Namely, the samples were homogenized in 2% acetic acid and centrifuged (5000g, 20 min, 277 K). The supernatant was combined and subjected to ammonium sulfate fractionation. The lysozyme was precipitated in 20–90% saturated ammonium sulfate. The precipitate was collected by centrifugation (5000g, 20 min, 277 K), dissolved in 50 mM phosphate buffer pH 7.0 to decrease the ammonium sulfate concentration to 0.1 M and subjected to chromatographic purification on a CM-Toyopearl 650M cation-exchange column (2 × 28 cm) equilibrated with 50 mM phosphate buffer pH 7.0. The column was washed with the same buffer and the bound protein was eluted by stepwise elution with 0.3 and 0.5 M NaCl in 50 mM phosphate buffer pH 7.0. The lysozyme fraction was purified using a C4 RP-HPLC column with a Jasco 800 series HPLC system (Japan Spectroscopic). The proteins were eluted by a gradient elution method using 0.1% trifluoroacetic acid (solvent A) and 60% acetonitrile in solvent A (solvent B). A gradient of 0–60% solvent B was achieved in 40 min. The eluted proteins were detected by monitoring the absorbance at 280 nm. One of the two obtained isozymes (MLL-B) was used as MLL. The resulting protein was lyophilized.
2.3. Protein crystallization and X-ray structure determination
Purified MLL was dissolved in distilled water containing 1.4 mM (GlcNAc)3 (10 mg ml−1). Conditions for crystallization of the complex were screened using screening kits such as Crystal Screen, Crystal Screen 2 and Wizard Cryo 1 and 2 at 293 K using the sitting-drop vapour-diffusion method. Initially, small crystals were grown using reagent No. 31 [35% 2-propanol, 5% polyethylene glycol (PEG) 1000, 0.1 M citrate buffer pH 5.5] of Wizard Cryo 1. This precipitant solution was taken as a starting point and was optimized by variation of the 2-propanol concentration; diffraction-quality crystals (maximum dimensions of 0.4 × 0.2 × 0.1 mm) were obtained within two weeks using a reservoir solution composed of 33% 2-propanol, 5% PEG 1000, 0.1 M citrate buffer pH 5.5.
Crystals were coated with a layer of viscous oil (Paratone-N) and transferred into a stream of nitrogen gas for data collection at 100 K. Diffraction data were collected at 1.78 Å resolution using monochromated radiation of wavelength 1.0 Å and an ADSC CCD detector system on beamline BL-17A at the Photon Factory, Tsukuba, Japan. The oscillation angle per image was set to 1°. The data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). The crystals belonged to the tetragonal space group P43212. A summary of the data statistics is presented in Table 1 ▶. The R merge value in the highest resolution shell (11.9%) indicates that the crystals diffracted to far better than 1.78 Å resolution. Refinement of the structure was performed by molecular replacement using VPL (PDB entry 2dqa; Goto et al., 2007 ▶) as the model, since VPL has 65% sequence identity to MLL. Further manual model building was accomplished using Coot (Emsley & Cowtan, 2004 ▶). Refinement was performed with REFMAC5 (Murshudov et al., 2011 ▶). The coordinates of (GlcNAc)3 were obtained from PDB entry 2dqa. The coordinates of the complex have been deposited in the Protein Data Bank as entry 3ab6. Molecular graphics were created using PyMOL (http://www.pymol.org/).
Table 1. Statistics of data collection, phase determination and refinement.
Values in parentheses are for the last resolution shell.
| Data collection | |
| Space group | P43212 |
| Unit-cell parameters (Å) | a = b = 41.6, c = 123.2 |
| Wavelength (Å) | 1.0 |
| X-ray source | BL-17A, Photon Factory |
| Resolution range (Å) | 50.0–1.78 |
| Total No. of reflections | 238430 |
| No. of unique reflections | 11004 |
| Multiplicity | 13.4 (12.8) |
| Completeness (%) | 99.2 (87.3) |
| R merge † (%) | 5.4 (11.9) |
| 〈I/σ(I)〉‡ | 23.1 |
| Refinement | |
| Resolution range (Å) | 39.4–1.78 |
| R cryst § (%) | 20.2 |
| R free ¶ (%) | 22.5 |
| No. of protein atoms | 925 |
| No. of water molecules | 185 |
| No. of (GlcNAc)3 molecules | 1 |
| R.m.s.d., bond lengths (Å) | 0.004 |
| R.m.s.d., bond angles (°) | 1.5 |
| Wilson B factor (Å2) | 15.5 |
| Overall B factor (Å2) | 17.5 |
| Average B factors (Å2) | |
| Protein atoms | 14.2 |
| Water molecules | 34.1 |
| (GlcNAc)3 molecule | 14.7 |
| Ramachandran quality†† | |
| Favoured regions (%) | 98.3 |
| Outliers (%) | 0.00 |
| PDB code | 3ab6 |
R
merge = 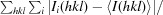
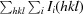 , where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is the mean intensity for this reflection.
