

The cloning, purification, crystallization and preliminary X-ray diffraction studies of S. aureus homoserine dehydrogenase, an enzyme that regulates the biosynthesis of several essential amino acids, are reported.
Keywords: homoserine dehydrogenase, essential amino-acid metabolism, potential drug target
Abstract
Staphylococcus aureus is a Gram-positive nosocomial pathogen. The prevalence of multidrug-resistant S. aureus strains in both hospital and community settings makes it imperative to characterize new drug targets to combat S. aureus infections. In this context, enzymes involved in cell-wall maintenance and essential amino-acid biosynthesis are significant drug targets. Homoserine dehydrogenase (HSD) is an oxidoreductase that is involved in the reversible conversion of l-aspartate semialdehyde to l-homoserine in a dinucleotide cofactor-dependent reduction reaction. HSD is thus a crucial intermediate enzyme linked to the biosynthesis of several essential amino acids such as lysine, methionine, isoleucine and threonine.
1. Introduction
Staphylococcus aureus is an opportunistic pathogen and is a leading cause of several hospital-borne infections. Effective therapy to deal with S. aureus infections is complicated by the ability of this nosocomial pathogen to rapidly acquire resistance to several currently administered antimicrobial drugs (Mwangi et al., 2007 ▶). This feature makes it imperative to understand the mechanisms of drug resistance and cell-wall synthesis in order to design effective therapeutics for S. aureus infections. An emerging theme in this context is the viability of enzymes involved in the biosynthesis of essential amino acids as suitable drug targets (Hutton et al., 2007 ▶). Aspartic acid is a common precursor for the synthesis of essential amino acids such as threonine, isoleucine and methionine. In addition to these amino acids, this amino-acid synthesis pathway also produces meso-diaminopimelate (m-DAP) as an intermediate. m-DAP is an essential component of the peptidoglycan in Gram-negative bacteria and is a precursor for the synthesis of l-lysine. l-Lysine is an essential component of Gram-positive peptidoglycan (Hutton et al., 2007 ▶). This biosynthetic route to l-lysine synthesis is absent in humans. It is in this context that the lysine-biosynthesis pathway of S. aureus has received substantial attention. Several enzymes from the lysine-biosynthesis pathway of S. aureus have been structurally characterized. These include DapA (PDB entries 3di0 and 3di1; Girish et al., 2008 ▶), DapB (PDB entry 3qy9; Girish et al., 2011 ▶) and DapE (PDB entries 3khx, 3khz and 3ki9; Girish & Gopal, 2010 ▶). These studies served to rationalize the aspects of feedback regulation in lysine biosynthesis that differ between Gram-positive and Gram-negative bacteria. Distinctive structural features, in addition to the finding that redox stimuli regulate the action of S. aureus DapE, provided a basis for the design of specific small-molecule inhibitors. Structural and mechanistic studies of the homoserine dehydrogenase enzyme acquire significance in the context of l-lysine biosynthesis owing to the role of this enzyme in the synthesis of l-homoserine. This step regulates the biosynthesis of several essential amino acids.
Homoserine dehydrogenase (HSD) catalyzes a reaction at the branch point of the pathway leading to lysine biosynthesis. This pathway is also referred to as the diaminopimelate (dap) pathway (Ejim et al., 2004 ▶). HSD is an oxidoreductase (EC 1.1.1.3) that catalyzes the reversible conversion of l-aspartate semialdehyde to l-homoserine in a nicotinamide cofactor (NADP)-dependent reduction reaction. l-Aspartate semialdehyde is an essential precursor for the biosynthesis of l-lysine. l-Homoserine, on the other hand, is a precursor for the synthesis of l-methionine, l-isoleucine and l-threonine. In this context, enzymes such as HSD involved in branch-point reactions are critical in deciding the course of the reaction intermediates. HSD is distributed widely across most kingdoms of life except for mammals, a feature that makes it a potential target for therapeutic intervention (DeLaBarre et al., 2000 ▶). To date, high-resolution crystal structures of HSDs from different organisms such as Thiobacillus denitrificans (PDB entry 3mtj; Midwest Center for Structural Genomics, unpublished work), Thermoplasma volcanium (PDB entries 3jsa and 3c8m; Midwest Center for Structural Genomics, unpublished work), Thermoplasma acidophilum (PDB entry 3ing; Joint Center for Structural Genomics, unpublished work), Archaeoglobis fulgidus (PDB entry 3do5; Joint Center for Structural Genomics, unpublished work), Thermus thermophilus (PDB entry 2ejw; R. Omi, M. Goto, I. Miyahara & K. Hirotsu, unpublished work) and Saccharomyces cerevisiae (PDB entries 1tve, 1q7g, 1ebf and 1ebu; Ejim et al., 2004 ▶; Jacques et al., 2003 ▶; DeLaBarre et al., 2000 ▶) have been determined. Of these, only the biochemical features and reaction mechanism of S. cerevisiae HSD have been extensively characterized (DeLaBarre et al., 2000 ▶). In an effort towards the structural and functional analysis of S. aureus HSD (SaHSD), we cloned, overexpressed and purified this enzyme on a scale suitable for structural studies. SaHSD could be crystallized in different conditions. It is anticipated that the crystal structure of SaHSD will provide a potential route to understand the catalytic mechanism from a conformational perspective. Here, we describe the purification, crystallization and preliminary crystallographic analysis of this protein.
