

Abstract
Human C-reactive protein (CRP) protects mice from lethality after infection with virulent Streptococcus pneumoniae type 3. For CRP-mediated protection, the complement system is required; however, the role of complement activation by CRP in the protection is not defined. Based on the in vitro properties of CRP, it has been assumed that protection of mice begins with the binding of CRP to pneumococcal C-polysaccharide on S. pneumoniae and subsequent activation of the mouse complement system. In this study, we explored the mechanism of CRP-mediated protection by utilizing two CRP mutants, F66A and F66A/E81A. Both mutants, unlike wild-type CRP, do not bind live virulent S. pneumoniae. We found that passively administered mutant CRP protected mice from infection as effectively as the wild-type CRP did. Infected mice injected with wild-type CRP or with mutant CRP lived longer and had lower mortality than mice that did not receive CRP. Extended survival was caused by the persistence of reduced bacteremia in mice treated with any CRP. We conclude that the CRP-mediated decrease in bacteremia and the resulting protection of mice are independent of an interaction between CRP and the pathogen and therefore are independent of the ability of CRP to activate mouse complement. It has been shown previously that the Fcγ receptors also do not contribute to such CRP-mediated protection. Combined data lead to the speculation that CRP acts on the effector cells of the immune system to enhance cell-mediated cytotoxicity and suggest investigation into the possibility of using CRP-loaded APC-based strategy to treat microbial infections.
C-reactive protein (CRP)4 is the prototypic human acute phase protein considered to be a multifunctional component of the acute phase response and innate host defense machinery (1–3). CRP displays Ca2+-dependent binding specificity for phosphocholine (PCh) residues present on C-polysaccharide (PnC) of the cell wall of Streptococcus pneumoniae (4). Once CRP is complexed with a ligand such as PnC, it activates the classical pathway of complement in human serum (5, 6). PnC-bound human CRP also activates mouse complement, although not through the classical pathway (7). The only other established in vitro effector function of CRP is to participate in phagocytosis that probably involves binding of CRP to FcγRs present on human and mouse phagocytic cells (8–13).
In mice, CRP is only a trace serum component and is not an acute phase protein (14). Therefore, mice have been used to define the in vivo functions of passively administered purified human CRP. In mouse models of infection, human CRP has been shown to be protective against lethal infection with S. pneumoniae (15–17). The mechanism of action of CRP in this protection is partially revealed. It perhaps involves the ability of CRP to recognize and bind to bacteria through PCh groups present on their surfaces and the subsequent activation of the complement system by pathogen-bound CRP (2). Using C3 knockout or cobra venom factor-treated decomplemented mice, it has been shown that a functioning complement system is required to observe full CRP-mediated protection (18–21). It is not known whether the activation of complement by CRP complexes in mouse serum is needed for CRP-mediated protection. The FcγRs do not play a role in CRP-mediated protection of mice from pneumococcal infection (21).
The purpose of this study was to determine whether the binding of CRP to bacteria was a prerequisite for the protection of mice from pneumococcal infection. We used site-directed mutagenesis and engineered two mutant forms of CRP, F66A and F66A/E81A, by mutating the PCh-binding site of CRP (22). Complete biochemical characterization of the CRP mutants has been reported previously, and their overall structure was found not to be different from the wild-type (WT) native and WT recombinant CRP (22). We used the CRP mutants that bind neither PnC nor live virulent S. pneumoniae (Ref. 22 and this article) in the protection experiments. If the binding of CRP to bacteria was required for the protection of mice from infection, then the mutant CRP should not be protective. Surprisingly, our results indicated that the protection was independent of binding of CRP to bacteria and hence was independent of the property of CRP to activate the complement system.
Materials and Methods
CRP mutants
The construction, expression, and characterization of the CRP mutants F66A and F66A/E81A have been described earlier (22). For stable expression of CRP mutants, Chinese hamster ovary cells were transfected with the mixture of 10 μg of mutant CRP cDNA-p91023 construct and 2 μg of pSV2neo vector (Invitrogen Life Technologies) using the FuGENE 6 reagent (Roche) as described previously (7). At 96 h posttransfection, stably transfected cells were selected by growth for another 2 wk in the culture medium supplemented with Geneticin sulfate. Chinese hamster ovary cell lines for both mutants were isolated by a series of subcloning steps.
Purification of CRP
The WT native human CRP was purified from pleural fluid by affinity chromatography on a PCh-Sepharose column followed by ion exchange chromatography and gel filtration chromatography, as described earlier (7). The F66A and F66A/E81A CRP were purified from cell culture supernatants by immunoaffinity chromatography with a polyclonal anti-CRP Ab-conjugated Sepharose column, as before (22, 23). Briefly, the culture medium containing mutant CRP was diluted in TBS and passed through the immunoaffinity column. After the column was washed with TBS, bound CRP was eluted with 50 mM glycine-HCl, pH 3.0, after which the pH was neutralized immediately with 1 M Tris, pH 9.0. Protein-containing fractions were pooled and dialyzed against TBS and subjected to ion exchange chromatography on the MonoQ column. CRP-containing fractions were pooled, concentrated, dialyzed against TBS, and judged to be pure based on denaturing SDS-PAGE. The native structure of the CRP mutants was confirmed by gel filtration.
