

The cholesterol-dependent cytolysin listeriolysin O from L. monocytogenes has been purified and crystallized. The orthorhombic crystals diffracted to beyond 2.2 Å resolution and contained one molecule in the asymmetric unit.
Keywords: cholesterol-dependent cytolysins, toxins, listeriolysin O, listeriolysis, Listeria monocytogenes
Abstract
The secreted pore-forming toxin listeriolysin O (LLO) from the intracellular pathogen Listeria monocytogenes is a member of the family of cholesterol-dependent cytolysins (CDC) with broad properties in pathogenesis. Its role as a virulence factor is enigmatic: it disrupts membranes and acts as an inductor of both pro- and anti-inflammatory responses in infected cells. In addition, LLO is also a potent target for immunogenicity during infection. Natively secreted LLO from a recombinant L. innocua strain was crystallized in its water-soluble monomeric form. The crystals obtained belonged to the orthorhombic space group P212121, with unit-cell parameters a = 26.7, b = 85.1, c = 230.0 Å, and diffracted to beyond 2.2 Å resolution. The Matthews coefficient and the solvent content were estimated to be 2.4 Å3 Da−1 and 49.2%, respectively. The structure with one molecule in the asymmetric unit was solved using Phaser employing the structure of the previously characterized CDC toxin perfringolysin O as a search model.
1. Introduction
Listeria monocytogenes (Lm) is a Gram-positive facultative intracellular food-borne pathogen that causes gastroenteritis, meningitis, encephalitis and foetal infections (Vázquez-Boland et al., 2001 ▶; Barbuddhe & Chakraborty, 2009 ▶). A primary virulence factor of Lm is listeriolysin O (LLO), a secreted haemolytic/cytolytic protein that is a sine qua non factor for infection (Gaillard et al., 1987 ▶; Berche et al., 1987 ▶; Kathariou et al., 1987 ▶). As a pore-forming toxin, LLO can generally disrupt host cell membranes and it is presumed that, following entry of the bacterium, the activity of this toxin promotes the escape of engulfed bacteria from the primary entry vacuole, rendering bacteria intracellular (Gaillard et al., 1987 ▶; Kathariou et al., 1987 ▶). In the host cytosol, LLO activity is controlled in such a way that it does not disrupt the endomembrane system of the host cell, which would release Lm into the extracellular environment (Hamon et al., 2012 ▶).
LLO is a member of the cholesterol-dependent cytolysins (CDC), a family of pore-forming toxins with 28 known members to date from various bacterial species (Heuck et al., 2010 ▶). CDCs are secreted in a soluble monomeric state and bind to the host membrane. Subsequently, they oligomerize on the membrane into a ring, which finally forms a large pore of around 30 nm (Bhakdi et al., 1993 ▶). Initial information on the conformational changes leading to membrane insertion has been based on single-particle electron cryo-microscopy, atomic force microscopy and modelling studies (Czajkowsky et al., 2004 ▶; Tilley et al., 2005 ▶). Upon oligomerization, an α-helical region undergoes conformational changes to form β-hairpins which insert into the membrane. To date, perfringolysin (PFO) from Clostridium perfringens (Rossjohn et al., 1997 ▶), intermedilysin (ILY) from Streptococcus intermedius (Polekhina et al., 2005 ▶), anthrolysin (ALO) from Bacillus anthracis (Bourdeau et al., 2009 ▶) and suilysin from S. suis (Xu et al., 2010 ▶) have been structurally characterized in their monomeric soluble forms. They exhibit highly similar tertiary structures with strong sequence homology (around 40–70%), thus suggesting a common mechanism of pore formation within the entire family of CDCs. Nonetheless, the different CDCs have slightly differing functional characteristics. For LLO its uniqueness is related to its activity, owing to the presence of an acidic triad located within a membrane-inserting domain that acts as a pH sensor and thereby regulates structural changes to promote pore formation at acidic pH (Schuerch et al., 2005 ▶). The pH optimum of LLO activity is around pH 5.5, which reflects the pH of the acidified phagosome. In addition, LLO is also involved in the induction of a plethora of cellular reactions ranging from stimulation of MAP kinase (Tang et al., 1996 ▶) to the induction of cytokine production (Nishibori et al., 1996 ▶) and adhesion factors (Kayal et al., 1999 ▶), as well as the stimulation of mucin exocytose in intestinal cells (Coconnier et al., 2000 ▶).
