

Abstract
Objective
Telmisartan, an angiotensin II type 1 (AT1) receptor blocker, and amlodipine, a calcium channel blocker, are antihypertensive agents clinically used as monotherapy or in combination. They exert beneficial cardiovascular effects independently of blood pressure lowering and classic mechanisms of action. In this study, we investigate molecular mechanisms responsible for the off-target effects of telmisartan and telmisartan-amlodipine in endothelial cells (EC), using an unbiased genomic approach.
Approach and Results
In human umbilical vein endothelial cells, microarray analysis of gene expression followed by pathway enrichment analysis and quantitative PCR validation revealed that telmisartan modulates the expression of key genes responsible for cell cycle progression and apoptosis. Amlodipine’s effect was similar to control. EC exposed to telmisartan, but not amlodipine, losartan or valsartan, exhibited a dose-dependent impairment of cell growth and failed to enter the S-phase of the cell cycle. Similarly, telmisartan inhibited proliferation in COS-7 cells lacking the AT1 receptor. In telmisartan-treated EC, phosphorylation and activation of Akt as well as MDM2 was reduced, leading to accumulation of p53 in the nucleus, where it represses the transcription of cell cycle promoting genes. Phosphorylation of GSK3β was also reduced, resulting in rapid proteolytic turnover of CyclinD1. Telmisartan induced downregulation of proapoptotic genes and protected EC from serum starvation- and 7-ketocholesterol-induced apoptosis.
Conclusions
Telmisartan exerts antiproliferative and antiapoptotic effects in EC. This may account for the improved endothelial dysfunction observed in the clinical setting.
Keywords: telmisartan, endothelial cells, proliferation, Akt pathway, apoptosis
Introduction
Hypertension represents a well-established risk factor for the development of cardiovascular disease (CVD), and randomized clinical trials in hypertensive patients clearly demonstrate that effective antihypertensive therapy resulting in blood pressure (BP) control reduces morbidity and mortality.1 Nonetheless, patients with controlled hypertension have a higher risk of developing CVD as compared to normotensive persons, due to the presence of other risk factors, including dyslipidemia, insulin resistance, glucose intolerance, and obesity.2
Many classes of drugs are currently used to treat hypertension as monotherapy or in combination: diuretics, beta-blockers, angiotensin receptor blockers (ARBs), renin inhibitors, angiotensin converting enzyme inhibitors, and calcium channel blockers (CCBs). Among the clinically relevant ARBs and CCBs, both telmisartan (TLM) and amlodipine (AML) have been shown to exert additional beneficial cardiovascular effects independently of BP reduction.3
Experimental and clinical evidence demonstrated that TLM induces regression of cardiac hypertrophy, enhances endothelium-dependent relaxation and improves endothelial function and arterial stiffness in hypertensive patients.4-6 Furthermore, growing evidence is establishing its antiatherosclerotic properties; TLM was shown to protect from development and progression of atherosclerosis, and to promote plaque stability.7-9 All of these actions are independent of BP lowering or angiotensin II type 1 (AT1) receptor antagonism, and in most of these studies, partial peroxisome proliferator-activated receptor gamma (PPARγ) agonism by TLM was proven accountable for the reported beneficial effects. Importantly, the cardiovascular effects of TLM have been linked also to increased nitric oxide (NO) availability, combined with anti-inflammatory and anti-oxidative properties on the endothelium.10-12
AML is a third generation L-type CCB, originally developed as potent and long-lasting vasodilator because of its ability to bind to and to block L-type calcium channels. The reduced calcium influx into vascular smooth muscle cells (VSMC) results in smooth muscle relaxation and vasodilation. Interestingly, AML exhibits additional biological or pleiotropic actions independent of the interaction with calcium channels, by acting on cell types other than VSMC or cells in which L-type calcium channels are not expressed or play only minor roles such as endothelial cells (EC).13 In clinical trials, AML was shown to reduce ischemia in patients with coronary artery disease and hospitalizations for unstable angina and revascularization, as well as the need for revascularization in patients with stable angina and to slow down the progression of existing atherosclerotic lesions and the onset of new ones.14-16 As for TLM, these effects are independent of BP lowering or calcium channel blockade and a link to NO biology has been suggested. Indeed, AML was shown to induce NO release and to reduce oxidative stress and inflammation, thus improving endothelial function. To explain this unexpected ability, it has been shown that AML alters caveolae integrity, increases caveolin-free endothelial nitric oxide synthase, leading to potentiated NO production in response to agonists.17
Clinical evidence demonstrated that the TLM-AML combination generates a dose-dependent BP-lowering effect significantly greater than that of either agent administered as monotherapy.18 Such a combination is likely to provide also a better cardiovascular protection compared to monotherapies, and clinical trials are currently corroborating this hypothesis.19 Improvement of endothelial dysfunction by therapy with TLM alone or TLM-AML combination represents an attractive strategy to reduce onset and progression of CVD. However, an exhaustive understanding of the molecular mechanisms underlying the beneficial effects of these drugs on the endothelium is missing. Therefore, the aim of the present study was to provide a comprehensive analysis of the potential pleiotropic actions of these agents on EC using an unbiased genomic approach. Here, we show that TML alone induces a state of EC quiescence by regulating networks of genes influencing cell growth and apoptosis.
Materials and Methods
Reagents
All reagents were purchased from Sigma-Aldrich unless otherwise specified.
