

A series of N-(2-phenylethyl)nitroaniline derivatives is presented, demonstrating that modest changes in the functional groups cause significant differences in molecular conformation, intermolecular interactions and packing.
Keywords: crystal structure, N-(2-phenylethyl)nitroaniline derivatives, secondary amines, nitric oxide release agents
Abstract
2,4-Dinitro-N-(2-phenylethyl)aniline, C14H13N3O4, (I), crystallizes with one independent molecule in the asymmetric unit. The adjacent amine and nitro groups form an intramolecular N—H⋯O hydrogen bond. The anti conformation about the ethyl C—C bond leads to the phenyl and aniline rings being essentially parallel. Molecules are linked into dimers by intermolecular N—H⋯O hydrogen bonds, such that each amine H atom participates in a three-centre interaction with two nitro O atoms. Though the dimers pack so that the arene rings of adjacent molecules are parallel, the rings are staggered and π–π interactions do not appear to be favoured. 4,6-Dinitro-N,N′-bis(2-phenylethyl)benzene-1,3-diamine, C22H22N4O4, (II), differs from (I) in the presence of a second 2-phenylethylamine group on the substituted ring. Compound (II) also crystallizes with one unique molecule in the asymmetric unit. Both amine groups are involved in intramolecular N—H⋯O hydrogen bonds with adjacent nitro groups. Although one ethyl group adopts an anti conformation as in (I), the other is gauche, with the result that the pendant phenyl rings are not parallel. The amine group that is part of the gauche conformation participates in a three-centre N—H⋯O hydrogen bond with the nitro group of a neighbouring molecule, leading to dimers as in (I). The other amine H atom does not form any intermolecular hydrogen bonds. The packing leads to separations of ca 3.4 Å of the parallel anti phenyl and aminobenzene rings. 2-Cyano-4-nitro-N-(2-phenylethyl)aniline, C15H13N3O2, (III), differs from (I) only in having a cyano group in place of the 2-nitro group. The absence of the adjacent nitro group eliminates the intramolecular N—H⋯O hydrogen bond. Molecules of (III) adopt the same anti conformation about the ethyl group as in (I), but crystallize in the higher-symmetry monoclinic space group P21/n. The molecules are linked into dimers via N—H⋯N amine–cyano hydrogen bonds, while the nitro groups are not involved in any N—H⋯O interactions. Owing to the different symmetry, the molecules pack in a herringbone pattern with fewer face-to-face interactions between the rings. The closest such interactions are about 3.5 Å between rings that are largely slipped past one another. 4-Methylsulfonyl-2-nitro-N-(2-phenylethyl)aniline, C15H16N2O4S, (IV), differs from (I) in having a methylsulfonyl group in place of the 4-nitro group. The intramolecular N—H⋯O hydrogen bond is present as in (I). However, unlike (I), the conformation about the ethyl group is gauche, so the two arene rings are nearly perpendicular rather than parallel. The packing is significantly different from the other three structures in that there are no intermolecular hydrogen bonds involving the N—H groups. The molecules are arranged in tetragonal columns running along the c axis, with the aniline rings mostly parallel and separated by ca 3.7 Å. Taken together, these structures demonstrate that modest changes in functional groups cause significant differences in molecular conformation, intermolecular interactions and packing.
Introduction
Nitric oxide (NO) is known for its significance as a messenger molecule in essentially every organ system across many life forms: mammals, plants, fish and insects (Giles, 2006 ▶). In humans, NO has important functions in the cardiovascular, nervous, respiratory and immune systems. It plays vital roles in protecting the heart, brain and kidneys. NO activities within these systems include facilitating endothelium-dependent vasodilation; regulating blood pressure, vascular tone and compliance; and inhibiting platelet aggregation, vascular inflammatory factors, leukocyte adhesion and smooth muscle cell proliferation (Mason & Cockcroft, 2006 ▶). Metabolic activity, sexual response, insulin release and the peripheral nervous system are affected by NO as well (Giles, 2006 ▶).