Purification of CRP by immunoaffinity chromatography followed by ion exchange chromatography did not affect the functional properties of CRP. There was no difference in the PCh-binding and fibronectin-binding properties of WT CRP purified by either PCh affinity or immunoaffinity chromatography and subsequent ion exchange chromatography (22).
For the protection experiment shown in Fig. 5, purified WT CRP and F66A/E81A mutant CRP were treated with the Detoxi-Gel Endotoxin Removing Gel according to manufacturer's instructions (Pierce). Then, the concentration of endotoxin in CRP preparations and buffers was determined by using the Limulus amebocyte lysate kit QCL-1000 according to manufacturer's instructions (Cambrex).
FIGURE 5.

Effect of endotoxin on the CRP-mediated protection of mice infected with S. pneumoniae. Mice were injected with 0.5 × 108 CFU bacteria, with 150 μg of either WT or F66A/E81A CRP. Control groups A and B received 150 μl of TBS. The endotoxin levels (EU) are given in the parentheses. TBS or CRP was injected first; bacteria were injected 30 min later. Deaths were recorded twice per day for 10 days. Six male mice were used in all groups. The p values for the differences in the survival curves among groups A and B, A and C, A and D, and C and D are 0.30, 0.01, 0.004, and 0.87, respectively.
Mice, bacteria, protection experiments, and bacteremia
C57BL/6J mice (The Jackson Laboratory) were brought up and maintained according to protocols approved by the University Committee on Animal Care. Both male and female mice were 8–10 wk old when used in experiments. Virulent S. pneumoniae type 3, strain WU2, was prepared for infection of mice as described previously (7). Mice were injected i.v. with 108 CFU bacteria in 100 μl of TBS, unless otherwise mentioned, as described earlier (7). Mice were injected i.v. with 150 μg of WT or mutant CRP in 150 μl of TBS, 30 min before injecting bacteria, unless otherwise mentioned. Survival of mice was noted twice per day for 10 days. The plotting of the survival curves was done using GraphPad Prism 4 software (GraphPad). To determine p values for the differences in the survival curves among various groups, the survival curves were compared using the log rank test contained in the software. To determine bacteremia (CFU per milliliter), blood was collected daily for 5 days from the tip of the tail vein of each mouse. Blood was diluted in normal saline, plated on blood agar plates, and incubated for 18 h at 37°C in candle extinction jars before the colonies were counted. The plotting and statistical analyses of the bacteremia data were done using GraphPad Prism 4 software and the Mann-Whitney two-tailed test.
PnC binding assays
Binding activity of CRP for PnC was assessed by three assays. The first assay utilized purified PnC (Statens Serum Institut) and was dependent on the complement-activating property of CRP-PnC complexes. Microtiter wells were coated with PnC (40 μg/ml) in PBS. After blocking with PBS plus 1% BSA and rinsing with buffer A (PBS plus 0.1% BSA plus 1 mM CaCl2), purified WT and mutant CRP diluted in buffer B (buffer A plus 0.01% Tween 20) were added to the wells. After 1 h at 37°C, the wells were washed with buffer B and rinsed with buffer C (PBS plus 1% BSA plus 0.15 mM CaCl2 plus 0.5 mM MgCl2). Binding of CRP to PnC was evaluated by the subsequent deposition of C3 from the serum to the CRP-PnC complexes, as described earlier (24). For that determination, normal human serum diluted 1/40 in chilled buffer C was added to each well and incubated for 30 min at 37°C. The serum was preabsorbed with PnC-conjugated agarose for 3 h at 0°C to remove any CRP and anti-PnC Abs. PnC was linked to Tresyl-activated agarose (Tosh Bioscience) according to the manufacturer's instructions. The wells were then washed with buffer C, and murine anti-human C3d mAb (Quidel), diluted to 1 μg/ml in buffer C, was added to each well. After 1 h at room temperature, the wells were washed and developed with HRP-conjugated goat anti-mouse IgG (Pierce). Color was developed with ABTS reagent and measured at 405 nm in a microtiter plate reader (Molecular Devices).
The second PnC binding assay utilized killed S. pneumoniae type 3, strain WU2 (25). Bacteria were grown for 18 h at 37°C in Todd-Hewitt broth supplemented with 0.5% yeast extract. Bacteria were killed with 0.5% formalin in normal saline for 1 h at 37°C with shaking. After a washing with normal saline, the pellet was resuspended in TBS and adjusted to 1.2 × 108 bacteria/ml using the McFarland opacity standard (26). Aliquots (100 μl) of this suspension were transferred to microtiter wells. After the plate was centrifuged at 2000 rpm for 10 min, 200 μl of ethanol were added to the supernatant in each well. This mixture of ethanol and supernatant was immediately discarded, and 100 μl of methanol were added. After 5 min of fixation, the methanol was discarded, and the plates were dried at 37°C and stored at 4°C until use. For the assay, the wells were blocked with TBS plus 0.1% BSA. The binding curves were constructed using serial dilutions of purified WT and mutant CRP in buffer D (TBS, pH 7.0, plus 0.1% BSA plus 5 mM CaCl2 plus 0.01% Tween 20). After overnight incubation at 4°C, the wells were washed with buffer D. Affinity-purified anti-CRP mAb HD2.4 (23), diluted to 0.5 μg/ml in buffer D, was added to each well. After 1 h at room temperature, the wells were developed as in the first assay.