Although many aspects of the biochemistry of LLO based on models have emerged, its three-dimensional structure has not yet been solved. Here, we purified and crystallized LLO in its water-soluble form in order to obtain further information on the specific features of this pore-forming toxin.
2. Experimental procedures
2.1. Protein overproduction and purification
The production and purification of untagged LLO using a recombinant L. innocua strain have been described previously (Darji et al., 1995 ▶). In brief, the recombinant strain was grown in 1 l Listeria minimal medium (Barbuddhe & Chakraborty, 2009 ▶) at 303 K for at least 48 h. The culture supernatant was obtained following centrifugation to remove bacteria and debris at 8000g for 30 min. A single cOmplete Protease Inhibitor Cocktail Tablet (Roche, Mannheim, Germany) was added to the supernatant, which was subsequently concentrated to 30 ml using a Pall Centramate system (Pall Corporation, Crailsheim, Germany).
Purification comprised two steps. In the first step the crude culture supernatant was incubated for 1 h at 277 K in the presence of 3 ml Q Sepharose Fast Flow (GE Healthcare, Germany) with gentle rotation. Following centrifugation, the non-absorbed fraction was filtered and desalted in 50 mM sodium pyrophosphate pH 6.2 on a HiPrep 26/10 desalting column (GE Healthcare, Germany). In the second purification step the concentrate was loaded onto a 1 ml Resource S column (GE Healthcare, Germany) with 50 mM NaH2PO4/Na2HPO4 pH 6.2 and eluted with NaH2PO4/K2HPO4 pH 5.6 and a gradient to 1 M NaCl. LLO eluted at a salt concentration of between 0.2 and 0.26 M NaCl as monitored by its haemolytic activity. Fractions containing the pure protein were pooled, dialysed against 25 mM Tris–HCl pH 7.7, concentrated to 1 mg ml−1 using a 50 kDa cutoff filter, flash-frozen in liquid nitrogen and stored at 193 K. The protein purity was estimated to be greater than 95% by SDS–PAGE analysis.
2.2. Crystallization
Before crystallization, the protein storage buffer was exchanged to 25 mM Tris–HCl pH 7.7 with 50 or 150 mM NaCl. For crystallization, the protein was further concentrated to 7 mg ml−1 as determined using the Bradford protein assay. Initial crystallization conditions were screened in 96-well plates by the hanging-drop vapour-diffusion technique. 400 nl of commercial crystallization solutions (Crystal Screen HT and Index HT; Hampton Research) were pipetted and mixed with 400 nl protein solution using a pipetting robot (Mosquito; Molecular Dimensions) and were incubated at 277, 285 or 291 K over 100 µl reservoir solution. The initial crystals were optimized using 24-well plates. For crystal optimization, 1 µl protein solution and 1 µl reservoir solution were mixed and incubated over 1 ml reservoir solution.
2.3. X-ray data collection and processing
Crystals were dipped briefly into reservoir solution, which served as a cryoprotectant, and flash-cooled in liquid nitrogen. Data were collected under a stream of nitrogen gas (100 K) on beamline ID23-2 at the European Synchrotron Research Facility (ESRF), Grenoble, France. Data processing, integration and scaling were performed using the XDS package (Kabsch, 2010 ▶). Molecular replacement was carried out using Phaser (McCoy, 2007 ▶) from the CCP4 package (Winn et al., 2011 ▶).