Numerous factors (including aging, hypercholesterolaemia, smoking and diabetes mellitus) can contribute to the loss of NO bioavailability and/or lack of NO production, resulting in endothelial dysfunction (Cai et al., 2005 ▶). Diseases and disorders based on NO malfunction include hypertension, atherosclerosis, cardiovascular disease, asthma, pulmonary hypertension, erectile dysfunction, preeclampsia and insulin resistance (Giles, 2006 ▶).
In order to ameliorate deleterious conditions arising out of NO shortfall, numerous NO donors have been developed, most of which release NO in a single high-concentration burst (Cai et al., 2005 ▶). We have reported a series of N-nitrosated secondary amines which release NO in a slow, sustained and rate-tunable manner (Wang et al., 2009 ▶; Yu et al., 2011 ▶; Curtis et al., 2013 ▶). Furthermore, the released NO has been shown to inhibit the proliferation of human aortic smooth muscle cells, a contributing factor to the progression of atherosclerosis (Yu et al., 2011 ▶; Curtis et al., 2013 ▶). As part of this continuing study, we report herein the syntheses and X-ray crystal structures of four secondary amines that are precursors to slow and sustained NO-releasing agents. These secondary amines, 2,4-dinitro-N-(2-phenylethyl)aniline, (I), 4,6-dinitro-N,N′-bis(2-phenylethyl)benzene-1,3-diamine, (II), 2-cyano-4-nitro-N-(2-phenylethyl)aniline, (III), and 4-methylsulfonyl-2-nitro-N-(2-phenylethyl)aniline, (IV), were prepared by the reactions of 2-phenylethylamine and four different activated aromatic mono- and difluorides, namely 2,4-dinitrofluorobenzene for (I), 1,5-difluoro-2,4-dinitrobenzene for (II), 2-cyano-4-nitrofluorobenzene for (III) and (3-nitro-4-fluorophenyl)methyl sulfone for (IV).
Experimental
Synthesis and crystallization
For the synthesis of 2,4-dinitro-N-(2-phenylethyl)aniline, (I), a 100 ml three-necked round-bottomed flask, equipped with a magnetic stirrer bar, nitrogen inlet, air condenser and thermometer, was charged with 2-phenylethylamine (PEA; 0.6370 g, 5.26 mmol), 2,4-dinitrofluorobenzene (0.9775 g, 5.25 mmol), potassium carbonate (1.4489 g, 10.48 mmol) and dimethylacetamide (DMAC, 50 ml). The vials used to weigh PEA and 2,4-dinitrofluorobenzene were washed with DMAC (5 ml) and the washes were transferred to the reaction vessel. The reaction vessel was heated using an oil bath until the temperature of the reaction mixture reached 343 K. The reaction was allowed to proceed with stirring for 12 h, after which the DMAC was removed by distillation at reduced pressure. The crude product was dissolved in dichloromethane (75 ml) and washed with a saturated sodium chloride solution (200 ml), followed by two washes with deionized water (200 ml). The organic layer was separated and anhydrous magnesium sulfate was added to dry the product. The resulting mixture was filtered and the filtrate was evaporated at reduced pressure using a rotary evaporator. Crystals of (I) suitable for X-ray diffraction were obtained by recrystallization from a dichloromethane–hexane solution (4:1 v/v) (yield 69%; m.p. 425–427 K). Spectroscopic analysis for (I): 1H NMR (300 MHz, DMSO-d 6): δ 8.8 (overlapping peaks, 2H), 8.2 (dd, J 1 = 10.00, J 2 = 3.33 Hz, 1H), 7.3–7.2 (overlapping peaks, 6H), 3.7 (q, J = 6.36 Hz, 2H), 2.9 (t, J = 7.58 Hz, 2H); IR (NaCl, ν, cm−1): 3361, 3124, 3070, 3036, 2969, 2938, 2860, 1622, 1592, 1550, 1525, 1501, 1452, 1418, 1380, 1337, 1313.