Cell culture
Human umbilical vein endothelial cells (HUVEC) were obtained from the Yale University Vascular Biology and Therapeutics Core facility, plated on 0.1% gelatin coated dishes in M199 media supplemented with endothelial cell growth supplement (ECGS), 10% fetal bovine serum (FBS), penicillin-streptomycin and glutamine and used between passages 2 and 4. Human umbilical artery endothelial cells (HUAEC, Clonetics Lonza) were plated on 0.1% gelatin coated dishes in EBM-2 supplemented with EGM2-MV SingleQuots (Lonza) and 5% FBS and used between passages 6 and 8. COS-7 cells were cultured in DMEM supplemented with 10% FBS, penicillin-streptomycin and glutamine. Cultures were kept in a humidified incubator at 37°C in an atmosphere containing 5% CO2. In all experiments, culture media were supplemented with the indicated concentrations of TLM (Sigma-Aldrich, cat. n. T8949 or Boehringer-Ingelheim, Ridgefield, CT), losartan (LOS, Cayman chemical, cat. n. 10006594), valsartan (VAL, Sigma-Aldrich, cat. n. SML0142) or AML (Sigma-Aldrich, cat. n. A5605) in dimethyl sulfoxide (DMSO) or an equivalent volume of DMSO alone, as control, for the indicated times. Media with drugs were replaced every 24 hours.
Gene expression analysis
HUVEC were cultured for 24 hours in complete media containing DMSO, 100 μmol/L TLM or both 100 μmol/L TLM and 5 μmol/L AML. Total RNA was isolated using the miRNeasy Mini kit (Qiagen) according to the manufacturer’s instructions. RNA samples were assessed with a NanoDrop 2000c spectrophotometer (Thermo Scientific) and run on a bioanalyzer to determine acceptable quality and quantity. In order to generate sense-strand cDNA, samples were processed using The Ambion® WT Expression Kit (Applied Biosystems), followed by the GeneChip® WT Terminal Labeling Kit (Affymetrix) for fragmentation and biotin labeling, according to the manufacturer’s protocol. Samples were then hybridized for 16 hours at 45°C to the Affymetrix Whole Transcript GeneChip Human Gene 1.0 ST array using the GeneChip® Hybridization, Wash, and Stain Kit (Affymetrix). Microarray analysis was carried out on two biological replicates per group. In accordance with MIAME (Minimum Information About a Micro-array Experiment) regulations, all data are available at the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE42808.
Pathway analysis and data mining of microarray data
Microarray data were filtered by signal intensity to exclude transcripts whose signal was below background level. Only transcripts with significantly different expression levels between groups were included in the pathway enrichment analysis, which was performed using the MetaCore database and software suite.
Validation of microarray data
Relevant microarray data were validated by quantitative RT-PCR. Total RNA samples collected from HUVEC treated with DMSO, 100 μmol/L TLM, 5 μmol/L AML or both TLM and AML for 12, 24, 48 or 72 hours were retro-transcribed using the RT2 First Strand Kit (SABiosciences) and then assayed using Custom Human RT2 Profiler PCR Arrays with proprietary wet-bench validated primers (SABiosciences), according to the manufacturer’s instructions. Alternatively, retro-transcription was done using the iScript cDNA Synthesis Kit (Bio-Rad), followed by quantitative PCR using the iQ SYBR Green Supermix (Bio-Rad) and primers designed according to published sequences: VDAC1 fwd-CGGAATAGCAGCCAAGTATCA, rev-CTGGCTTTAGAGTCTGAGTGTATC; VDAC2 fwd-GGTTCAGCTGTCTTTGGTTATG, rev-GTAGCCCACTGCAAAGTTATTC; SLC25A4 fwd-GTCTCTGTCCAAGGCATCATTA, rev-TCACACTCTGGGCAATCATC; DIABLO fwd-CGCAGATCAGGCCTCTATAAC, rev-CCAGCTTGGTTTCTGCTTTC; BCL2L2 fwd-CTATAGGTGTGGGCACATGAAA, rev-CGTTCCCTAAATCCCACTCATC. Validation experiments were carried out on three biological replicates and averaged.
Immunoblot analysis
HUVEC were serum starved (0.5% FBS, no ECGS) overnight. During the last 30 min before stimulation, cells were pre-incubated with DMSO or 100 μmol/L TLM, followed by stimulation with complete media containing 10% FBS and ECGS for the indicated times in the presence of DMSO or 100 μmol/L TLM. Cells were then washed with ice-cold PBS and immediately resuspended in lysis buffer (50 mmol/L Tris-HCl, 1% NP-40, 0.1% SDS, 0.1% Deoxycholic Acid, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, protease and phosphatase inhibitors) with the aid of cell scrapers, sonicated and incubated for 30-45 min on ice. Protein extracts (30-100 μg) were separated by SDS-PAGE and then transferred to 0.45 micron nitrocellulose membranes (Bio-Rad). Membranes were probed with primary antibodies against phospho-Ser473 (clone D9E, cat n.4060), phospho-Thr308 (clone L32A4, cat. n. 5106) and total Akt (cat. n. 9272), phospho-Ser166 MDM2 (cat. n. 3521), p53 (clone 7F5, cat. n. 2527), Histone H3 (clone D1H2, cat. n. 4499), phospho-Ser9 (cat. n. 9336) and total GSK3β (clone 27C10, cat. n. 9315, Cyclin D1 (clone DCS6, cat. n.2926) (all from Cell Signaling Technology) and Hsp90 (Santa Cruz Biotechnology, cat. n. 13119), followed by species specific secondary antibodies anti-IgG conjugated with either AlexaFluor 680 (Invitrogen) or IRDye800 (Rockland). Blots were washed and visualized using a LI-COR Odyssey imager. Each Immunoblot analysis was carried out on five biological replicates. Densitometric analyses were performed using the Image J software.