2. Materials and methods
2.1. Cloning, expression and purification of SaHSD
The gene encoding SaHSD was PCR-amplified from the genomic DNA of S. aureus strain COL using the primers 5′-CACGGCTAGCATGAAAAAATTAAATATA-3′ and 5′-CGACCTCGAGAACTCCTTCTACTGGGTA-3′. The PCR product was subsequently cloned between the NheI and XhoI sites of the pET expression vectors pET-28b and pET-15b (Novagen). In the case of the recombinant protein obtained using the pET-15b construct, an additional polypeptide stretch (MGSSHHHHHHSSGLVPRGSH) was added at the N-terminal end of the recombinant protein. The integrity of the clones was confirmed by DNA sequencing. The expression clones were transformed into Escherichia coli Rosetta (DE3) pLysS competent cells. The transformed cells were grown at 310 K to an optical density of 0.5 at 600 nm. Expression of the recombinant protein was induced using 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Post-induction, the cells were grown at 291 K for 12 h. The cells were harvested by centrifugation at 5000 rev min−1 for 15 min. The harvested cells were resuspended in lysis buffer (40 mM HEPES pH 7.5, 300 mM NaCl, 3% glycerol) and homogenized using a sonicator. During sonication, protease-inhibitor cocktail tablets were added to the lysis buffer to prevent nonspecific proteolysis. The lysate was further centrifuged at 14 000 rev min−1 for 40 min at 277 K and the resultant supernatant was incubated with Co2+–NTA affinity beads (Sigma–Aldrich) for 90 min at 277 K on an end-to-end rotor. The recombinant protein containing a hexahistidine tag was eluted from the Co2+–NTA affinity beads using an imidazole gradient (0–300 mM). The imidazole was desalted from the partially purified protein solution by subjecting it to size-exclusion chromatography (HiPrep 25/10 desalting column; GE Biosciences). The fractions containing the pure protein were concentrated to 8 mg ml−1 for crystallization trials. The purity of the protein was analysed by SDS–PAGE and mass spectrometry (Fig. 1 ▶).
Figure 1.

Purification of S. aureus homoserine dehydrogenase. (a) A 12% SDS–PAGE profile of purified SaHSD. Lane M contains molecular-mass marker (labelled in kDa; Thermo Scientific), while lane P corresponds to freshly purified SaHSD. (b) Mass spectrum of purified SaHSD. Of the two peaks in this spectrum obtained on a MALDI–TOF/TOF mass spectrometer (Bruker Daltonics), the mass of the smaller peak is consistent with a dimeric species. The calculated molecular weight of the SaHSD monomer is 51 203 Da.
2.2. Crystallization
Preliminary crystallization trials for SaHSD were performed by the microbatch crystallization method using crystallization screens from Hampton Research. The recombinant protein obtained using the pET-28b expression vector (with polyhistidine tags at both the N- and the C-termini) yielded microcrystals in over half a dozen conditions containing mainly calcium or magnesium salts. Further optimization of these microcrystals gave rise to crystals with a long hollow cylinder-like morphology that appeared within 3–4 d but diffracted poorly (∼4 Å; Fig. 2 ▶ a). Different approaches were examined to improve the crystal quality. The most significant involved changing the expression construct to pET-15b in addition to screening for additives, streak-seeding, variation of oil ratios, variation of the pH of the crystallization buffer (in the range pH 6–8.5) and several variations of the composition of the initial crystallization conditions. Both hanging-drop and sitting-drop methods of vapour diffusion were also examined. Addition of glycerol followed by streak-seeding with diluted seed solution (∼1:50) yielded single large rod-shaped crystals within 1 or 2 d of setting up crystallization experiments (Fig. 2 ▶ b; Stura & Wilson, 1991 ▶). The initial data were collected using a crystal from a condition consisting of 0.2 M magnesium acetate tetrahydrate, 0.1 M Tris–HCl pH 7.5, 18%(w/v) PEG 8000, 5% glycerol. These crystals were also incubated with ligands together with co-crystallization trials with the protein–ligand complex. To obtain larger crystals for a room-temperature data set, in addition to these strategies crystallization by experiments in microbridges (Hampton Research) was also examined at a protein-to-mother liquor ratio of 5 µl:5 µl. However, the size and quality of the crystals were similar to those obtained by other crystallization methods.