Compounds (II)–(IV) were prepared in a similar manner, using 1,5-difluoro-2,4-dinitrobenzene for (II) (yield 62%; m.p. 408–409 K), 2-cyano-4-nitrofluorobenzene for (III) (yield 73%; m.p. 427–429 K) and (3-nitro-4-fluorophenyl)methyl sulfone for (IV) (yield 63%; m.p. 414–416 K) in place of 2,4-dinitrofluorobenzene. Spectroscopic analysis for (II): 1H NMR (300 MHz, DMSO-d 6): δ 8.9 (s, 1H), 8.4 (t, J = 5.45 Hz, 2H), 7.3–7.2 (overlapping peaks, 10H), 5.8 (s, 1H), 3.6 (q, J = 6.97 Hz, 4H), 3.0 (t, J = 7.27 Hz, 4H); IR (NaCl, ν, cm−1): 3360, 1617, 1540, 1474, 1456, 1348, 1308. Spectroscopic analysis for (III): 1H NMR (300 MHz, DMSO-d 6): δ 8.4 (d, J = 3.33 Hz, 1H), 8.1 (dd, J 1 = 9.70, J 2 = 2.73 Hz, 1H), 7.6 (t, J = 5.76 Hz, 1H), 7.3–7.2 (overlapping peaks, 5H), 6.9 (d, J = 9.39 Hz, 1H), 3.5 (q, J = 6.36 Hz, 2H), 2.9 (t, J = 7.58 Hz, 2H); IR (NaCl, ν, cm−1): 3353, 2222, 1610, 1587, 1541, 1507, 1497, 1456, 1337, 1321. Spectroscopic analysis for (IV): 1H NMR (300 MHz, DMSO-d 6): δ 8.6 (t, J = 5.78 Hz, 1H), 8.5 (d, J = 2.42 Hz, 1H), 7.9 (dd, J 1 = 7.58, J 2 = 2.12 Hz, 1H), 7.3–7.2 (overlapping peaks, 6H), 3.7 (q, J = 5.78 Hz, 2H), 3.2 (s, 3H), 2.9 (t, J = 7.58 Hz, 2H); IR (NaCl, ν, cm−1): 3369, 3088, 3063, 3023, 2927, 2869, 1616, 1569, 1521, 1466, 1456, 1431, 1411, 1359, 1307.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 1 ▶. All H atoms were located from difference Fourier syntheses and refined isotropically without any restraints [N—H = 0.866 (15)–0.900 (17) Å and C—H = 0.898 (13)–1.006 (16) Å].
Table 1. Experimental details.
| (I) | (II) | (III) | (IV) | |
|---|---|---|---|---|
| Crystal data | ||||
| Chemical formula | C14H13N3O4 | C22H22N4O4 | C15H13N3O2 | C15H16N2O4S |
| M r | 287.27 | 406.44 | 267.28 | 320.36 |
| Crystal system, space group | Triclinic, P

|
Triclinic, P
|
Monoclinic, P21/n | Tetragonal, I41/a |
| Temperature (K) | 100 | 100 | 100 | 100 |
| a, b, c (Å) | 7.235 (2), 7.282 (2), 13.512 (4) | 8.1121 (7), 10.1819 (8), 13.1018 (11) | 7.4871 (5), 16.270 (1), 10.8432 (7) | 20.4639 (13), 20.4639 (13), 14.3952 (14) |
| α, β, γ (°) | 88.714 (5), 85.131 (5), 67.239 (5) | 93.466 (1), 106.595 (1), 106.932 (1) | 90, 94.384 (1), 90 | 90, 90, 90 |
| V (Å3) | 654.0 (4) | 979.88 (14) | 1317.00 (15) | 6028.3 (10) |
| Z | 2 | 2 | 4 | 16 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα | Mo Kα |
| μ (mm−1) | 0.11 | 0.10 | 0.09 | 0.24 |
| Crystal size (mm) | 0.20 × 0.10 × 0.02 | 0.20 × 0.10 × 0.05 | 0.40 × 0.20 × 0.10 | 0.40 × 0.20 × 0.20 |
| Data collection | ||||
| Diffractometer | Bruker APEX DUO CCD area-detector diffractometer | Bruker APEX DUO CCD area-detector diffractometer | Bruker APEX DUO CCD area-detector diffractometer | Bruker APEX DUO CCD area-detector diffractometer |
| Absorption correction | Multi-scan (SADABS; Sheldrick, 1996 ▶) | Multi-scan (SADABS; Sheldrick, 1996 ▶) | Multi-scan (SADABS; Sheldrick, 1996 ▶) | Multi-scan (SADABS; Sheldrick, 1996 ▶) |