The third assay utilized live S. pneumoniae type 2, strain R36A (obtained from American Type Culture Collection). Bacteria (107 CFU) were incubated with increasing amounts of WT and mutant CRP in a final volume of 200 μl of Todd-Hewitt broth. The bacteria, in the presence of CRP, were grown overnight at 37°C. Then the bacteria were pelleted, washed with TBS three times, and resuspended in TBS containing 10 mM EDTA to elute the bound CRP. After 10 min, the supernatants were recovered by centrifugation. ELISA (23) was used to determine the amount of CRP in the supernatant.
Binding of CRP to live virulent S. pneumoniae type 3, strain WU2
Virulent bacteria (107 CFU) were incubated with WT and mutant CRP in a final volume of either 200 μl of Todd-Hewitt broth or 200 μl of mouse serum (1/2 diluted). The bacteria, in the presence of CRP, were grown for 4 h at 37°C. The bacteria were then pelleted, washed with TBS three times, and resuspended in TBS containing 10 mM EDTA to elute the bound CRP. After 10 min, the supernatants were recovered by centrifugation. ELISA (23) was used to determine the amount of CRP in the supernatant. In a separate assay, after growing bacteria in the presence of CRP for 4 h at 37°C, an aliquot of the culture was plated to determine the effect of CRP on the growth of bacteria.
Results
F66A and F66A/E81A mutant CRP, although incapable of binding to PnC, protect mice from infection
Combined results of three separate protection experiments using WT CRP and mutant CRP are shown in Fig. 1. The median survival time (the time taken for the death of 50% of mice) for mice injected with bacteria alone (control group) was 3 days. During this period, survival was 94 and 92% in the infected mice treated with WT CRP and F66A/E81A CRP, respectively. No death occurred in 3 days in the infected mice treated with F66A CRP. By the end of the 6th day, survival was 61% in WT CRP-treated mice, 83% in F66A CRP-treated mice, and 75% in F66A/E81A CRP-treated mice compared with only 6% of the mice that survived in the control group. More than 60% mice survived for up to 10 days in mice treated with any CRP. Thus, both WT and mutant CRP decreased mortality and prolonged survival of infected mice. These results suggested that the protection of mice conferred by CRP was independent of its PnC recognition activity and that the direct interaction of CRP with bacteria was not required for the protection of mice from infection.
FIGURE 1.

Survival curves of mice infected with S. pneumoniae. Mice were injected with 108 CFU of bacteria, with or without 150 μg of either WT, or F66A, or F66A/E81A CRP. CRP was injected first; bacteria were injected 30 min later. Deaths were recorded twice per day for 10 days. The data are combined from three separate experiments with six mice in each group: Experiment 1 was performed with six male mice in each of group A, B, and C; Experiment 2 was performed with six male mice in each group A, B, and D; Experiment 3 was performed with six female mice in each group A, B, and D. The p values for the differences in the survival curves among groups A and B, A and C, and A and D are 0.0001, 0.001, and 0.0001, respectively. The p values for the differences in the survival curves among groups B and C, B and D, and C and D are 0.33, 0.42, and 0.73, respectively.
Without binding to bacteria, CRP reduces bacteremia in the infected mice
The bacteremia values, determined every day for 5 days in each mouse from the protection experiment shown in Fig. 1, are plotted as scatter diagram (Fig. 2). Based on the median bacteremia values, in all four groups, bacteremia was <1000 CFU/ml of blood 1 day postinfection. In the control group, the bacteremia increased after day 1 and remained high until day 4. Mice died once bacteremia approached ~107 CFU/ml. In the mice treated with either WT or any of the CRP mutants, there was no significant increase in bacteremia past day 1. Statistically significant differences in bacteremia were observed between the control and CRP-treated groups by day 2, and the differences persisted until day 4. Thus, the positive effects of CRP treatment were realized within 2 days of infection. Based on the median bacteremia values, CRP (WT or mutants)-treated mice had 100- to 1000-fold fewer bacteria on any day compared with the control group. These results indicated that the increased resistance to infection in CRP-treated mice was associated with the maintenance of a reduced bacteremia and that the binding specificity of CRP for PnC had no role in maintaining the reduced bacteremia.
FIGURE 2.

Bacteremia in mice treated with WT and mutant CRP. Blood samples were collected from each surviving mouse shown in Fig. 1 for the first 5 days postinfection. Bacteremia was determined by plating. Each dot represents one mouse. The horizontal line in each group of mice represents the median value of bacteremia in that group. A bacteremia value of >107 indicates a dead mouse. The p values for the differences among groups A and B, A and C, and A and D, on days 2, 3, and 4 are <0.008. The p values for the differences among groups B and C, B and D, and C and D on days 2, 3, and 4 are >0.05.
