

The catalytic domain of the histidine kinase of the ethylene receptor ETR1 from A. thaliana was expressed and crystallized. The crystals belonged to space group I222 or I212121.
Keywords: ETR1, Arabidopsis thaliana
Abstract
Ethylene is a gaseous plant hormone which controls many aspects of plant growth and development. It is perceived by membrane-bound receptors with a similarity to bacterial two-component systems. The catalytic and ATP-binding domain of the histidine kinase domain of ETR1 from Arabidopsis thaliana has been cloned, overexpressed and crystallized. The protein was crystallized together with various nucleotides. Crystals obtained in the presence of ADP belonged to space group I222 or I212121 with one molecule per asymmetric unit. They diffracted X-ray radiation to beyond 1.85 Å resolution.
1. Introduction
Plants employ ethylene to regulate many developmental processes such as seed germination, root growth, fruit ripening and senescence (Bleecker & Kende, 2000 ▶). Arabidopsis thaliana perceives ethylene using a group of five partly redundant membrane-bound receptors (ETR1, ETR2, ERS1, ERS2 and EIN4). Sequence analysis of ethylene receptors (ERs) indicated a similarity to bacterial two-component systems (TCS) in their C-terminal cytosolic histidine kinase (HK) and receiver domains (Chang et al., 1993 ▶). Based on the integrity of signature motifs in the catalytic domain of the HK, the receptors are further assigned to two subfamilies. The members of subfamily 1 (ETR1 and ERS1) possess all of the sequence motifs of canonical HK domains and also histidine kinase activity. Subfamily 2 receptors (ETR2, ERS2 and EIN4) have incomplete motifs and lack histidine kinase activity (Moussatche & Klee, 2004 ▶). While all receptors participate in ethylene signal transduction (Hall & Bleecker, 2003 ▶), the members of subfamily 1 seem to play a predominant role.
As in bacterial TCS, the activity of the ethylene receptors is controlled by their signalling molecule. Interestingly, the receptors are active in the absence of ethylene as shown by the autophosphorylation activity of their respective HK in vivo, which is abolished upon ethylene binding (Hua & Meyerowitz, 1998 ▶). At the same time the HK domain, but not its activity, is needed for ethylene signalling (Qu & Schaller, 2004 ▶).
One confirmed target of ethylene receptor activity in A. thaliana is CTR1, a Raf-like mitogen-activated protein kinase kinase kinase (MAPKKK; Kieber et al., 1993 ▶). CTR1 associates with ethylene receptors (Gao et al., 2003 ▶) and just like the ethylene receptors it is active in the absence of ethylene. Active CTR1 is a negative regulator of ethylene signalling, as shown in vivo by mutants with abolished protein kinase activity, which show a constitutive triple response (CTR; Huang et al., 2003 ▶). Thus, ethylene acts as an inverse agonist which inhibits activity of its receptors, thereby removing the inhibition of ethylene signalling by CTR1.
In our continued attempts to functionally characterize this important receptor class, we have cloned, overexpressed and crystallized the catalytic and ATP-binding domain of ETR1 from A. thaliana.
2. Materials and methods
2.1. Cloning
Based on secondary-structure and domain-organization analysis (Punta et al., 2012 ▶), the catalytic and ATP-binding domain (CA) of ETR1 (ETR1CA, residues 407–589) was amplified by PCR from a cDNA library obtained from the Arabidopsis Biological Research Center at Ohio State University (Kieber et al., 1993 ▶). The sequences of the forward and reverse primers were 5′-TACTTCCAATCCAATGCCGATGGAAGTCTTCAACTTGAACTTGGGAC-3′ and 5′-TTATCCACTTCCAATGTTATTCGTTTGAACGTTCTGAGATCCCAAG-3′, respectively.