| T min, T max | 0.978, 0.998 | 0.981, 0.995 | 0.964, 0.991 | 0.912, 0.955 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 9039, 3158, 2288 | 17364, 6868, 5289 | 23007, 4732, 4189 | 52424, 5524, 5155 |
| R int | 0.027 | 0.018 | 0.018 | 0.018 |
| (sin θ/λ)max (Å−1) | 0.661 | 0.767 | 0.765 | 0.765 |
| Refinement | ||||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.040, 0.104, 1.03 | 0.043, 0.121, 1.03 | 0.038, 0.113, 1.04 | 0.029, 0.086, 1.06 |
| No. of reflections | 3158 | 6868 | 4732 | 5524 |
| No. of parameters | 242 | 359 | 233 | 263 |
| H-atom treatment | All H-atom parameters refined | All H-atom parameters refined | All H-atom parameters refined | All H-atom parameters refined |
| Δρmax, Δρmin (e Å−3) | 0.29, −0.26 | 0.57, −0.21 | 0.56, −0.20 | 0.51, −0.31 |
Results and discussion
2,4-Dinitro-N-(2-phenylethyl)aniline, (I), crystallizes in the triclinic space group P
with one independent molecule in the asymmetric unit (Fig. 1 ▶). As we (Payne et al., 2010 ▶) and others (Panunto et al., 1987 ▶; Clegg et al., 1994 ▶) have observed previously, the adjacent amine and nitro groups form an intramolecular N—H⋯O hydrogen bond (Table 2 ▶). The ethyl group adopts an anti conformation [N1—C7—C8—C9 = −173.75 (12)°], with the result that the phenyl and aniline rings are nearly parallel. This is the same overall molecular conformation that was observed in the related compound N-[2-(2-formylphenyl)ethyl]-2-nitroaniline (Clegg et al., 1994 ▶).
Figure 1.
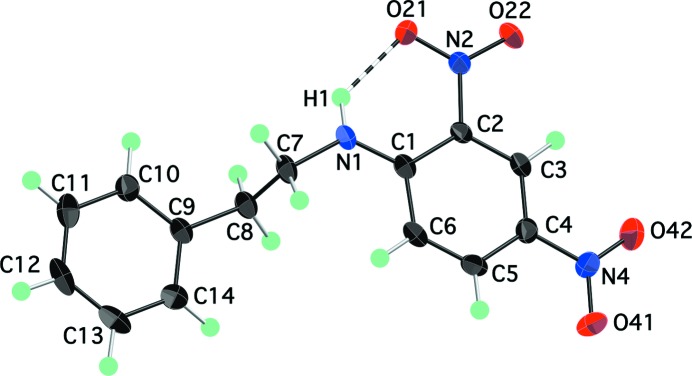
The molecular structure of (I), showing atom labels and 70% probability displacement ellipsoids for non-H atoms. The intramolecular hydrogen bond is shown as a dashed line.
Table 2. Hydrogen-bond geometry (Å, °) for (I) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O21 | 0.867 (19) | 1.983 (19) | 2.6377 (16) | 131.5 (16) |
| N1—H1⋯O21i | 0.867 (19) | 2.381 (19) | 3.0823 (18) | 138.3 (16) |
Symmetry code: (i)  .
.
Neighbouring molecules of (I) are linked into dimers across centres of inversion by an intermolecular N—H⋯O hydrogen bond between the amine group of one molecule and the nitro group of the other (Fig. 2 ▶). The amine H1 atom thus participates in a three-centre hydrogen bond with two nitro O21 atoms, and each O21 atom serves as an acceptor for both intra- and intermolecular hydrogen bonds (Table 2 ▶). The intermolecular H1⋯O21i distance of 2.381 (19) Å [symmetry code: (i) −x + 1, −y, −z + 1] is within the typical range for this type of N—H⋯O amine–nitro interaction (Panunto et al., 1987 ▶). The absence of a more extended hydrogen-bond network (i.e. chains) is a departure from what we had found in an earlier study of a related set of arenes with similar functional groups (Payne et al., 2010 ▶). Owing to the triclinic symmetry of (I), all of the arene rings in the crystal structure are parallel to one another and they are approximately parallel to the (110) plane. Although the plane-to-plane distance (ca 3.4 Å) is short enough for π–π interactions, the rings are staggered rather than face-to-face, suggesting that such interactions are not important in this structure.