3. Results
Native LLO (molecular mass 56 kDa) from the culture medium was purified in two steps using Q Sepharose and Resource S ion-exchange columns (Fig. 1 ▶ a). The protein buffer for crystallization consisted of 25 mM Tris–HCl pH 7.7, 150 mM NaCl. Initial crystals were found in conditions B4 (100 mM HEPES pH 7.5, 1.5 M lithium sulfate) and G7 (100 mM HEPES pH 7.5, 20% Jeffamine M-600) of Crystal Screen HT. However, these crystals diffracted to a resolution of not better than 15 Å and neither condition could be further optimized. In subsequent crystallization experiments the salt concentration in the protein buffer was reduced. At protein concentrations of greater than 3 mg ml−1 a salt concentration below 50 mM NaCl led to precipitation of LLO. At a NaCl concentration of 50 mM and a protein concentration of 7 mg ml−1 crystal growth was observed in three other conditions (G9 of Crystal Screen HT and D2 and F6 of Index HT), one of which could be optimized.
Figure 1.
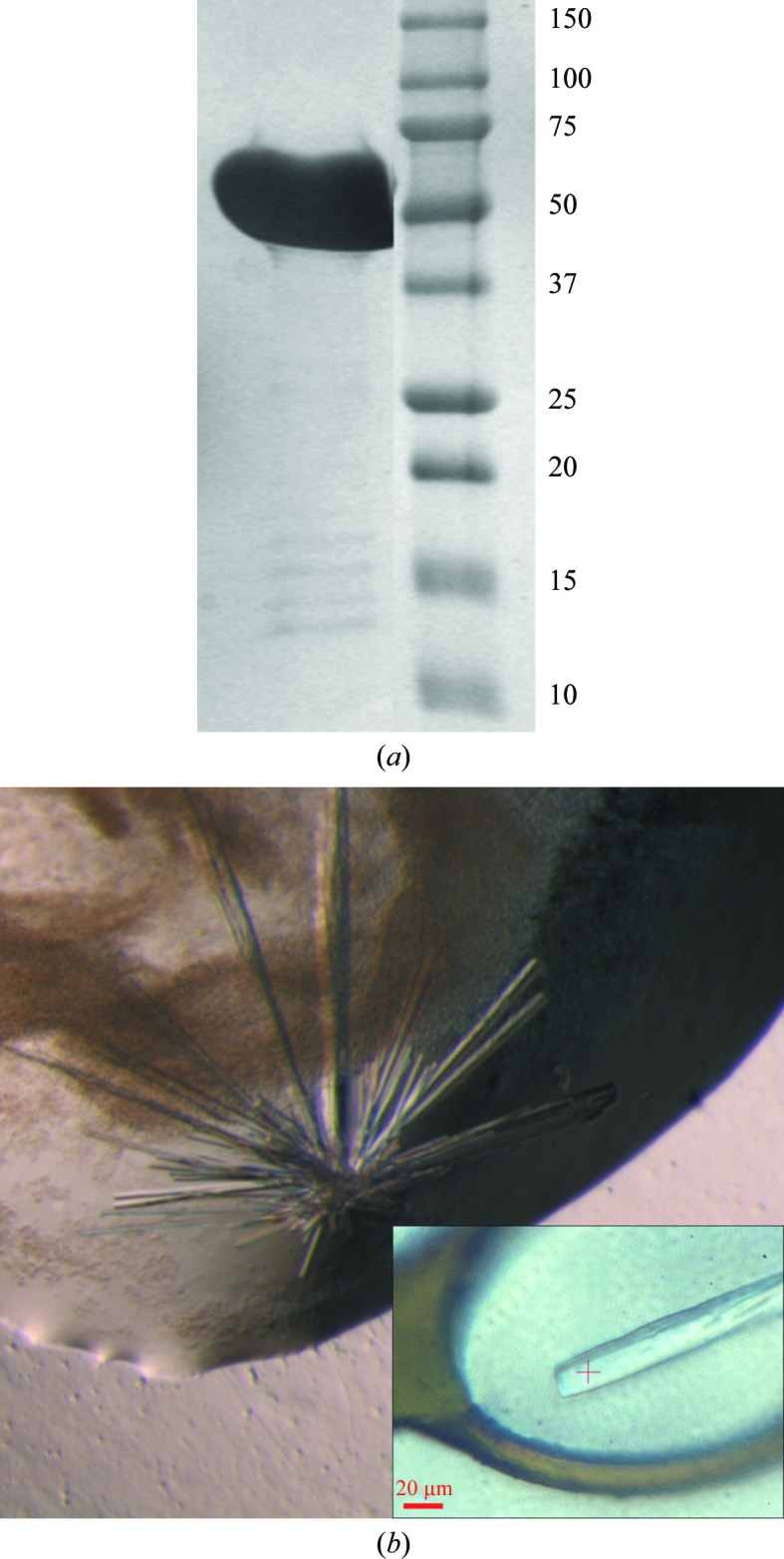
Purification and crystallization of LLO. (a) 12% SDS–PAGE of the purified LLO protein (56 kDa) used for crystallization. The molecular masses of the marker are indicated in kDa. (b) Crystal cluster of LLO obtained by the hanging-drop vapour-diffusion technique, with an average crystal thickness of 15 µm. Crystals were obtained using 100 mM bis-tris pH 5.5, 200 mM ammonium acetate, 25%(w/v) PEG 3350 as the crystallization solution. The inset shows a single crystal from the cluster in a CryoLoop before data collection.