Comparison of binding of WT and mutant CRP to PnC
The finding that F66A and F66A/E81A mutant CRP protected mice from infection was unexpected. We therefore reassessed the PnC-binding activity of these CRP mutants using three assays. In the first assay, the PnC-binding ability of CRP was determined by the extent of complement activation by CRP-PnC complexes. The WT CRP bound to PnC in a dose-dependent manner as reflected by the dose-dependent increase in C3 deposition (Fig. 3A). The F66A CRP was ~50 times less efficient than WT CRP in activating complement, indicating drastically reduced binding of F66A CRP to PnC compared with WT CRP. In contrast, negligible C3 deposition was detected with F66A/E81A CRP reflecting the loss of PnC-binding activity of the double mutant. In the second assay, utilizing extracts from killed bacteria (Fig. 3B), the F66A mutant CRP exhibited some binding to killed bacteria, but the F66A/E81A CRP failed to bind killed bacteria throughout the dose-response range. The results of the third assay using live S. pneumoniae strain R36A are shown in Fig. 3C. As shown, neither F66A nor F66A/E81A CRP bound to R36A bacteria.
FIGURE 3.

Binding of WT and mutant CRP to PnC. A representative experiment is shown. A, Deposition of C3 reflecting the formation of CRP-PnC complexes as a function of CRP dose. Microtiter wells were coated with PnC. Increasing concentrations of purified CRP were then added to the wells. Normal human serum was then added as the source of complement. Deposited C3 was detected by using anti-human C3d mAb. Values on the y-axis represent binding of anti-C3d to C3 deposited on PnC-CRP complexes. B, Binding of CRP to killed strain WU2 bacteria. Microtiter wells were coated with killed bacteria. Increasing concentrations of purified CRP were then added to the wells. Bound CRP was detected using anti-CRP mAb HD2.4 as a reporter. C, Binding of CRP by live S. pneumoniae strain R36A. A fixed number of bacteria were grown in the presence of increasing amount of CRP, as shown on the x-axis. Then, CRP that bound to bacteria was eluted and measured by ELISA. The amount of bound CRP is shown on the y-axis.
Binding of WT and mutant CRP by live virulent S. pneumoniae strain WU2
We next determined the binding of WT and mutant CRP to bind to S. pneumoniae strain WU2 that we used in the protection experiments in mice. Fig. 4A shows the results of the binding assay performed in Todd-Hewitt broth. As shown, neither F66A nor F66A/E81A CRP was bound to live bacteria. Fig. 4B shows the results of the binding assay performed in mouse serum. The mutant CRP did not bind to bacteria even in the presence of mouse serum. Both the WT CRP and the mutant CRP themselves did not kill pneumococci in culture, suggesting that the antibiotics were not present in CRP preparations (data not shown). The data obtained from these assays confirmed that the protection of mice seen with the mutant CRP in Fig. 1 did not involve direct interaction between CRP and bacteria.
FIGURE 4.
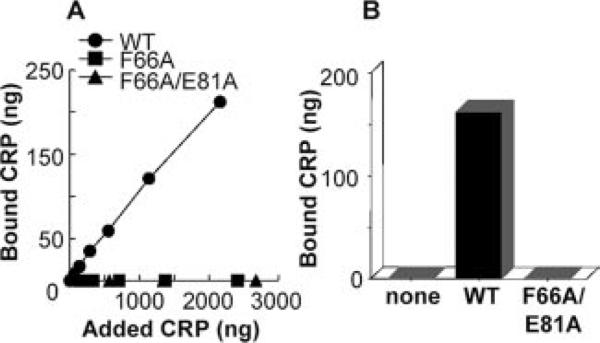
Binding of CRP by live virulent S. pneumoniae strain WU2. A, Binding in Todd-Hewitt broth. A fixed number of live bacteria were grown in the presence of increasing amount of CRP, as shown on the x-axis. Then, CRP that bound to bacteria was eluted and measured by ELISA. Amount of bound CRP is shown on the y-axis. B, Binding in mouse serum. A fixed number of live bacteria were grown in the presence of 2000 ng of WT or mutant CRP. Then, CRP that bound to bacteria was eluted and measured by ELISA. The amount of bound CRP is shown on the y-axis.
The endotoxin, present in the CRP preparations, was not responsible for the protection of mice from infection
We performed another protection experiment in which endotoxin-free TBS (group A) and endotoxin-containing TBS (group B) were included as controls (Fig. 5). The concentrations of endotoxin in TBS, in WT CRP, and in F66A/E81A CRP mutant were comparable. The median survival time for mice in both control groups A and B was 2.5 days. This result indicated that the amount of endotoxin given to mice was not sufficient to confer protection from infection. Similar to the results shown in Fig. 1, there was no significant difference in the protection mediated by WT and mutant CRP.
Administration of CRP 36 h postinfection does not confer protection
To determine whether CRP was required during the first 24 h of infection to protect mice, we injected CRP 36 h after infection and compared the protection seen in the mice treated with CRP 30 min before infection. As shown in Fig. 6, mice preinjected with CRP were protected. In contrast, the survival curves for the mice treated with CRP 36 h postinfection and for the control group were overlapping. Thus, the actions of CRP required for the protection of mice from infection required the presence of CRP in the early stages of infection.
FIGURE 6.

Survival curves of mice infected with S. pneumoniae. Mice were injected with 108 CFU bacteria, either alone (●), or with 150 μg of WT CRP. CRP was administered either 30 min before infection (■) or 36 h after infection (□). Six male mice were used in all groups. Deaths were recorded twice per day for 10 days.