, where I
i(hkl) is the ith intensity measurement of reflection hkl and 〈I(hkl)〉 is the mean intensity for this reflection.
For the highest resolution shell, the number of reflections with I/σ(I) < 1 was 18 (3.3%).
R
cryst = 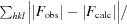
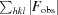 .
.
R free was calculated using randomly selected reflections (10%).
MolProbity (Chen et al., 2010 ▶) was used to monitor and validate the structural model.
3. Results and discussion
3.1. Crystal structure
Details of the purification of MLL have been reported in Kuwano et al. (2013 ▶) and the determined amino-acid sequence of MLL is shown in Fig. 1 ▶ together with that of VPL (UniProt accession No. Q9BLE0).
Figure 1.

Amino-acid sequence comparison of VPL and MLL. The proposed catalytic residues of VPL are indicated by filled triangles and the amino-acid residues contributing to dimer formation of VPL are indicated by unfilled triangles.
In order to compare the structure of MLL with that of VPL, the crystal structure of MLL was solved and analysed (Fig. 2 ▶).
Figure 2.

The overall structure of MLL with (GlcNAc)3. (GlcNAc)3 is indicated in purple. The two deduced catalytic residues (Glu18 and Asp29) from comparison with VPL and the substrate-binding subsites (A, B and C) from comparison with HEL are also indicated.
The established tertiary structure of MLL, together with those of HEL and VPL, is shown in Fig. 3 ▶. The catalytic Glu residue, which is essential as a proton donor for hydrolysis, is conserved at an equivalent position in all three lysozymes. However, the position of another catalytic residue, Asp, was not consistent in the three lysozymes. MLL and VPL have no Asp residue at the position corresponding to Asp52 of HEL; instead, they have Asp29 or Asp30 in a neighbouring β-sheet at a distance of 6.89 or 7.49 Å from the Cα atom, respectively (Fig. 3 ▶ a). Furthermore, the orientation of the side chain of Asp29 in MLL differs from that in VPL (Fig. 3 ▶ b). Glu35 of HEL can function as a general acid–base catalyst to donate a proton to the glycosyl O atom with a high pK a value of 6.2. In contrast, Asp52 stabilizes the oxocarbonium ion by interacting electrostatically with a normal pK a value of 3.5 (Ito, Kuroki et al., 1999 ▶). Therefore, the observed difference may be owing to the hydrophilic environment of this Asp residue in these lysozymes. In addition to the formation of the catalytic carboxylate, the location of the substrate analogue (GlcNAc)3 was not equivalent in the three lysozymes. The sugar at the nonreducing end of MLL showed a difference in relative position compared with that of HEL (located in subsite A; Fig. 4 ▶ a). Compared with VPL, a similar minor difference was found in MLL: the distance between the hydroxyls of the GlcNAc in subsite A in MLL and VPL was 1.07 Å. These shifts may be owing to amino-acid substitutions in the subsites corresponding to subsites A–C of HEL, which interact with (GlcNAc)3. In these subsites, two amino acids of MLL, Ala43 and Lys102, are replaced by Pro44 and Ser103, respectively, in VPL. In particular, the bulky side chain of the Lys residue at this position is likely to cause the shift of the (GlcNAc)3. The substituted Ala residue appeared to interact with the GlcNAc moieties at subsites A and B through the Cα atom. This substitution may also be responsible for the difference in the orientation of the (GlcNAc)3 (Fig. 4 ▶ a).
Figure 3.

Comparison of the local structure of MLL with HEL and VPL. MLL, green; HEL (PDB entry 1hew; Cheetham et al., 1992 ▶), purple; VPL (PDB entry 2dqa; Goto et al., 2007 ▶), cyan. (a) Comparison of the structure of HEL with that of MLL. (b) Comparison of the structure of VPL with that of MLL. The (GlcNAc)3 in MLL, HEL and VPL is shown in green, purple and cyan, respectively.
Figure 4.

(a) Interaction of (GlcNAc)3 with binding sites. (GlcNAc)3 in MLL and VPL is shown in green and cyan, respectively. (b) Comparison of the amino acids that contribute to dimer formation. Amino acids in MLL and VPL are shown in green and cyan, respectively. The amino acids derived from the second subunit of the VPL dimer are underlined.