The best quality crystals grew after five weeks at 277 K in a 2 µl hanging drop consisting of a 1:1 mixture of protein solution and 100 mM bis-tris pH 5.5, 200 mM ammonium acetate, 25%(w/v) PEG 3350. Interestingly, the pH of the crystallization condition reflects the pH optimum of LLO activity. These crystals were rod-shaped, with a thickness of approximately 15 µm, and formed clusters (Fig. 1 ▶ b). Using an acupuncture needle, single crystals could be isolated and cooled in liquid nitrogen after transfer into reservoir solution. Dissected single crystals diffracted to 2.2 Å resolution on the microfocus beamline ID23.2 at the ESRF (Fig. 2 ▶). A full 180° data set was collected from a single crystal using a crystal-to-detector distance of 266 mm with 1° oscillation and 9 s exposure per image. The calculated Matthews coefficient V M (Matthews, 1968 ▶) of 2.4 Å3 Da−1 (solvent content 49.2%) suggested the presence of one molecule per asymmetric unit. These crystals belonged to space group P212121, with unit-cell parameters a = 26.7, b = 85.1, c = 230.0 Å (Table 1 ▶).
Figure 2.

X-ray diffraction pattern collected from a single LLO crystal with 1° oscillation per image displayed using iMosflm (Battye et al., 2011 ▶). The purple circles and numbers correspond to the resolution shells (labelled in Å). The pattern displays a maximum resolution of 2.15 Å.
Table 1. Crystal parameters and data-collection statistics for LLO.
Values in parentheses are for the outermost resolution shell.
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 26.72, b = 85.15, c = 229.90 |
| Matthews coefficient (Å3 Da−1) | 2.4 |
| Solvent content (%) | 49.2 |
| No. of molecules per asymmetric unit | 1 |
| Resolution (Å) | 50–2.15 (2.30–2.15) |
| Wavelength (Å) | 0.8726 |
| X-ray source | ID23-2, ESRF |
| R meas (%) | 10.9 (114.5) |
| R mrgd-F (%) | 14.2 (92.5) |
| 〈I/σ(I)〉 | 15.76 (1.7) |
| Completeness (%) | 99.9 (99.4) |
| No. of observed reflections | 305038 (39986) |
| No. of unique reflections | 29894 (5336) |
| Multiplicity | 10.2 (7.5) |
Molecular replacement using Phaser (McCoy, 2007 ▶) from the CCP4 package (Winn et al., 2011 ▶) and perfringolysin O from C. perfringens (PDB entry 1pfo; Rossjohn et al., 1997 ▶; 66% sequence identity) as a polyalanine search model gave a clear solution with a log-likelihood gain of 132 and Z-scores for the rotation and translation functions of 6.8 and 11.4, respectively. The resulting electron-density maps were of high quality and no clashes were found between molecules. The initial R factors for the solution with the polyalanine model were very high (R work = 44%, R free = 51%). However, after fitting the correct sequence the R factors decreased to below 30%. The structure is currently being further refined and the final structural details will be described in a separate paper.