Immunofluorescence
HUVEC were seeded on 8-well chamber slides coated with 0.1% gelatin at a density of 5×103 cells/well. Once adherent, cells were treated as described in the immunoblot analysis section (see above). Cells were then fixed in 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.3% Trinton X-100 in PBS for 30 min at RT. Cells were then incubated in 5% goat serum diluted in 0.3% Triton X-100 for 1 hour at RT, followed by incubation overnight at +4°C with a primary antibody against p53 (Cell Signaling Technology, clone 1C12, cat. n. 2524) diluted in PBS/0.3% Triton X-100. Cells were then incubated with a goat anti-mouse AlexaFluor 568 secondary antibody (Invitrogen) for 1 hour at RT, in the dark. A matched isotype control was included in each staining to check for unspecific binding of the primary antibody. Slides were mounted with fluorescence mounting medium containing DAPI and analyzed using a ZEISS Axiovert 200M fluorescence microscope. Nuclear fluorescence intensity was measured for each cell using the ‘count density (red)’ tool in the Image-Pro Plus software and then normalized by the number of nuclei analyzed in each group.
Functional assays on cultured cells
Cell growth
HUVEC, HUAEC or COS-7 cells (4 to 15×104 cells/well) were seeded in 6-well plates and serum starved (HUVEC: 0.5% FBS, no ECGS overnight; HUAEC: EBM-2 media, no FBS for 3 hours; COS-7: 0.5% FBS overnight). Cells were then stimulated with complete media containing 10% FBS and ECGS (HUVEC), EGM2-MV media containing 5% FBS (HUAEC), or 10% FBS (COS-7) for the indicated times in the presence of DMSO, various concentrations of TLM (0.1 to 100 μmol/L), 5 μmol/L AML, both TLM and AML or various concentrations of LOS or VAL (1 to 100 μmol/L). After trypsinization, the number of cells was counted with a hemocytometer. Three replicates of each condition were performed in each of three independent experiments.
Cell proliferation
Cells (5×103 cells/well) were seeded in 96-well plates until adherent , serum starved and treated as described above (cell growth). In the last 20 hours before assay, cells were incubated in the presence of BrdU. BrdU incorporation was detected using the Cell Proliferation ELISA, BrdU colorimetric kit (Roche, cat. n. 11647229001) according to the manufacturer’s instructions. Five replicates of each condition were performed in each of three independent experiments.
Apoptosis assay
Cells were pre-incubated with DMSO or TLM for 24 hours in complete media containing 10% FBS and ECGS (HUVEC) or 5% FBS (HUAEC) and then treated with 1) growth factors- and serum-free media containing 0.1% albumin for 4 hours or 2) complete media containing 10% FBS and ECGS supplemented with either DMSO (vehicle) or 20 μg/mL 7-ketocholesterol (7-KC) for 20 hours (HUVEC only), in the presence of DMSO or TLM. The extent of apoptosis was immediately assessed by measuring Caspase-3 activity by using the Caspase-3 Colorimetric Assay kit (R&D Systems, cat. n. BF3100), according to the manufacturer’s instruction. To confirm cell death by apoptosis, HUVEC treated as described above were stained with the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit, following the manufacturer’s instruction, and observed using a ZEISS Axiovert 200M fluorescence microscope. Three replicates of each condition were performed in each of three independent experiments.
Statistical analysis
Microarray datasets were compared by one tailed unpaired t test. Results are presented as means±SEM. All differences which returned a P≤0.05 were considered significant and taken in consideration for further analysis. All experiments where the effects of two variables (e.g. time and treatment or two different treatments) were tested were analyzed by 2-way ANOVA followed by Bonferroni post hoc tests. Differences between two groups were compared by unpaired Student t test. P≤0.05 was considered significant.
Results
Telmisartan modulates endothelial gene expression
In order to assess the effects of TLM, AML and TLM+AML on endothelial gene expression in an unbiased manner, we used an Affymetrix human whole transcript microarray which includes 30000 genes. Total RNA samples were obtained from primary cultures of human umbilical vein endothelial cells (HUVEC) treated with DMSO, TLM or TLM+AML for 24 hours. TLM significantly modulated the expression of about 1700 genes, while TLM and AML together significantly modulated about 1500 genes (http://www.ncbi.nlm.nih.gov/geo, accession number GSE42808). Interestingly, MetaCore pathway enrichment analysis revealed that several cell cycle and proapoptotic genes were downregulated by TLM alone or in combination with AML as compared to vehicle DMSO (Table 1).
Table 1.