Discussion
We investigated the mechanism of CRP-mediated protection of mice from S. pneumoniae infection. To test the hypothesis that the protection is due to the binding of CRP to bacteria, we compared the protective ability of WT CRP that binds S. pneumoniae with that of F66A and F66A/E81A CRP mutants that do not bind to bacteria in mice infected with a lethal dose of S. pneumoniae. Our major findings were: 1) all CRP species, WT, F66A, and F66A/E81A, decreased mortality and prolonged survival of infected mice. The binding ability of CRP for bacteria was irrelevant for the protective function of CRP in the infected mice; 2) increased resistance to infection in CRP (WT or mutant)-treated mice was associated with the maintenance of reduced bacteremia. Reduction in bacteremia and the maintenance of reduced bacteremia occurred without the binding of CRP to the pathogen; 3) the protective function of CRP was not seen when mice received CRP 36 h postinfection.
Our data indicate that CRP has no role in the decrease in bacteremia during the first 24 h of infection. Instead, CRP contributes by reducing bacteremia after day 1 and also by preventing an increase in the already reduced bacteremia thereafter. This contribution of CRP to the protection of mice from infection did not require binding of CRP to pneumococci. These results are consistent with the finding that CRP also protects mice from infection with Salmonella typhimurium, a pathogen to which CRP does not bind (27). The CRP-mediated protection of mice from LPS challenge (28–30) has also been shown to be independent of binding of CRP to LPS (31).
CRP activates the complement system only when it is bound to a ligand (5, 6, 32). If the binding of CRP to bacteria is not involved in the protection of mice from infection, then the property of CRP complexes to activate complement is also not involved. Because it is established that complement plays a key role in the protection of mice from S. pneumoniae infection (33), our data suggest that activation of complement by bacteria participates in the protection but activation of complement by CRP does not. The protection involves combined but independent effects of complement and CRP. We hypothesize that complement serves as the first line of defense and that CRP protects mice from those bacteria that escape complement attack.
In this study, our aim was to compare the protective ability of WT and mutant CRP. For that, it was necessary that we used a system where WT CRP showed maximum protection. It has been shown previously that complement is required to observe maximum protection mediated by WT CRP (19–21); for those reasons, we did not pursue experiments with C3 knockout or cobra venom factor-treated decomplemented mice.
Under certain experimental conditions, CRP binds to FcγRs (9–13). However, because FcγRs have been shown not to participate in the CRP-mediated protection of mice from S. pneumoniae infection (21), we did not investigate the role of FcγR-bearing cells as the mechanism for the protective function of CRP observed with the CRP mutants. Thus, presumably both effector functions of CRP, complement activation phagocytosis, do not constitute the mechanism of CRP-mediated protection of mice from pneumococcal infection.
A single prior injection of CRP is sufficient to protect mice from infection (Ref. 15 and this article). CRP exerts its effect only when injected 6 h before to 2 h after the infection (34). If CRP acted by binding to bacteria and by subsequent complement activation, then CRP should have been able to protect even when supplied at later stages of infection. This was not the case. Combined data therefore support our previous proposal that CRP possibly contributes to protection of mice from infection by binding to factor H-coated S. pneumoniae (7). Factor H-coated S. pneumoniae are capable of escaping the attack by the pathogen-activated alternative pathway of complement (35). Because CRP binds to factor H (36, 37), CRP may be binding to factor H-coated bacteria to prevent them from escaping the complement attack (7). To test this hypothesis, the construction of a CRP mutant incapable of binding to both, bacteria and factor H, is in progress.
Because CRP also protects mice from infectious agents that do not interact with factor H, an alternate hypothesis therefore is that CRP contributes to the protection of mice from infection by acting directly on the effector cells of the immune system. Most likely, APCs would take up CRP and present it in the processed form on their surfaces. APCs exposing CRP peptides may recognize the pathogenic polysaccharides, in addition to their recognition by TLRs, and enhance cell-mediated cytotoxicity to reduce bacteremia. It has been shown previously that a synthetic peptide derived from CRP that did not contain either Phe66 or Glu81 binds PCh (38). Two lines of evidences support the notion that CRP can become an autoantigen: 1) upon induction of CRP in mice transgenic for human CRP, T cells have been found that recognize CRP epitopes on thymic and splenic APCs (39); and 2) anti-CRP autoantibodies directed toward nonnative forms of CRP have been found to be present in some patients with systemic autoimmune diseases (40, 41). The hypotheses that we propose for the mechanisms of action only involve the role of human CRP in mouse pneumococcal infection.
The in vitro PCh-binding and bacteria-binding properties of CRP would be expected to correlate to its in vivo protective functions. Although these properties of CRP did not contribute to protection of mice in our i.v. infection model, we cannot rule out the possibility that the binding of CRP to bacteria may be relevant to prevent colonization of bacteria in the intranasal infection model. CRP has been shown to block the attachment of PCh-expressing bacteria to the receptors for platelet-activating factor on the host cells (42). In addition, the PCh-binding ability of CRP was required for the protection of mice from challenge with platelet-activating factor (43).