Both primers contain the necessary extensions for ligation-independent cloning (LIC). PCR mixtures consisted of 1–5 ng template DNA, 1 µM primers and 0.5 µl Phusion High Fidelity DNA polymerase (Thermo Scientific, Germany). The PCR amplification programme included an initial denaturation step of 3 min at 367 K, 30 cycles of amplification at 30 s at 367 K and 45 s at 328 K, 1 min extension at 341 K and a final extension for 10 min at 341 K. The PCR product was inserted into the expression vector pMCSG7 using ligation-independent cloning as described in Eschenfeldt et al. (2009 ▶). The expression vector adds an N-terminal hexahistidine tag followed by a TEV cleavage site. Owing to cloning, three additional amino acids (SNA) are added to the native N-terminus after TEV digestion. The final expression vector sequences were verified by DNA sequencing.
2.2. Expression and purification
The expression vector containing ETR1CA was transformed into Escherichia coli strain BL21 cells co-expressing chaperones DnaK, DnaJ, GrpE, ClpB, GroEL and GroES (CC4 cell lines, courtesy of A. Geerlof, Helmholtz Zentrum München). Freshly transformed cells were used to prepare a 5 ml overnight pre-culture at 310 K in Luria–Bertani (LB) medium. 2 l of autoinduction medium (Studier, 2005 ▶) was inoculated with the pre-culture and incubated at 310 K. The cells were grown to an OD600 of ∼0.8 and incubated for a further 18 h at a lower temperature of 293 K. The cells were harvested by centrifugation at 5000 rev min−1 in a JLA-8.1000 rotor (Beckman Coulter GmbH, Germany) for 30 min at 277 K. The cell pellets were resuspended in lysis buffer [50 mM HEPES pH 7.0, 250 mM NaCl, 5% glycerol, 2 mM MgSO4, 0.1%(w/v) CHAPS] supplemented with 1 mM EDTA-free protease inhibitors (Roche Applied Science, Germany). The resuspended cells were lysed by sonication on ice for two cycles each of 2 min at a power of 2.5 kJ with an interval of 3 min. The lysate was centrifuged at 18 000 rev min−1 in an SS-34 rotor (Thermo Fisher Scientific Instruments, Germany) at 277 K for 45 min.
ETR1CA was isolated from the supernatant in three steps: Ni–NTA affinity chromatography, TEV protease cleavage to remove the His tag and gel-filtration chromatography. The supernatant was passed twice through a 5 ml Ni–NTA column (Qiagen, Germany) which had first been equilibrated against 20 mM NiSO4 in water and then against buffer A (50 mM HEPES pH 7.0, 250 mM NaCl, 5% glycerol, 2 mM MgSO4). After sample loading, the column was washed with 15 column volumes (CV) of buffer A followed by 3 CV of buffer A with 10% buffer B (buffer A with 500 mM imidazole). The proteins were eluted with a gradient of 10–60% of buffer B diluted into buffer A (50–300 mM imidazole) within 8 CV.
Fractions containing ETR1CA were pooled and incubated with purified TEV protease at a molar ratio of 1:20 to remove the His tag. The incubated solution was dialyzed overnight against buffer C (50 mM HEPES pH 7.0, 250 mM NaCl, 5% glycerol, 2 mM MgSO4, 1 mM DTT, 0.5 mM EGTA). Around 2 ml of Ni–NTA beads were added to the dialyzed protein to remove His-tagged TEV protease and uncleaved ETR1CA. The cleaved protein was concentrated using Ultrafree-30 concentrators (Amicon Bioseparation, Millipore, Billerica, Massachusetts, USA) and applied onto a gel-filtration column (Hiload 16/60 Superdex G75, GE Healthcare, Germany) equilibrated with buffer D (50 mM HEPES pH 7.0, 250 mM NaCl, 5% glycerol, 2 mM MgSO4). Protein fractions were checked with SDS–PAGE for homogeneity. Pure protein fractions were pooled and concentrated to ∼20 mg ml−1. The concentrated protein solution was then aliquoted in 0.2 ml PCR tubes, shock-frozen in liquid nitrogen and stored at 193 K.