MetaCore Pathway Analysis
| Biological Process |
Probe Set ID |
Gene Symbol |
Transcript description | Fold change TLM (% of DMSO) |
p-value | Fold change TLM+AML (% of DMSO) |
p-value |
|---|---|---|---|---|---|---|---|
| Cell cycle | 8124492 | HIST1H2BK | histone cluster 1, H2bk | −46 | 0.002 | −39 | 0.01 |
| 7968637 | CCNA1 | Cyclin A1 | −17 | 0.02 | −24 | 0.01 | |
| 7983969 | CCNB2 | Cyclin B2 | −10 | ns | −27 | 0.01 | |
| 8043602 | NCAPH | non-SMC condensin I complex, subunit H |
−12 | 0.03 | −22 | 0.01 | |
| 8067167 | AURKA | Aurora-A | −13 | 0.06 | −19 | 0.02 | |
| 8156290 | CKS2 | CDC28 protein kinase regulatory subunit 2 |
−14 | ns | −18 | 0.01 | |
| 7987636 | OIP5 | Opa interacting protein 5 | −11 | 0.03 | −17 | 0.02 | |
| 7899688 | KPNA6 | Importin (karyopherin)-alpha 6 | −8 | 0.03 | −9 | 0.0001 | |
| 8053610 | ANAPC1 | anaphase promoting complex subunit 1 | 9 | 0.01 | 7 | 0.01 | |
|
| |||||||
|
Apoptosis
and survival |
8098576 | SLC25A4 | solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 4 |
−12 | 0.01 | −14 | 0.07 |
| 8114145 | VDAC1 | Voltage-dependent anion channel 1 | −12 | 0.02 | −14 | 0.02 | |
| 8042335 | VDAC2 | Voltage-dependent anion channel 2 | −9 | ns | −13 | 0.01 | |
| 7967230 | DIABLO | Smac/Diablo | −7 | 0.04 | −12 | 0.02 | |
| 7973377 | BCL2L2 | Apoptosis regulator Bcl-W | 10 | 0.02 | 5 | 0.06 | |
Telmisartan prevents the cyclic expression of cell cycle related genes thus inhibiting endothelial cell proliferation
On the basis of functional relevance of genes regulating cell cycle progression, we evaluated the differential expression of selected genes by real-time PCR over a time course of 72 hours. Notably, TLM prevented the typical cyclic expression of Cyclin A1 (CCNA1), Cyclin B2 (CCNB2), Opa interacting protein 5 (OIP5), Aurora-A (AURKA), CDC28 protein kinase regulatory subunit 2 (CKS2), and Histone cluster 1, H2bk (HIST1H2BK) at multiple time points (Figure 1A-F). AML alone had no effect compared to vehicle and did not influence the actions of TLM when used in combination. As a result of TLM reducing the expression of cell cycle related genes, serum and growth factor-induced cell growth was inhibited by TLM (Figure 2A, i) and HUVEC failed to enter the S-phase of the cell cycle as shown by the prominent decrease in BrdU incorporation at 48 hours after stimulation (Figure 2A, ii). As expected, AML alone did not affect cell growth or BrdU incorporation, consistent with the idea that the above effects on gene expression are solely TLM-dependent, thus prompting us to focus only on this agent. To test the dose-dependent effect of TLM on cell proliferation, HUVEC were treated with TLM at various concentrations from 0.1 to 100 μmol/L. TLM inhibited cell growth at a starting concentration of 10 μmol/L, whereas lower concentrations had no effect (Figure 2B, i). Accordingly, BrdU incorporation measured at 48 hours after treatment was significantly decreased already at 10 μmol/L, with no significant effect at lower concentrations (Figure 2B, ii). Interestingly, TLM inhibited also the serum and growth factor-induced cell growth of human umbilical artery endothelial cells (HUAEC) in a dose-dependent manner, thus suggesting that its antiproliferative action is not cell type specific (Figure 2C, i-ii).
Figure 1.

TLM prevents the expression of cell cycle related genes. Bar graphs illustrate quantitative PCR analyses of serum and growth factor-induced expression of genes regulating cell cycle progression in HUVEC treated with 100 μmol/L TLM, 5 μmol/L AML or TLM+AML, over a time course of 72 hours. n=3; *P<0.05 vs DMSO.
Figure 2.

TLM inhibits endothelial cell proliferation. A, Growth curve (i) and BrdU incorporation assay (ii) in HUVEC treated with 100 μmol/L TML, 5 μmol/L AML or TLM+AML for the indicated times. B, Dose-dependent effects of TLM on cell growth (i) and BrdU incorporation (ii) in HUVEC. C, Dose-dependent effects of TLM on cell growth (i) and BrdU incorporation (ii) in HUAEC. n=3; *P<0.05 vs DMSO.
TLM inhibits cell cycle progression independently of AT1 receptor blockade
Although all experiments were performed in the absence of exogenously added angiotensin II, we asked whether the observed antiproliferative effect could be due to AT1 receptor blockade by TLM. We therefore exposed HUVEC to two other ARBs, losartan (LOS) and valsartan (VAL), using concentrations similar to those used in the experiments involving TLM. While LOS did not influence cell proliferation even at the highest concentration tested, VAL seemed to induce a delay in cell growth at aconcentration of 100 μmol/L (Figure 3A, i-ii). However, BrdU incorporation at 48 hours after treatment with 100 μmol/L VAL did not differ from vehicle, suggesting that the difference in cell number was due to cell toxicity rather than an actual effect on cell cycle (Figure 3A, iii). In addition, we treated fibroblast-like COS-7 cells, which do not express AT1 receptor20, with 100 μmol/L TLM and evaluated serum-induced cell growth. Similar to HUVEC and HUAEC, TLM treatment resulted in a marked inhibition of cell proliferation and S-phase entry, as documented by significantly reduced BrdU incorporation at 72 hours after stimulation (Figure 3B, i-ii).
Figure 3.