CRP binds a wide variety of bacteria including several serotypes of S. pneumoniae (44, 45), Haemophilus influenzae (46), and Neisseriae spp. (47). Although protection experiments have not been performed with all of them, current data indicate nonspecific resistance conferred by CRP to bacterial infection. In conclusion, although alternative interpretations may be possible, combined data suggest that CRP functions in vivo as a surveillance molecule for pathogens that escape complement, or that escape TLRs, and probably for those bacteria that escape killing mediated by CD4+ T cells (48, 49). Dendritic cell-based vaccinations are in use for malignant diseases (50, 51), and our findings raise the possibility of utilizing CRP-sensitized APC-based treatment strategy to treat microbial infections. Adoptive transfer experiments must be performed to validate the use of CRP in cell-based vaccinations.
Acknowledgments
We are grateful to Dr. David E. Briles (University of Alabama, Birmingham, AL) for the gift of S. pneumoniae type 3, strain WU2. We thank the staff of our Division of Laboratory Animal Resources for help with the mouse experiments.
Footnotes
This work was supported by National Institutes of Health Grant RO1HL71233.
Abbreviations used in this paper: CRP, C-reactive protein; PnC, pneumococcal C-polysaccharide; PCh, phosphocholine; WT, wild type.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Kushner I. The phenomenon of the acute phase response. Ann. NY Acad. Sci. 1982;389:39–48. doi: 10.1111/j.1749-6632.1982.tb22124.x. [DOI] [PubMed] [Google Scholar]
- 2.Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol. Immunol. 2001;38:189–197. doi: 10.1016/s0161-5890(01)00042-6. [DOI] [PubMed] [Google Scholar]
- 3.Agrawal A. CRP after 2004. Mol. Immunol. 2005;42:927–930. doi: 10.1016/j.molimm.2004.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Volanakis JE, Kaplan MH. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc. Soc. Exp. Biol. Med. 1971;136:612–614. doi: 10.3181/00379727-136-35323. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan MH, Volanakis JE. Interaction of C-reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal C-polysaccharide and with the choline phosphatides, lecithin, and sphingomyelin. J. Immunol. 1974;112:2135–2147. [PubMed] [Google Scholar]
- 6.Siegel J, Rent R, Gewurz H. Interactions of C-reactive protein complexes with the complement system. I. Protamine-induced consumption of complement in acute phase sera. J. Exp. Med. 1974;140:631–647. doi: 10.1084/jem.140.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suresh MV, Singh SK, Ferguson DA, Jr., Agrawal A. Role of the property of C-reactive protein to activate the classical pathway of complement in protecting mice from Streptococcus pneumoniae infection. J. Immunol. 2006;176:4369–4374. doi: 10.4049/jimmunol.176.7.4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mold C, Du Clos TW, Nakayama S, Edwards KM, Gewurz H. C-reactive protein reactivity with complement and effects on phagocytosis. Ann. NY Acad. Sci. 1982;389:251–262. doi: 10.1111/j.1749-6632.1982.tb22141.x. [DOI] [PubMed] [Google Scholar]
- 9.Bharadwaj D, Stein M-P, Volzer M, Mold C, Du Clos TW. The major receptor for C-reactive protein on leucocytes is Fcγ receptor II. J. Exp. Med. 1999;190:585–590. doi: 10.1084/jem.190.4.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodman-Smith KB, Melendex AJ, Campbell I, Harrison PT, Allen JM, Raynes JG. C-reactive protein-mediated phagocytosis and phospholipase D signalling through the high-affinity receptor for immunoglobulin G (FcγRI). Immunology. 2002;107:252–260. doi: 10.1046/j.1365-2567.2002.01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi M, Tridandapani S, Zhong W, Coggeshall KM, Mortensen RF. C-reactive protein induces signaling through FcγRIIa on HL-60 granulocytes. J. Immunol. 2002;168:1413–1418. doi: 10.4049/jimmunol.168.3.1413. [DOI] [PubMed] [Google Scholar]
- 12.Heuertz RM, Schneider GP, Potempa LA, Webster RO. Native and modified C-reactive protein bind different receptors on human neutrophils. Int. J. Biochem. Cell Biol. 2005;37:320–335. doi: 10.1016/j.biocel.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Stein M-P, Mold C, Du Clos TW. C-reactive protein binding to murine leukocytes requires Fcγ receptors. J. Immunol. 2000;164:1514–1520. doi: 10.4049/jimmunol.164.3.1514. [DOI] [PubMed] [Google Scholar]