Figure 2.

Molecules of (I) are linked into dimers across centres of inversion by N—H⋯O amine–nitro hydrogen bonds (dashed lines). The view is onto the (100) plane. [Symmetry codes: (#) x, y + 1, z; ($) −x + 1, −y + 1, −z + 1.]
4,6-Dinitro-N,N′-bis(2-phenylethyl)benzene-1,3-diamine, (II), differs from (I) only in having a second 2-phenylethylamine group in the 5-position on the nitro ring. As in (I), both amine H atoms form intramolecular N—H⋯O hydrogen bonds with the adjacent nitro groups (Table 3 ▶). Although the two halves of the molecule through the central ring are chemically identical, in the crystal structure the molecule possesses no internal symmetry (Fig. 3 ▶) because the two 2-phenylethyl groups adopt different conformations. One, as in (I), is anti [N1—C7—C8—C9 = 172.60 (8)°], while the other is gauche [N5—C15—C16—C17 = −66.94 (10)°]. As a result, the C1–C6 and C9–C14 rings are nearly parallel, but the C17–C22 ring is approximately orthogonal to the other rings.
Table 3. Hydrogen-bond geometry (Å, °) for (II) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O21 | 0.891 (15) | 1.935 (15) | 2.6318 (11) | 133.8 (13) |
| N5—H5⋯O41 | 0.900 (17) | 1.967 (16) | 2.6358 (11) | 129.8 (14) |
| N5—H5⋯O41i | 0.900 (17) | 2.800 (17) | 3.5877 (11) | 146.8 (13) |
Symmetry code: (i)  .
.
Figure 3.

The molecular structure of (II), showing atom labels and 70% probability displacement ellipsoids for non-H atoms. Intramolecular hydrogen bonds are shown as dashed lines.
The packing of (II) has features in common with that of (I). Compound (II) also crystallizes in the P
space group with one independent molecule in the asymmetric unit, and the molecules are linked into dimers across centres of inversion via three-centre N—H⋯O amine–nitro hydrogen bonds (Fig. 4 ▶ and Table 3 ▶). The intermolecular H5⋯O41i distance of 2.800 (17) Å in (II) [symmetry code: (i) −x + 2, −y + 2, −z + 1] is significantly longer than that in (I), but it is still in the observed range for a second amine–nitro contact (Panunto et al., 1987 ▶). Although there are two amine groups in (II), only that involving the gauche 2-phenylethyl group participates in intermolecular hydrogen bonds. Therefore, as was the case in (I), there are no hydrogen-bonded chains in (II). The molecules pack with the nitro and anti-phenyl rings approximately parallel to (10). Neighbouring rings are ca 3.5 Å apart and overlap more than in (I), suggesting greater π–π interaction.
Figure 4.

Molecules of (II) are linked into dimers across centres of inversion by weak N—H⋯O amine–nitro hydrogen bonds (dashed lines). The view is onto the (100) plane. [Symmetry codes: (#) x, y − 1, z; ($) −x + 2, −y + 1, −z + 1.]
2-Cyano-4-nitro-N-(2-phenylethyl)aniline, (III), differs from (I) only in having a cyano group in place of the 2-nitro group. Since the 2-nitro group adjacent to the amine is absent, there is no intramolecular hydrogen bond in (III). The molecule adopts the same anti conformation seen in (I) [N1—C7—C8—C9 = −171.53 (6)°] (Fig. 5 ▶). Molecules of (III) are linked into dimers across centres of inversion by N—H⋯N amine–cyano hydrogen bonds (Fig. 6 ▶ and Table 4 ▶). The nitro groups are not involved in any hydrogen bonds. A similar amine–cyano dimerization was observed in 2,6-bis(ethylamino)-3-nitrobenzonitrile (Payne et al., 2010 ▶), though in that case the molecules were also joined by three-centre N—H⋯O amine–nitro hydrogen bonds involving the other amine group. In (III), the single amine H atom is not available for additional interactions.