Acknowledgments
This work was funded in part by the German Research Foundation, Collaborative Research Centre (SFB628 and SFB807) and an ERANET Pathogenomics Network SPATELIS grant to TC. We thank the beamline staff at the ESRF Grenoble for excellent facilities and assistance during data collection, as well as the beamline staff of PXII at SLS for the excellent facility and for assistance with crystal screening.
References
- Barbuddhe, S. B. & Chakraborty, T. (2009). Curr. Top. Microbiol. Immunol. 337, 173–195. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Berche, P., Gaillard, J. L. & Sansonetti, P. J. (1987). J. Immunol. 138, 2266–2271. [PubMed]
- Bhakdi, S., Weller, U., Walev, I., Martin, E., Jonas, D. & Palmer, M. (1993). Med. Microbiol. Immunol. 182, 167–175. [DOI] [PubMed]
- Bourdeau, R. W., Malito, E., Chenal, A., Bishop, B. L., Musch, M. W., Villereal, M. L., Chang, E. B., Mosser, E. M., Rest, R. F. & Tang, W.-J. (2009). J. Biol. Chem. 284, 14645–14656. [DOI] [PMC free article] [PubMed]
- Coconnier, M.-H., Lorrot, M., Barbat, A., Laboisse, C. & Servin, A. L. (2000). Cell. Microbiol. 2, 487–504. [DOI] [PubMed]
- Czajkowsky, D. M., Hotze, E. M., Shao, Z. & Tweten, R. K. (2004). EMBO J. 23, 3206–3215. [DOI] [PMC free article] [PubMed]
- Darji, A., Chakraborty, T., Niebuhr, K., Tsonis, N., Wehland, J. & Weiss, S. (1995). J. Biotechnol. 43, 205–212. [DOI] [PubMed]
- Gaillard, J. L., Berche, P., Mounier, J., Richard, S. & Sansonetti, P. (1987). Infect. Immun. 55, 2822–2829. [DOI] [PMC free article] [PubMed]
- Hamon, M. A., Ribet, D., Stavru, F. & Cossart, P. (2012). Trends Microbiol. 20, 360–368. [DOI] [PubMed]
- Heuck, A. P., Moe, P. C. & Johnson, B. B. (2010). Subcell. Biochem. 51, 551–577. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kathariou, S., Metz, P., Hof, H. & Goebel, W. (1987). J. Bacteriol. 169, 1291–1297. [DOI] [PMC free article] [PubMed]
- Kayal, S., Lilienbaum, A., Poyart, C., Memet, S., Israel, A. & Berche, P. (1999). Mol. Microbiol. 31, 1709–1722. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- Nishibori, T., Xiong, H., Kawamura, I., Arakawa, M. & Mitsuyama, M. (1996). Infect. Immun. 64, 3188–3195. [DOI] [PMC free article] [PubMed]
- Polekhina, G., Giddings, K. S., Tweten, R. K. & Parker, M. W. (2005). Proc. Natl Acad. Sci. USA, 102, 600–605. [DOI] [PMC free article] [PubMed]
- Rossjohn, J., Feil, S. C., McKinstry, W. J., Tweten, R. K. & Parker, M. W. (1997). Cell, 89, 685–692. [DOI] [PubMed]
- Schuerch, D. W., Wilson-Kubalek, E. M. & Tweten, R. K. (2005). Proc. Natl Acad. Sci. USA, 102, 12537–12542. [DOI] [PMC free article] [PubMed]
- Tang, P., Rosenshine, I., Cossart, P. & Finlay, B. B. (1996). Infect. Immun. 64, 2359–2361. [DOI] [PMC free article] [PubMed]
- Tilley, S. J., Orlova, E. V., Gilbert, R. J., Andrew, P. W. & Saibil, H. R. (2005). Cell, 121, 247–256. [DOI] [PubMed]
- Vázquez-Boland, J. A., Kuhn, M., Berche, P., Chakraborty, T., Domínguez-Bernal, G., Goebel, W., González-Zorn, B., Wehland, J. & Kreft, J. (2001). Clin. Microbiol. Rev. 14, 584–640. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Xu, L., Huang, B., Du, H., Zhang, X. C., Xu, J., Li, X. & Rao, Z. (2010). Protein Cell, 1, 96–105. [DOI] [PMC free article] [PubMed]