- 14.Whitehead AS, Zahedi K, Rits M, Mortensen RF, Lelias JM. Mouse C-reactive protein: generation of cDNA clones, structural analysis, and induction of mRNA during inflammation. Biochem. J. 1990;266:283–290. doi: 10.1042/bj2660283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mold C, Nakayama S, Holzer TJ, Gewurz H, Du Clos TW. C-reactive protein is protective against Streptococcus pneumoniae infection in mice. J. Exp. Med. 1981;154:1703–1708. doi: 10.1084/jem.154.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yother J, Volanakis JE, Briles DE. Human C-reactive protein is protective against fatal Streptococcus pneumoniae infection in mice. J. Immunol. 1982;128:2374–2376. [PubMed] [Google Scholar]
- 17.Szalai AJ, Briles DE, Volanakis JE. Human C-reactive protein is protective against fatal Streptococcus pneumoniae infection in transgenic mice. J. Immunol. 1995;155:2557–2563. [PubMed] [Google Scholar]
- 18.Nakayama S, Mold C, Gewurz H, Du Clos TW. Opsonic properties of C-reactive protein in vivo. J. Immunol. 1982;128:2435–2438. [PubMed] [Google Scholar]
- 19.Horowitz J, Volanakis JE, Briles DE. Blood clearance of Streptococcus pneumoniae by C-reactive protein. J. Immunol. 1987;138:2598–2603. [PubMed] [Google Scholar]
- 20.Szalai AJ, Briles DE, Volanakis JE. Role of complement in C-reactive protein-mediated protection of mice from Streptococcus pneumoniae. Infect. Immun. 1996;64:4850–4853. doi: 10.1128/iai.64.11.4850-4853.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mold C, Rodic-Polic B, Du Clos TW. Protection from Streptococcus pneumoniae infection by C-reactive protein and natural antibody requires complement but not Fcγ receptors. J. Immunol. 2002;168:6375–6381. doi: 10.4049/jimmunol.168.12.6375. [DOI] [PubMed] [Google Scholar]
- 22.Agrawal A, Simpson MJ, Black S, Carey MP, Samols D. A C-reactive protein mutant that does not bind to phosphocholine and pneumococcal C-polysaccharide. J. Immunol. 2002;169:3217–3222. doi: 10.4049/jimmunol.169.6.3217. [DOI] [PubMed] [Google Scholar]
- 23.Suresh MV, Singh SK, Agrawal A. Interaction of calcium-bound C-reactive protein with fibronectin is controlled by pH: In vivo implications. J. Biol. Chem. 2004;279:52552–52557. doi: 10.1074/jbc.M409054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agrawal A, Shrive AK, Greenhough TJ, Volanakis JE. Topology and structure of the C1q-binding site on C-reactive protein. J. Immunol. 2001;166:3998–4004. doi: 10.4049/jimmunol.166.6.3998. [DOI] [PubMed] [Google Scholar]
- 25.Barka N, Tomasi J-P, Stadtsbaeder S. Use of whole Streptococcus pneumoniae cells as a solid phase sorbent for C-reactive protein measurement by ELISA. J. Immunol. Methods. 1985;82:57–63. doi: 10.1016/0022-1759(85)90224-8. [DOI] [PubMed] [Google Scholar]
- 26.Paik G. Reagents, stains, and miscellaneous test procedures. In: Balows A, Hausler WJ Jr., Traunt JP, Lennette EH, editors. Manual of Clinical Microbiology. 3rd Ed. American Society for Microbiology; Washington, DC: 1980. pp. 1000–1024. [Google Scholar]
- 27.Szalai AJ, VanCott JL, McGhee JR, Volanakis JE, Benjamin WH., Jr. Human C-reactive protein is protective against fatal Salmonella enterica serovar typhimurium infection in transgenic mice. Infect. Immun. 2000;68:5652–5656. doi: 10.1128/iai.68.10.5652-5656.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia D, Samols D. Transgenic mice expressing rabbit C-reactive protein are resistant to endotoxemia. Proc. Natl. Acad. Sci. USA. 1997;94:2575–2580. doi: 10.1073/pnas.94.6.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noursadeghi M, Bickerstaff MCM, Gallimore JR, Herbert J, Cohen J, Pepys MB. Role of serum amyloid P component in bacterial infection: protection of the host or protection of the pathogen. Proc. Natl. Acad. Sci. USA. 2000;97:14584–14589. doi: 10.1073/pnas.97.26.14584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mold C, Rodriguez W, Rodic-Polic B, Du Clos TW. C-reactive protein mediates protection from lipopolysaccharide through interactions with FcγR. J. Immunol. 2002;169:7019–7025. doi: 10.4049/jimmunol.169.12.7019. [DOI] [PubMed] [Google Scholar]
- 31.Hirschfield GM, Herbert J, Kahan MC, Pepys MB. Human C-reactive protein does not protect against acute lipopolysaccharide challenge in mice. J. Immunol. 2003;171:6046–6051. doi: 10.4049/jimmunol.171.11.6046. [DOI] [PubMed] [Google Scholar]
- 32.Kishore U, Ghai R, Greenhough TJ, Shrive AK, Bonifati DM, Gadjeva M, Waters P, Kojouharova MS, Chakraborty T, Agrawal A. Structural and functional anatomy of the globular domain of the complement protein C1q. Immunol. Lett. 2004;95:113–128. doi: 10.1016/j.imlet.2004.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown JS, Hussell T, Gilliland SM, Holden DW, Paton JC, Ehrenstein MR, Walport MJ, Botto M. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc. Natl. Acad. Sci. USA. 2002;99:16969–16974. doi: 10.1073/pnas.012669199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakayama S, Gewurz H, Holzer T, Du Clos TW, Mold C. The role of the spleen in the protective effect of C-reactive protein in Streptococcus pneumoniae infection. Clin. Exp. Immunol. 1983;54:319–326. [PMC free article] [PubMed] [Google Scholar]