Figure 5.

The molecular structure of (III), showing atom labels and 70% probability displacement ellipsoids for non-H atoms.
Figure 6.

Molecules of (III) are linked into dimers across centres of inversion by N—H⋯N hydrogen bonds (dashed lines) between the amine H and cyano N atom. [Symmetry code: (#) −x + 1, −y, −z + 2.]
Table 4. Hydrogen-bond geometry (Å, °) for (III) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯N2i | 0.866 (15) | 2.300 (15) | 3.0287 (10) | 141.9 (12) |
Symmetry code: (i) 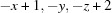 .
.
The packing in (III) shows some notable differences from that seen in (I). The compound crystallizes in the monoclinic space group P21/n (Z = 4), which does not require all of the rings to be oriented in the same direction. Instead, we see more of a herringbone pattern (Fig. 6 ▶) in which most of the nearby arene rings are not parallel to each other. The closest contacts between the aniline rings (ca 3.4 Å) involve rings that are mostly slipped past each other. In this, i.e. the apparent lack of strong π–π interactions, the structure of (III) is consistent with (I) and (II).
4-Methylsulfonyl-2-nitro-N-(2-phenylethyl)aniline, (IV), is modified from (I) in having a methylsulfonyl group in place of the 4-nitro group. The amine and 2-nitro groups form the expected intramolecular hydrogen bond but, unlike (I) and (III), the 2-phenylethyl group adopts a gauche conformation [N1—C7—C8—C9 = −61.20 (8)°] (Fig. 7 ▶). Such an arrangement was also observed in a 2,4-dinitroaniline derivative in which the 2-phenylethyl group is functionalized at the α-ethyl C atom (Williams et al., 2011 ▶). Unlike the other compounds studied here, there are no intermolecular hydrogen bonds of any significance in the structure of (IV). The closest H⋯O contact, H1⋯O22(−y +  , x −
, x −  , z − ) [3.26 (2) Å], is well beyond a reasonable H⋯A distance for a normal two-centre hydrogen bond. The absence of amine–sulfone interactions is surprising, as such hydrogen bonds have been shown to be favoured in similar systems (Glidewell et al., 2001 ▶; Glidewell & Ferguson, 1996 ▶; Bertolasi et al., 1993 ▶).
, z − ) [3.26 (2) Å], is well beyond a reasonable H⋯A distance for a normal two-centre hydrogen bond. The absence of amine–sulfone interactions is surprising, as such hydrogen bonds have been shown to be favoured in similar systems (Glidewell et al., 2001 ▶; Glidewell & Ferguson, 1996 ▶; Bertolasi et al., 1993 ▶).
Figure 7.

The molecular structure of (IV), showing atom labels and 70% probability displacement ellipsoids for non-H atoms. The intramolecular hydrogen bond is shown as a dashed line.
In addition to the lack of intermolecular hydrogen bonding, the crystal packing in (IV) is substantially different from that in (I)–(III). First, the compound crystallizes in the tetragonal system, space group I41/a (Z = 16). According to a previous study of over 29000 organic compounds (Mighell et al., 1983 ▶), only ca 2.9% are tetragonal. Of the 68 tetragonal space groups, I41/a is the second most popular among organic compounds, with over 11% of the occurrences being in that system. However, this is still only ca 0.34% of the total number of surveyed organic structures. As required by the 41 axis, molecules of (IV) are arranged in columns along the c axis, with successive molecules rotated by 90° with respect to each other (Fig. 8 ▶). The aniline rings are approximately parallel and slipped somewhat. The separation between the planes is ca 3.7 Å, but this is apparently sufficiently favourable to take precedence over hydrogen bonding. As the views in Fig. 8 ▶ suggest, the gauche conformation of the ethyl group may be a key factor in making this motif possible, as the phenyl rings are positioned conveniently around the periphery of the columns.
Figure 8.

(a) A packing diagram for (IV), showing half of the 16 molecules belonging to the I-centred tetragonal unit cell. Note the absence of any intermolecular N—H⋯O hydrogen-bonding interactions. (b) The same eight molecules shown in part (a) form columns along [001] with fourfold symmetry.