Figure 2.

Crystals of homoserine dehydrogenase. (a) Long hollow crystals of SaHSD. (b) Crystals obtained after optimization of the recombinant SaHSD protein construct.
2.3. Data collection
SaHSD crystals were flash-cooled in a cryoprotectant consisting of 15% DMSO added to the mother liquor. Diffraction data were collected from the cryocooled crystals on a CCD detector with 0.75° oscillation per image on beamline BM14 of the European Synchrotron Radiation Facility (ESRF), Grenoble. To obtain room-temperature diffraction data, the crystals were mounted in CryoLoops, which were then inserted into glass capillaries and sealed (Mac Sweeney & D’Arcy, 2003 ▶; Li et al., 2005 ▶). A room-temperature data set was collected using a MAR 345 image plate mounted on a Rigaku MicroMax-007 HF rotating-anode X-ray generator. The data-collection statistics for the cryocooled and room-temperature data sets are compiled in Table 1 ▶. The diffraction data were integrated using iMosflm (Battye et al., 2011 ▶) and scaled using SCALA (Evans, 2006 ▶).
Table 1. Diffraction data statistics for the cryocooled (at different pH values) and room-temperature data sets.
Values in parentheses are for the outer shell.
| Condition | pH 8.5 | pH 7.5 | pH 7.0 | pH 6.5 | pH 6.0 | Room temperature (pH 7.5) |
|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.953 | 0.953 | 0.953 | 0.953 | 0.953 | 1.54 |
| Resolution (Å) | 2.2 (2.32–2.20) | 2.1 (2.21–2.10) | 2.2 (2.32–2.20) | 2.2 (2.32–2.20) | 2.2 (2.32–2.20) | 3.0 (3.16–3.00) |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| Unit-cell parameters (Å) | a = 72.87, b = 117.51, c = 119.53 | a = 73.04, b = 117.66, c = 119.48 | a = 72.64, b = 116.88, c = 118.62 | a = 72.48, b = 115.43, c = 118.23 | a = 72.46, b = 116.32, c = 118.35 | a = 74.28, b = 117.92, c = 120.72 |
| No. of observations | 294024 (42519) | 434629 (56962) | 433742 (62873) | 440608 (62547) | 411939 (55716) | 52793 (7471) |
| No. of unique observations | 52569 (7591) | 59396 (8141) | 52050 (7494) | 51107 (7340) | 50394 (6957) | 19340 (2804) |
| Mean I/σ(I) | 15.3 (3.6) | 19.2 (4.2) | 17.4 (4.7) | 20.5 (4.8) | 17.0 (4.8) | 5.0 (2.8) |
| R merge † (%) | 7.5 (48.6) | 5.7 (43.8) | 7.5 (44.2) | 6.5 (49.2) | 7.1 (43.5) | 17.1 (36.4) |
| Multiplicity | 5.6 (5.6) | 7.3 (7.0) | 8.3 (8.4) | 8.6 (8.5) | 8.2 (8.0) | 2.7 (2.7) |
| Completeness (%) | 99.6 (100) | 97.8 (93.4) | 100 (100) | 100 (100) | 98.0 (94.1) | 89.0 (89.8) |
R
merge = 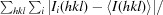
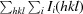 , where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity.
3. Results and discussion
Single rod-shaped crystals of ∼0.3 × 0.1 × 0.1 mm in size were obtained by changing the recombinant protein construct with two polyhistidine tags (obtained from the pET-28b expression vector) to one with a single, albeit longer, polyhistidine tag at the N-terminus (using the pET-15b expression vector). Furthermore, streak-seeding the drops with nucleant (crushed microcrystals) immediately after setting up the crystallization experiment substantially improved the crystal quality. The protein could be crystallized in buffers at five different pH values (pH 6–8.5; Table 2 ▶). The crystals appeared at a similar time across these conditions. While all of the cryocooled crystals diffracted to a resolution of ∼2.0–2.2 Å, the diffraction was relatively poor (∼3.0–3.2 Å) from the crystals from which data were collected at room temperature. The crystals belonged to a primitive orthorhombic space group. Based on the space group and the protein sequence composition, two molecules in the asymmetric unit are anticipated with a solvent content of around 50% (corresponding to a Matthews coefficient of 2.51 Å3 Da−1; Matthews, 1968 ▶). Examination of the diffraction data using SFCHECK (from the CCP4 suite; Winn et al., 2011 ▶) suggests that the diffraction data are devoid of crystal pathologies such as twinning or translational pseudo-symmetry. A molecular-replacement strategy using A. fulgidus homoserine dehydrogenase (NCBI Reference Sequence NP_069768.1; PDB entry 3do5; Joint Center for Structural Genomics, unpublished work; 38% sequence identity to SaHSD) provided preliminary phase information. These calculations were performed using Phaser (Storoni et al., 2004 ▶; McCoy et al., 2005 ▶). The SaHSD model is currently being rebuilt and refined with noncrystallographic symmetry (NCS) averaging to improve the phase information.