2.3. Crystallization
Prior to crystallization, the protein solution was diluted to 10 mg ml−1 with buffer D. ADP was added to a final concentration of 5 mM and the solution was incubated for 30 min at 277 K. Initial crystallization experiments were carried out with six commercial crystallization screens (The Classics, Classics II, PEGs, PEGs II and JCSG+ Suites from Qiagen and Index from Hampton Research) at the EMBL Hamburg high-throughput crystallization facility (Mueller-Dieckmann, 2006 ▶). All initial screens were performed using the sitting-drop vapour-diffusion method at 292 K in 96-well Greiner plates. 300 nl protein solution was mixed with an equal volume of reservoir solution and equilibrated against 50 µl reservoir solution. Only one lead condition was found with condition No. 39 of The Classics Suite (Qiagen), consisting of 50 mM cadmium sulfate, 0.1 M HEPES pH 7.5, 1.0 M sodium acetate. Crystals were reproduced under identical conditions. During manual optimization in hanging-drop vapour-diffusion experiments at 292 K the sample concentration was increased to 12 mg ml−1 and the drop volumes to 2 µl reservoir solution plus 2 µl sample. Single crystals appeared within 1 week (Fig. 1 ▶).
Figure 1.

Crystals of ETR1CA from A. thaliana. The large crystal shown here has approximate dimensions of 120 × 50 × 40 µm.
2.4. X-ray diffraction data collection and processing
Prior to data collection, a single crystal of ETR1CA and ADP was immersed for about 5 s in mother liquor augmented with 15% glycerol as a cryoprotectant. The crystals were then flash-cooled to 77 K in liquid nitrogen. X-ray diffraction data sets were collected on beamline X06DA of the Swiss Light Source (SLS), Villigen, Switzerland at 100 K at a wavelength of 0.998 Å using a PILATUS 2M detector. The best crystal diffracted X-ray radiation to 1.85 Å resolution. A total of 800 images were collected with an oscillation range of 0.45°. The data were indexed and integrated using XDS (Kabsch, 2010a ▶) and scaled with XSCALE (Kabsch, 2010b ▶). Table 1 ▶ summarizes the data-collection and processing statistics.
Table 1. X-ray data-collection and processing statistics.
Values in parentheses are for the outermost resolution shell.
| Protein | ETR1CA |
| X-ray source | X06DA, SLS |
| Wavelength (Å) | 0.998 |
| Detector | PILATUS 2M-F |
| Temperature (K) | 100 |
| Crystal-to-detector distance (mm) | 180 |
| Rotation range per image (°) | 0.45 |
| Total rotation range (°) | 360 |
| Space group | I222 or I212121 |
| Unit-cell parameters (Å) | a = 76.32, b = 83.17, c = 91.96 |
| Resolution range (Å) | 45–1.85 (1.95–1.85) |
| Observed reflections | 329812 (46194) |
| Unique reflections | 25476 (3615) |
| R merge † (%) | 6.5 (47.1) |
| R p.i.m. (%) | 1.9 (13.5) |
| Completeness (%) | 99.7 (98.3) |
| 〈I/σ(I)〉 | 22.5 (5.2) |
| Multiplicity | 12.9 (12.8) |
| Mosaicity (°) | 0.089 |
| Wilson B factor (Å2) | 37 |
R
merge = 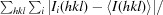
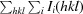 .
.
3. Results and discussion
The catalytic domain of the histidine kinase of ETR1 from A. thaliana was successfully cloned and expressed in E. coli BL21 cells co-expressing the chaperones DnaK, DnaJ, GrpE, ClpB, GroEL and GroES. Purification by affinity and gel-filtration chromatography resulted in 15–20 mg of pure protein per litre of culture. After gel-filtration chromatography, the protein eluted as a single peak corresponding to the monomeric state. The samples were at least 95% pure as estimated by SDS–PAGE.