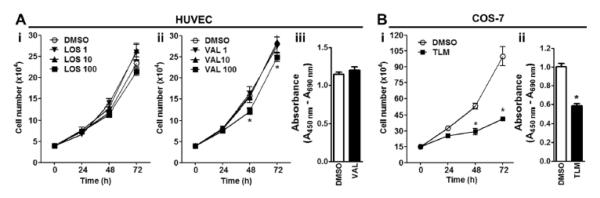
TLM-induced inhibition of cell proliferation is independent of AT1 receptor blockade. A, Dose-dependent effects of LOS or VAL on cell growth (i and ii, respectively) and BrdU incorporation (iii) in HUVEC. B, Growth curve (i) and BrdU incorporation assay (ii) in COS-7 cells treated with 100 μmol/L TML for the indicated times. n=3; *P<0.05 vs DMSO.
Telmisartan negatively modulates the Akt signaling pathway
To uncover the molecular mechanisms underlying the antiproliferative effect of TLM, we performed a bioinformatic analysis of the cell cycle related genes modulated by TLM using GeneWeb analysis of gene interactions (SABioscience platform). The expression of some genes (e.g. CCNA1, AURKA and CKS2) is reportedly repressed by p53,21-23 while others (e.g. HIST1H2BK) are under the positive transcriptional control of E2F. Both p53 and E2F stability and thus activity can be regulated by the Akt pathway via MDM2 and CyclinD1, respectively. It is, indeed, well established that the Akt pathway regulates proliferation and cell survival.24-26 We therefore investigated the activation of Akt and downstream signaling over a time course spanning from 10 min to 72 hours after serum stimulation. Western blot analyses showed that TLM treatment significantly inhibited the full activation of Akt by reducing phosphorylation at the mammalian target of rapamycin complex 2 (mTORC2) dependent site S473 and the PDK1 dependent site, T308, throughout the time course (Figure 4A and quantified in Figure 4B, i-ii). Interestingly, Akt-dependent phosphorylation of MDM2 at the S166 residue was also decreased, particularly at the later time points, rendering the ubiquitin ligase less active (Figure 4A and quantified in 4B, iii). MDM2 mediates p53 ubiquitination and degradation.26 We therefore hypothesized that since MDM2 was less active, p53 would accumulate in the nucleus and act as a repressor for genes important for cell cycle progression. Consistent with this hypothesis, immunofluorescence and Western blot analyses showed that TLM-treated HUVEC retained higher amounts of p53 in the nucleus as compared to control, starting at 24 hours until 72 hours after serum stimulation (Figure 4C i-iii and quantified in 4C, ii-iv). In addition, Akt-dependent phosphorylation of GSK3β (S9 residue) was assessed. As expected, TLM induced a decrease of GSK3β phosphorylation, resulting in higher kinase activity (Figure 4A and quantified in 4B, iv). GSK3β is known to trigger rapid proteolytic CyclinD1 turnover.24 Consistent with higher GSK3β activity in TLM-treated HUVEC, CyclinD1 protein levels increased only slightly following serum stimulation and remained low throughout the time course (Figure 4A and quantified in 4B, v).
Figure 4.

Negative modulation of Akt signaling accounts for antiproliferative action of TLM. Immunoblot analyses (A) and averaged densitometric data (B) show the effects of 100 μmol/L TLM on the dynamic phosphorylation of Akt (i and ii), MDM2 (iii), GSK3β (iv) and on CyclinD1 levels (v) over a time course of 72 hours in HUVEC. C, Representative immunofluorescence images (i), immunoblot analyses (iii) and respective averaged fluorescence intensity quantification (ii) and averaged densitometric data (iv) of nuclear p53 in HUVEC treated as in A. Scale bar: 50μm. n=5; for all data in B and C, P<0.05 vs DMSO by 2-way ANOVA for both the effect of time and treatment on fold-change.
Telmisartan protects endothelial cells from serum starvation- and 7-ketocholesterol-induced apoptosis
TLM treatment resulted in the downregulation of Voltage-dependent anion channel 1 (VDAC1), Voltage-dependent anion channel 2 (VDAC2), Solute carrier family 25 (mitochondrial carrier; adenine nucleotide translocator), member 4 (SLC25A4) and Direct inhibitor of apoptosis-binding protein with low pI (DIABLO), involved in the induction of apoptosis via the mitochondrial pathway and in the upregulation of BCL2-like 2 (BCL2L2), which reportedly promotes cell survival27 (Table 1). First, we validated the microarray results by real-time PCR (Figure 5A) and then investigated whether TLM could influence the response of EC to apoptotic stimuli. TLM significantly repressed Caspase-3 activation following serum-starvation and growth factor deprivation in both HUVEC and HUAEC (Figure 5B, i and 5C, respectively). Moreover, serum-starved cells undergoing apoptosis in the control group exhibited characteristic apoptotic bodies formation and stained positively for annexin V whereas TLM-treated cells did not (Figure 5B, ii-iii). Oxysterols constitute the major toxic component in oxidized low-density lipoprotein (ox-LDL) and are present in human atheromatous lesions.28 Oxysterols, such as 7-ketocholesterol (7-KC) contribute to endothelial dysfunction and induce endothelial cell apoptosis via the mitochondrial pathway.29, 30 Accordingly, control HUVEC treated with 7-KC showed a significant increase in Caspase-3 activity, while TLM exerted a protective effect (Figure 5D).
Figure 5.