Conclusions
The results of this study, taken together with those of a previous study of amine- and nitro-substituted arenes (Payne et al., 2010 ▶), indicate that the introduction of changes in functionalization at a single point in the molecule can result in substantial changes in the molecular and crystal structures. In the earlier work, molecules with two amine groups and one nitro group formed hydrogen-bonded chains, while in the present work we find that molecules with a single amine group and one or two nitro groups form at most hydrogen-bonded dimers. In general, it appears that reducing the number of amine groups from two to one correlates with a reduction in the dimensionality of the hydrogen-bonding network from two (chains) to one (dimers). It is true, though, that the addition of the phenyl rings to the 2-phenylethyl groups could have played a role in this tendency towards reduced dimensionality. The structure of (III) demonstrates that N—H⋯N amine–cyano hydrogen bonds are preferred over N—H⋯O amine–nitro hydrogen bonds, so the introduction of a cyano group in these systems will result in a completely different set of intermolecular interactions. Structures (I) and (IV) show that changing a single functional group, in this case in the 4-position on the main ring, can lead to a change in the preferred conformation of the molecule, a disruption in the hydrogen-bonding pattern and a dramatically different packing structure.
Transformation of the precursors into NO release agents involves nitrosation of the amine groups, which would eliminate the intramolecular hydrogen bonding. Also, NO release agents are expected to function in an aqueous solution environment (i.e. in vivo). Thus, it is probably unlikely that inter- and intramolecular interactions in the solid-state structures of the precursors would correlate with the NO release behaviour. Our recent findings suggest that the characteristics of the organic group (length of alkyl chain, presence of alkyl or aryl substituents) on the former amine N atom bearing the NO group have the primary effect on the NO release rate (Yu et al., 2011 ▶; Curtis et al., 2013 ▶). Given this observation, it would be interesting to see how the NO release rate changes when one uses aliphatic cyclic amines of different ring sizes. Since NO leaves as a radical, it will also be of interest to see how the release rate changes when one moves from benzene derivatives to aromatic heterocycles and to larger naphthalene and anthracene-based compounds.
Supplementary Material
Crystal structure: contains datablock(s) C14H13N3O4, C22H22N4O4, C15H13N3O2, C15H16N2O4S, global. DOI: 10.1107/S0108270113025869/cu3041sup1.cif
Structure factors: contains datablock(s) C14H13N3O4. DOI: 10.1107/S0108270113025869/cu3041C14H13N3O4sup2.hkl
Structure factors: contains datablock(s) C22H22N4O4. DOI: 10.1107/S0108270113025869/cu3041C22H22N4O4sup3.hkl
Structure factors: contains datablock(s) C15H13N3O2. DOI: 10.1107/S0108270113025869/cu3041C15H13N3O2sup4.hkl
Structure factors: contains datablock(s) C15H16N2O4S. DOI: 10.1107/S0108270113025869/cu3041C15H16N2O4Ssup5.hkl
Supplementary material file. DOI: 10.1107/S0108270113025869/cu3041C14H13N3O4sup6.cml
Supplementary material file. DOI: 10.1107/S0108270113025869/cu3041C22H22N4O4sup7.cml
Supplementary material file. DOI: 10.1107/S0108270113025869/cu3041C15H13N3O2sup8.cml
Supplementary material file. DOI: 10.1107/S0108270113025869/cu3041C15H16N2O4Ssup9.cml
Table 5. Hydrogen-bond geometry (Å, °) for (IV) .
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯O21 | 0.882 (16) | 1.951 (16) | 2.6364 (9) | 133.4 (14) |
Acknowledgments
DKM acknowledges financial support from the National Institutes of Health (NIH) through Award No. R15HL106600 from the National Heart, Lung, and Blood Institute (NHLBI). The content is solely the responsibility of the authors and does not represent the official views of NHLBI or NIH. The authors thank the College of Natural Science and Mathematics of the University of Toledo for generous financial support of the X-ray diffraction facility.
Footnotes
Supplementary data for this paper are available from the IUCr electronic archives (Reference: CU3041). Services for accessing these data are described at the back of the journal.