Initial crystallization screening was carried out at the EMBL high-throughput crystallization facility. A sample incubated with ADP produced crystals in a single crystallization condition. Manual optimization of the initial condition resulted in final conditions with a reservoir solution consisting of 50 mM CdSO4, 0.1 M HEPES pH 7.5, 1.0 M sodium acetate and a protein concentration of 12 mg ml−1.
Crystals of ETR1CA and ADP diffracted X-ray radiation to a resolution of 1.85 Å. A complete data set was collected on beamline X06DA of the Swiss Light Source (SLS), Villigen, Switzerland. The crystals belonged to space group I222 or I212121, with unit-cell parameters a = 76.32, b = 83.17, c = 91.96 Å. Calculation of the Matthews coefficient (V M = 3.61 Å3 Da−1; molecular mass 19.8 kDa; Matthews, 1968 ▶) indicated the presence of a single molecule per asymmetric unit and an approximate solvent content of 65%. Histidine kinases are known to adopt different conformations dependent on the state of the bound nucleotide and/or their interaction with their corresponding dimerization domain (Dutta et al., 1999 ▶; Bilwes et al., 2001 ▶; Marina et al., 2001 ▶). We will therefore attempt to phase the diffraction data by molecular replacement using the structures of various catalytic domains of bacterial histidine kinases.
Acknowledgments
We thank the Arabidopsis Biological Resource Center for providing us with an A. thaliana cDNA library. We acknowledge the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland for provision of synchrotron-radiation facilities.
References
- Bilwes, A. M., Quezanda, C. M., Croal, L. R., Crane, B. R. & Simon, M. I. (2001). Nature Struct. Biol. 8, 353–360. [DOI] [PubMed]
- Bleecker, A. B. & Kende, H. (2000). Annu. Rev. Cell Dev. Biol. 16, 1–18. [DOI] [PubMed]
- Chang, C., Kwok, S. F., Bleecker, A. B. & Meyerowitz, E. M. (1993). Science, 262, 539–544. [DOI] [PubMed]
- Dutta, R., Qin, L. & Inouye, M. (1999). Mol. Microbiol. 34, 633–640. [DOI] [PubMed]
- Eschenfeldt, W. H., Lucy, S., Millard, C. S., Joachimiak, A. & Mark, I. D. (2009). Methods Mol. Biol. 498, 105–115. [DOI] [PMC free article] [PubMed]
- Gao, Z., Chen, Y.-F., Randlett, M. D., Zhao, X.-C., Findell, J. L., Kieber, J. J. & Schaller, G. E. (2003). J. Biol. Chem. 278, 34725–34732. [DOI] [PubMed]
- Hall, A. E. & Bleecker, A. B. (2003). Plant Cell, 15, 2032–2041. [DOI] [PMC free article] [PubMed]
- Hua, J. & Meyerowitz, E. M. (1998). Cell, 94, 261–271. [DOI] [PubMed]
- Huang, Y., Li, H., Hutchison, C. E., Laskey, J. & Kieber, J. J. (2003). Plant J. 33, 221–233. [DOI] [PubMed]
- Kabsch, W. (2010a). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kieber, J. J., Rothenberg, M., Roman, G., Feldmann, K. A. & Ecker, J. R. (1993). Cell, 72, 427–441. [DOI] [PubMed]
- Marina, A., Mott, C., Auyzenberg, A., Hendrickson, W. A. & Waldburger, C. D. (2001). J. Biol. Chem. 276, 41182–41190. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Moussatche, P. & Klee, H. J. (2004). J. Biol. Chem. 279, 48734–48741. [DOI] [PubMed]
- Mueller-Dieckmann, J. (2006). Acta Cryst. D62, 1446–1452. [DOI] [PubMed]
- Punta, M. et al. (2012). Nucleic Acids Res. 40, D290–D301. [DOI] [PMC free article] [PubMed]
- Qu, X. & Schaller, G. E. (2004). Plant Physiol. 136, 2961–2970. [DOI] [PMC free article] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]