TLM protects EC from apoptosis. A, Bar graph illustrates quantitative RT-PCR analyses of pro- and anti-apoptotic genes after treatment with 100 μmol/L TML for 24 hours in HUVEC. B to D, Bar graphs document Caspase-3 activity in extracts of HUVEC and HUAEC that were serum-starved and growth factors-deprived (A, i and B) or exposed to 7-KC (D) in the presence of DMSO or 100 μmol/L TLM. Representative microphotographs illustrate the characteristic apoptotic bodies formation (A ii, scale bar: 50μm) and Annexin V positivity (A, iii, scale bar: 200μm) of serum-starved and growth factors-deprived HUVEC. n=3; *P<0.05 vs DMSO; #P<0.05 vs DMSO+0% FBS or 7-KC.
Discussion
Although an increasing numbers of clinical and experimental results indicate the effectiveness of TLM and AML in cardiovascular protection, independently of their BP-lowering action, little is known about the molecular mechanisms responsible for the observed pleiotropic effects. Given the importance of the endothelium in the maintenance of vascular homeostasis, in the current study we investigated the effects of TLM and TLM+AML combination on a well-defined model of endothelium using an unbiased genomic approach.
In the absence of angiotensin II stimulation, TLM alone or in combination with AML significantly blunted the expression of several genes, whose encoded products are involved in the regulation of the cell cycle. Among these, CyclinA1 and CyclinB2 are involved in the control of the cell cycle at the G1/S (start) and G2/M (mitosis) transitions, respectively.31, 32 Knockdown of OIP5 expression has been reported to inhibit cell growth of several cancer cell lines.33, 34 The serine/threonine kinase Aurora-A plays a pivotal role in several mitotic events including the establishment of mitotic spindle, centrosome duplication, centrosome separation as well as maturation, chromosomal alignment, spindle assembly checkpoint, and cytokinesis.35 CKS2 associates with cyclin-dependent kinases and has been shown to play a direct role in cell cycle regulation. Its knockdown causes cessation of proliferation and cell cycle arrest due to impaired transcription of key genes involved in cell cycle progression.36 Finally, Histone cluster 1, H2bk is a core component of the nucleosome and influences the compaction of chromatin into higher order structures. Our results revealed that the contribution of AML to the overall downregulation of the cell cycle machinery is negligible, while TLM is solely accountable for inducing a state of endothelial cell quiescence. As expected from our gene expression studies, we found that TLM inhibited cell proliferation in HUVEC as well as HUAEC in a dose-dependent manner beginning at a concentration of 10 μmol/L, while AML had no effect. Moreover, COS-7 cells, which lack the AT-1 receptor, displayed cell growth impairment when treated with TLM, underscoring that the antiproliferative effect of TLM relies neither on AT-1 receptor blockade nor on the cell-type used. This finding is in line with previous studies demonstrating a dose-dependent antiproliferative action of TLM on human vascular smooth muscle cells, cardiac fibroblasts and murine macrophages, by activating PPARγ-dependent and independent pathways.8, 37, 38 Interestingly, our gene expression analysis did not highlight any significant changes in the expression of neither PPARγ itself nor genes downstream of PPARγ, suggesting that TLM influences endothelial cell physiology via PPARγ-independent mechanisms. In addition, our data support the notion that TLM activation of PPARγ is cell type-specific.
In this study, we interrogated potential mechanisms responsible for the antiproliferative effects of TLM in EC by investigating its impact on Akt activation and downstream signaling. In line with another study showing that TLM inhibits vascular smooth muscle cell proliferation by inhibiting Akt activation,37 we found that TLM attenuates Akt phosphorylation and activation, ultimately resulting in accumulation of p53 in the nucleus, where it represses the transcription of genes necessary for cell cycle progression. In addition, TLM-treated EC displayed lower amounts of CyclinD1 as compared to control, which suggests diminished activation of E2F-dependent G1/S-phase gene expression. Taken together, we demonstrate a novel mechanism of action of TLM in EC, beyond its ability to block AT1 receptor or activate PPARγ. How TLM interferes with Akt activation remains speculative, although the time course of our experiments would point to interference with the initial steps of the pathway, i.e. diminished generation of phosphoinositides through reduction of phosphatidylinositol 3-kinase (PI3K) activity or enhancement of phosphatase and tensin homolog (PTEN) activity, or perhaps via direct inhibition of mTORC2 or PDK1.
Despite the negative modulation of the Akt signaling pathway, TLM treatment did not result in elevated baseline cell death as compared to control. On the contrary, TLM protected EC from apoptosis induced by serum/growth factors withdrawal or 7-KC treatment. Given that the Akt pathway is not completely abrogated by TLM, and the extent of its activation seems to be enough to support cell survival, the antiapoptotic effect seems to be due to a direct downregulation of key components of the apoptotic machinery in mitochondria, i.e. the channels participating in the formation of the permeability transition pore complex, responsible for the release of cytochrome c into the cytosol, leading to activation of caspases: Adenine nucleotide translocator, ANT (encoded by the gene SLC25A4), VDAC1, VDAC2.39, 40 Second mitochondria-derived activator of caspases (Smac)/Diablo, another mitochondrial gene whose expression is downregulated by TLM, promotes apoptosis by binding and blocking the activity of Inhibitor of apoptosis protein (IAP), a negative regulator of apoptosis.41 On the other hand, TLM promotes endothelial cell survival by upregulating the BCL2L2 gene expression, which encodes the antiapoptotic Bcl-2 family member Bcl-W.27
Given the higher proliferation rate of the endothelium in atherosclerotic susceptible regions42 and the role that endothelial proliferation and apoptosis play in the stability of the atherosclerotic plaque,43, 44 the finding that TLM promotes endothelial cell quiescence and survival, together with its known anti-inflammatory and anti-oxidative effect on the endothelium, may have important implications for the anti-atherogenic and plaque stabilizing actions of this agent.