Table 2. Crystallization conditions for SaHSD.
| pH | Condition | Cryoprotectant | Method of crystallization |
|---|---|---|---|
| 8.5 | 0.2 M magnesium acetate, 14%(w/v) PEG 8000, 0.2 M Bicine, 5% glycerol (200 µl of a 1:1 ratio of paraffin and silicone oil was overlaid on the mother liquor in the reservoir well) | 15% DMSO | Hanging-drop vapour diffusion |
| 7.5 | 0.2 M magnesium acetate, 16%(w/v) PEG 8000, 0.1 M Tris–HCl, 3% glycerol (crystal soaked with 5 mM lysine and 4 mM NADP for 2 min) | 15% DMSO | Hanging-drop vapour diffusion |
| 7.0 | 0.2 M magnesium acetate, 16%(w/v) PEG 3350, 0.1 M HEPES, 20% 2-propanol, 5% glycerol (crystal soaked with 5 mM serine for 2 min) | 15% DMSO | Sitting-drop vapour diffusion |
| 6.5 | 0.2 M magnesium acetate, 16%(w/v) PEG 3350, 0.1 M sodium cacodylate, 5% glycerol (crystal soaked with 5 mM serine and 2 mM NADP for 2 min) | 15% DMSO | Sitting-drop vapour diffusion |
| 6.0 | 0.2 M magnesium acetate, 22%(w/v) PEG 8000, 0.1 M bis-tris, 1 mM serine, 5% glycerol | 15% DMSO | Microbatch |
Acknowledgments
We acknowledge Dr Hassan Belrhali and Dr Babu A. Manjasetty for their help in data collection on beamline BM14 of the European Synchrotron Radiation Facility (ESRF), Grenoble.
References
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- DeLaBarre, B., Thompson, P. R., Wright, G. D. & Berghuis, A. M. (2000). Nature Struct. Mol. Biol. 7, 238–244. [DOI] [PubMed]
- Ejim, L., Mirza, I. A., Capone, C., Nazi, I., Jenkins, S., Chee, G. L., Berghuis, A. M. & Wright, G. D. (2004). Bioorg. Med. Chem. 12, 3825–3830. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Girish, T. S. & Gopal, B. (2010). J. Biol. Chem. 285, 29406–29415. [DOI] [PMC free article] [PubMed]
- Girish, T. S., Navratna, V. & Gopal, B. (2011). FEBS Lett. 585, 2561–2567. [DOI] [PubMed]
- Girish, T. S., Sharma, E. & Gopal, B. (2008). FEBS Lett. 582, 2923–2930. [DOI] [PubMed]
- Hutton, C. A., Perugini, M. A. & Gerrard, J. A. (2007). Mol. Biosyst. 3, 458–465. [DOI] [PubMed]
- Jacques, S. L., Mirza, I. A., Ejim, L., Koteva, K., Hughes, D. W., Green, K., Kinach, R., Honek, J. F., Lai, H. K., Berghuis, A. M. & Wright, G. D. (2003). Chem. Biol. 10, 989–995. [DOI] [PubMed]
- Li, S. J., Suzuki, M. & Nakagawa, A. (2005). J. Cryst. Growth, 281, 592–595.
- Mac Sweeney, A. & D’Arcy, A. (2003). J. Appl. Cryst. 36, 165–166.
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Mwangi, M. M., Wu, S. W., Zhou, Y., Sieradzki, K., de Lencastre, H., Richardson, P., Bruce, D., Rubin, E., Myers, E., Siggia, E. D. & Tomasz, A. (2007). Proc. Natl Acad. Sci. USA, 104, 9451–9456. [DOI] [PMC free article] [PubMed]
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]
- Stura, E. A. & Wilson, I. A. (1991). J. Cryst. Growth, 110, 270–282.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.