- 35.Jarva H, Janulczyk R, Hellwage J, Zipfel PF, Björck L, Meri S. Streptococcus pneumoniae evades complement attack and opsonophagocytosis by expressing the pspC locus-encoded Hic protein that binds to short consensus repeats 8–11 of factor H. J. Immunol. 2002;168:1886–1894. doi: 10.4049/jimmunol.168.4.1886. [DOI] [PubMed] [Google Scholar]
- 36.Jarva H, Jokiranta TS, Hellwage J, Zipfel PF, Meri S. Regulation of complement activation by C-reactive protein: targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8–11. J. Immunol. 1999;163:3957–3962. [PubMed] [Google Scholar]
- 37.Giannakis E, Jokiranta TS, Male DA, Ranganathan S, Ormsby RJ, Fischetti VA, Mold C, Gordon DL. A common site within factor H SCR7 responsible for binding heparin, C-reactive protein and streptococcal M protein. Eur. J. Immunol. 2003;33:962–969. doi: 10.1002/eji.200323541. [DOI] [PubMed] [Google Scholar]
- 38.Swanson SJ, Mortensen RF. Binding and immunological properties of a synthetic peptide corresponding to the phosphorylcholine-binding region of C-reactive protein. Mol. Immunol. 1990;27:679–687. doi: 10.1016/0161-5890(90)90011-n. [DOI] [PubMed] [Google Scholar]
- 39.Klein TC, Döffinger R, Pepys MB, Rüther U, Kyewski B. Tolerance and immunity to the inducible self antigen C-reactive protein in transgenic mice. Eur. J. Immunol. 1995;25:3489–3495. doi: 10.1002/eji.1830251242. [DOI] [PubMed] [Google Scholar]
- 40.Bell SA, Faust H, Schmid A, Meurer M. Autoantibodies to C-reactive protein (CRP) and other acute-phase proteins in systemic autoimmune diseases. Clin. Exp. Immunol. 1998;113:327–332. doi: 10.1046/j.1365-2249.1998.00655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sjowall C, Bengtsson T, Skogh T. CRP and anti-CRP autoantibodies in systemic lupus erythematosus. Curr. Rheumatol. Rev. 2005;1:81–89. [Google Scholar]
- 42.Gould JM, Weiser JN. The inhibitory effect of C-reactive protein on bacterial phosphorylcholine platelet-activating factor receptor-mediated adherence is blocked by surfactant. J. Infect. Dis. 2002;186:361–371. doi: 10.1086/341658. [DOI] [PubMed] [Google Scholar]
- 43.Black S, Wilson A, Samols D. An intact phosphocholine binding site is necessary for transgenic rabbit C-reactive protein to protect mice against challenge with platelet-activating factor. J. Immunol. 2005;175:1192–1196. doi: 10.4049/jimmunol.175.2.1192. [DOI] [PubMed] [Google Scholar]
- 44.Mold C, Rodgers CP, Kaplan RL, Gewurz H. Binding of human C-reactive protein to bacteria. Infect. Immun. 1982;38:392–395. doi: 10.1128/iai.38.1.392-395.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Beaufort AJ, Langermans JAM, Matze-van der Lans AM, Hiemstra PS, Vossen JM, van Furth R. Difference in binding of killed and live Streptococcus pneumoniae serotypes by C-reactive protein. Scand. J. Immunol. 1997;46:597–600. doi: 10.1046/j.1365-3083.1997.d01-171.x. [DOI] [PubMed] [Google Scholar]
- 46.Weiser JN, Pan N, McGowan KL, Musher D, Martin A, Richards J. Phosphorylcholine on the lipopolysaccharide of Haemophilus influenzae contributes to persistence in the respiratory tract and sensitivity to serum killing mediated by C-reactive protein. J. Exp. Med. 1998;187:631–640. doi: 10.1084/jem.187.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serino L, Virji M. Genetic and functional analysis of the phosphorylcholine moiety of commensal Neisseria lipopolysaccharide. Mol. Microbiol. 2002;43:437–448. doi: 10.1046/j.1365-2958.2002.02755.x. [DOI] [PubMed] [Google Scholar]
- 48.Van Kooyk Y, Engering A, Lekkerkerker AN, Ludwig IS, Geijtenbeek TB. Pathogens use carbohydrates to escape immunity induced by dendritic cells. Curr. Opin. Immunol. 2004;16:488–493. doi: 10.1016/j.coi.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 49.Malley R, Trzcinski K, Srivastava A, Thompson CM, Anderson PW, Lipsitch M. CD4+ T cells mediate antibody-independent acquired immunity to pneumococcal colonization. Proc. Natl. Acad. Sci. USA. 2005;102:4848–4853. doi: 10.1073/pnas.0501254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moll H. Dendritic cells and host resistance to infection. Cell Microbiol. 2003;5:493–500. doi: 10.1046/j.1462-5822.2003.00291.x. [DOI] [PubMed] [Google Scholar]
- 51.Grunebach F, Muller MR, Brossart P. New developments in dendritic cell-based vaccinations: RNA translated into clinics. Cancer Immunol. Immunother. 2005;54:517–525. doi: 10.1007/s00262-004-0605-x. [DOI] [PMC free article] [PubMed] [Google Scholar]