In line with most of the studies in the literature comparing the effects of TLM to those of other ARBs in multiple cell types,8, 11, 37, 38 two other ARBs tested in our study, LOS and VAL, failed to inhibit endothelial cell growth, underscoring the fact that TLM holds unique antiproliferative properties, not shared by other drugs belonging to the same family and that this effect is entirely independent of AT1 receptor blockade. Surprisingly, despite evidence that AML directly regulates EC functions,13, 17 we could not identify any major effect of this agent on isolated EC implying that AML may modify post-translational processes that do not result in quantitative changes in gene expression.
In summary, by analyzing global changes in EC gene expression, we have shed light into novel mechanisms by which TLM may help prevent endothelial dysfunction, thus protecting against development and progression of CVD in patients with hypertension.
Significance.
Telmisartan, an angiotensin II type 1 (AT1) receptor blocker, is a clinically used antihypertensive agent which exerts beneficial cardiovascular effects independently of blood pressure lowering and classic mechanism of action. This is the first study investigating the molecular mechanisms responsible for the pleiotropic actions of telmisartan on primary endothelial cells, using a genome-wide approach. We show that telmisartan negatively modulates the expression of key genes involved in cell cycle progression and induces a state of endothelial cell quiescence by affecting the Akt/MDM2/p53 and Akt/ GSK3β/CyclinD1 signaling pathways. Moreover, telmisartan promotes endothelial cell survival by inducing downregulation of proapoptotic genes. Thus, our data support the idea that telmisartan can uniquely protect and preserve the endothelium beyond AT1 receptor antagonism.
Acknowledgements
We wish to thank Gwendolyn Davis-Arrington for assistance with HUVEC isolation.
Sources of funding: This work was supported in part by a sponsored research agreement with Boehringer-Ingelheim International GmbH and the National Institutes of Health (HL64793, HL61371, HL081190, HL096670, PO1 1070205).
Non-standard Abbreviations
- AML
amlodipine
- LOS
losartan
- TLM
telmisartan
- VAL
valsartan
- 7-KC
7-ketocholesterol
Footnotes
Disclosures: none
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, Jr., Jones DW, Materson BJ, Oparil S, Wright JT, Jr., Roccella EJ. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure: The jnc 7 report. JAMA : the journal of the American Medical Association. 2003;289:2560–2572. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 2.Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: An update. Hypertension. 2001;37:1053–1059. doi: 10.1161/01.hyp.37.4.1053. [DOI] [PubMed] [Google Scholar]
- 3.Weintraub HS, Basile J. The pleiotropic effects of antihypertensive agents: Do they account for additional cardiovascular benefit beyond bp reduction? Southern medical journal. 2008;101:818–823. doi: 10.1097/SMJ.0b013e31817b6622. [DOI] [PubMed] [Google Scholar]
- 4.Krieger MH, Di Lorenzo A, Teutsch C, Kauser K, Sessa WC. Telmisartan regresses left ventricular hypertrophy in caveolin-1-deficient mice. Laboratory investigation; a journal of technical methods and pathology. 2010;90:1573–1581. doi: 10.1038/labinvest.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung AD, Kim W, Park SH, Park JS, Cho SC, Hong SB, Hwang SH. The effect of telmisartan on endothelial function and arterial stiffness in patients with essential hypertension. Korean circulation journal. 2009;39:180–184. doi: 10.4070/kcj.2009.39.5.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siarkos I, Urso V, Sinagra T, Drago F, Salomone S. Endothelium-dependent vasomotor effects of telmisartan in isolated rat femoral arteries. Pharmacological research : the official journal of the Italian Pharmacological Society. 2011;63:199–206. doi: 10.1016/j.phrs.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Blessing E, Preusch M, Kranzhofer R, Kinscherf R, Marx N, Rosenfeld ME, Isermann B, Weber CM, Kreuzer J, Grafe J, Katus HA, Bea F. Anti-atherosclerotic properties of telmisartan in advanced atherosclerotic lesions in apolipoprotein e deficient mice. Atherosclerosis. 2008;199:295–303. doi: 10.1016/j.atherosclerosis.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 8.Matsumura T, Kinoshita H, Ishii N, Fukuda K, Motoshima H, Senokuchi T, Taketa K, Kawasaki S, Nishimaki-Mogami T, Kawada T, Nishikawa T, Araki E. Telmisartan exerts antiatherosclerotic effects by activating peroxisome proliferator-activated receptor-gamma in macrophages. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1268–1275. doi: 10.1161/ATVBAHA.110.222067. [DOI] [PubMed] [Google Scholar]