VPL forms dimers in conditions of low salt concentration (Goto et al., 2007 ▶). The crystal structure of VPL also indicates a dimeric structure, and the interface of the two monomers is maintained by electrostatic interactions between two catalytic residues: Asp30 and Glu18. This dimeric structure suppresses the activity of VPL at low concentrations of NaCl, and gel-filtration analysis of VPL provided empirical evidence of dimer formation at low ionic strength (Goto et al., 2007 ▶). However, the enzymatic activities of MLL were not affected by the ionic strength (Kuwano et al., 2013 ▶). We therefore compared the residues at the interface of the dimer in VPL with those of MLL (Fig. 4 ▶ b). The amino acid electrostatically interacting with Asp30 in VPL, Lys108, was substituted by Gly in MLL and no other possible amino acid that could contribute to the interaction was found in MLL. It therefore seems that amino-acid substitutions at the interface of the VPL dimer are likely to change the oligomeric states of these lysozymes. Bivalve lysozymes are believed to be involved in digestion and are stored in a crystalline state (Van Herreweghe & Michiels, 2012 ▶). Therefore, at high salt concentrations (of about 500 mM) VPL is thought to be converted from an inactive VPL dimer to an active VPL monomer because of disruption of the electrostatic interactions at the dimer interface (Goto et al., 2007 ▶). However, the similar bivalve lysozyme MLL was found to be in a monomeric state (active state) at low concentrations of NaCl. This suggests that the dimer-to-monomer conversion of lysozymes is not a general process within the organs of bivalves.
Supplementary Material
PDB reference: Meretrix lusoria lysozyme, 3ab6
Acknowledgments
We thank Professor Sergei Strelkov and Dr Seppe Leysen at the Laboratory for Biocrystallography, Department of Pharmaceutical Sciences, Katholieke Universiteit Leuven for their critical reviews of the manuscript.
References
- Blake, C. C. F., Koenig, D. F., Mair, G. A., North, A. C. T., Phillips, D. C. & Sarma, V. R. (1965). Nature (London), 206, 757–761. [DOI] [PubMed]
- Cheetham, J. C., Artymiuk, P. J. & Phillips, D. C. (1992). J. Mol. Biol. 224, 613–628. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fukamizo, T., Goto, S., Torikata, T. & Araki, T. (1989). Agric. Biol. Chem. 53, 2641–2651.
- Goto, T., Abe, Y., Kakuta, Y., Takeshita, K., Imoto, T. & Ueda, T. (2007). J. Biol. Chem. 282, 27459–27467. [DOI] [PubMed]
- Inoue, M., Yamada, H., Yasukochi, T., Miki, T., Horiuchi, T. & Imoto, T. (1992). Biochemistry, 31, 10322–10330. [DOI] [PubMed]
- Ito, Y., Kuroki, R., Ogata, Y., Hashimoto, Y., Sugimura, K. & Imoto, T. (1999). Protein Eng. 12, 327–331. [DOI] [PubMed]
- Ito, Y., Yoshikawa, A., Hotani, T., Fukuda, S., Sugimura, K. & Imoto, T. (1999). Eur. J. Biochem. 259, 456–461. [DOI] [PubMed]
- Jollès, J. & Jollès, P. (1975). Eur. J. Biochem. 54, 19–23. [DOI] [PubMed]
- Kelly, J. A., Sielecki, A. R., Sykes, B. D., James, M. N. G. & Phillips, D. C. (1979). Nature (London), 282, 875–878. [DOI] [PubMed]
- Kuhara, S., Ezaki, E., Fukamizo, T. & Hayashi, K. (1982). J. Biochem. 92, 121–127. [DOI] [PubMed]
- Kuwano, Y., Yoneda, K., Kawaguchi, Y., Araki, N. & Araki, T. (2013). In the press.
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Olsen, Ø. M., Nilsen, I. W., Sletten, K. & Myrnes, B. (2003). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 136, 107–115. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pincus, M. R. & Scheraga, H. A. (1981). Biochemistry, 20, 3960–3965. [DOI] [PubMed]
- Pincus, M. R., Zimmerman, S. S. & Scheraga, H. A. (1977). Proc. Natl Acad. Sci. USA, 74, 2629–2633. [DOI] [PMC free article] [PubMed]
- Pollock, J. J. & Sharon, N. (1970). Biochemistry, 9, 3913–3925. [DOI] [PubMed]
- Smith-Gill, S. J., Rupley, J. A., Pincus, M. R., Carty, R. P. & Scheraga, H. A. (1984). Biochemistry, 23, 993–997. [DOI] [PubMed]
- Takeshita, K., Hashimoto, Y., Ueda, T. & Imoto, T. (2003). Cell. Mol. Life Sci. 60, 1944–1951. [DOI] [PMC free article] [PubMed]
- Van Herreweghe, J. M. & Michiels, C. W. (2012). J. Biosci. 37, 327–348. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Meretrix lusoria lysozyme, 3ab6