References
- Bertolasi, V., Ferretti, V., Gilli, P. & Benedetti, P. G. (1993). J. Chem. Soc. Perkin Trans. 2, pp. 213–219.
- Bruker (2005). APEX2 Bruker AXS Inc., Madison, Wisconsin, USA.
- Cai, T. B., Wang, P. J. & Holder, A. A. (2005). Nitric Oxide Donors for Pharmaceutical and Biological Applications, edited by P. G. Wang, T. B. Cai & N. Taniguchi, pp. 3–31 and 59–60. Weinheim: Wiley-VCH.
- Clegg, W., Stanforth, S. P., Hedley, K. A., Raper, E. S. & Creighton, J. R. (1994). Acta Cryst. C50, 583–585.
- Curtis, B., Payne, T. J., Ash, D. E. & Mohanty, D. K. (2013). Bioorg. Med. Chem. 21, 1123–1135. [DOI] [PMC free article] [PubMed]
- Giles, T. D. (2006). J. Clin. Hypertens. 8 (Suppl. s12), 2–16. [DOI] [PMC free article] [PubMed]
- Glidewell, C. & Ferguson, G. (1996). Acta Cryst. C52, 2528–2530.
- Glidewell, C., Harrison, W. T. A., Low, J. N., Sime, J. G. & Wardell, J. L. (2001). Acta Cryst. B57, 190–200. [DOI] [PubMed]
- Mason, R. P. & Cockcroft, J. R. (2006). J. Clin. Hypertens. 8 (Suppl. s12), 40–52. [DOI] [PMC free article] [PubMed]
- Mighell, A. D., Himes, V. L. & Rodgers, J. R. (1983). Acta Cryst. A39, 737–740.
- Palmer, D. (2013). CrystalMaker CrystalMaker Software Ltd, Yarnton, Oxfordshire, England.
- Panunto, T. W., Urbanczyk-Lipkowska, Z., Johnson, R. & Etter, M. C. (1987). J. Am. Chem. Soc. 109, 7786–7797.
- Payne, T. J., Thurman, C. R., Yu, H., Sun, Q., Mohanty, D. K., Squattrito, P. J., Giolando, M.-R., Brue, C. R. & Kirschbaum, K. (2010). Acta Cryst. C66, o369–o373. [DOI] [PubMed]
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wang, J., Teng, Y.-H., Yu, H., Oh-Lee, J. & Mohanty, D. K. (2009). Polym. J. 41, 715–725.
- Williams, D. E., Dalisay, D. S., Patrick, B. O., Matainaho, T., Andrusiak, K., Deshpande, R., Myers, C. L., Piotrowski, J. S., Boone, C., Yoshida, M. & Andersen, R. J. (2011). Org. Lett. 13, 3936–3939. [DOI] [PMC free article] [PubMed]
- Yu, H., Payne, T. J. & Mohanty, D. K. (2011). Chem. Biol. Drug Des. 78, 527–534. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) C14H13N3O4, C22H22N4O4, C15H13N3O2, C15H16N2O4S, global. DOI: 10.1107/S0108270113025869/cu3041sup1.cif
Structure factors: contains datablock(s) C14H13N3O4. DOI: 10.1107/S0108270113025869/cu3041C14H13N3O4sup2.hkl
Structure factors: contains datablock(s) C22H22N4O4. DOI: 10.1107/S0108270113025869/cu3041C22H22N4O4sup3.hkl
Structure factors: contains datablock(s) C15H13N3O2. DOI: 10.1107/S0108270113025869/cu3041C15H13N3O2sup4.hkl
Structure factors: contains datablock(s) C15H16N2O4S. DOI: 10.1107/S0108270113025869/cu3041C15H16N2O4Ssup5.hkl
Supplementary material file. DOI: 10.1107/S0108270113025869/cu3041C14H13N3O4sup6.cml
Supplementary material file. DOI: 10.1107/S0108270113025869/cu3041C22H22N4O4sup7.cml
Supplementary material file. DOI: 10.1107/S0108270113025869/cu3041C15H13N3O2sup8.cml
Supplementary material file. DOI: 10.1107/S0108270113025869/cu3041C15H16N2O4Ssup9.cml