- 9.Fukuda D, Enomoto S, Hirata Y, Nagai R, Sata M. The angiotensin receptor blocker, telmisartan, reduces and stabilizes atherosclerosis in apoe and at1ar double deficient mice. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2010;64:712–717. doi: 10.1016/j.biopha.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Mason RP, Jacob RF, Kubant R, Jacoby A, Louka F, Corbalan JJ, Malinski T. Effects of angiotensin receptor blockers on endothelial nitric oxide release: The role of enos variants. British journal of clinical pharmacology. 2012;74:141–146. doi: 10.1111/j.1365-2125.2012.04189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cianchetti S, Del Fiorentino A, Colognato R, Di Stefano R, Franzoni F, Pedrinelli R. Anti-inflammatory and anti-oxidant properties of telmisartan in cultured human umbilical vein endothelial cells. Atherosclerosis. 2008;198:22–28. doi: 10.1016/j.atherosclerosis.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 12.Dandona P, Dhindsa S, Ghanim H, Chaudhuri A. Angiotensin ii and inflammation: The effect of angiotensin-converting enzyme inhibition and angiotensin ii receptor blockade. Journal of human hypertension. 2007;21:20–27. doi: 10.1038/sj.jhh.1002101. [DOI] [PubMed] [Google Scholar]
- 13.Mason RP, Marche P, Hintze TH. Novel vascular biology of third-generation l-type calcium channel antagonists: Ancillary actions of amlodipine. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:2155–2163. doi: 10.1161/01.ATV.0000097770.66965.2A. [DOI] [PubMed] [Google Scholar]
- 14.Deanfield JE, Detry JM, Lichtlen PR, Magnani B, Sellier P, Thaulow E. Amlodipine reduces transient myocardial ischemia in patients with coronary artery disease: Double-blind circadian anti-ischemia program in europe (cape trial) Journal of the American College of Cardiology. 1994;24:1460–1467. doi: 10.1016/0735-1097(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 15.Pitt B, Byington RP, Furberg CD, Hunninghake DB, Mancini GB, Miller ME, Riley W. Effect of amlodipine on the progression of atherosclerosis and the occurrence of clinical events. Prevent investigators. Circulation. 2000;102:1503–1510. doi: 10.1161/01.cir.102.13.1503. [DOI] [PubMed] [Google Scholar]
- 16.Jorgensen B, Simonsen S, Endresen K, Forfang K, Vatne K, Hansen J, Webb J, Buller C, Goulet G, Erikssen J, Thaulow E. Restenosis and clinical outcome in patients treated with amlodipine after angioplasty: Results from the coronary angioplasty amlodipine restenosis study (capares) Journal of the American College of Cardiology. 2000;35:592–599. doi: 10.1016/s0735-1097(99)00599-9. [DOI] [PubMed] [Google Scholar]
- 17.Batova S, DeWever J, Godfraind T, Balligand JL, Dessy C, Feron O. The calcium channel blocker amlodipine promotes the unclamping of enos from caveolin in endothelial cells. Cardiovascular research. 2006;71:478–485. doi: 10.1016/j.cardiores.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 18.Suarez C. Single-pill telmisartan and amlodipine: A rational combination for the treatment of hypertension. Drugs. 2011;71:2295–2305. doi: 10.2165/11594510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 19.Siragy HM. Improving vascular function in hypertension: Potential benefits of combination therapy with amlodipine and renin-angiotensin-aldosterone system blockers. Journal of hypertension. 2010;28:2–8. doi: 10.1097/HJH.0b013e328332bcf0. [DOI] [PubMed] [Google Scholar]
- 20.Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RA, Thaiss F. Angiotensin ii activates nuclear transcription factor-kappab through at1 and at2 receptors. Kidney international. 2002;61:1986–1995. doi: 10.1046/j.1523-1755.2002.00365.x. [DOI] [PubMed] [Google Scholar]
- 21.Yang D, Qi Y, Chen Q, Wang Z, Jin X, Gao J, Fu J, Xiao X, Zhou Z. The over-expression of p53 h179y residue mutation causes the increase of cyclin a1 and cdk4 expression in helf cells. Molecular and cellular biochemistry. 2007;304:219–226. doi: 10.1007/s11010-007-9503-9. [DOI] [PubMed] [Google Scholar]
- 22.Chen SS, Chang PC, Cheng YW, Tang FM, Lin YS. Suppression of the stk15 oncogenic activity requires a transactivation-independent p53 function. The EMBO journal. 2002;21:4491–4499. doi: 10.1093/emboj/cdf409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rother K, Dengl M, Lorenz J, Tschop K, Kirschner R, Mossner J, Engeland K. Gene expression of cyclin-dependent kinase subunit cks2 is repressed by the tumor suppressor p53 but not by the related proteins p63 or p73. FEBS letters. 2007;581:1166–1172. doi: 10.1016/j.febslet.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 24.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin d1 proteolysis and subcellular localization. Genes & development. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/akt cell survival pathway. The Journal of biological chemistry. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 26.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/akt pathway promotes translocation of mdm2 from the cytoplasm to the nucleus. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson L, Holmgreen SP, Huang DC, Bernard O, Copeland NG, Jenkins NA, Sutherland GR, Baker E, Adams JM, Cory S. Bcl-w, a novel member of the bcl-2 family, promotes cell survival. Oncogene. 1996;13:665–675. [PubMed] [Google Scholar]
- 28.Iuliano L, Micheletta F, Natoli S, Ginanni Corradini S, Iappelli M, Elisei W, Giovannelli L, Violi F, Diczfalusy U. Measurement of oxysterols and alpha-tocopherol in plasma and tissue samples as indices of oxidant stress status. Analytical biochemistry. 2003;312:217–223. doi: 10.1016/s0003-2697(02)00467-0. [DOI] [PubMed] [Google Scholar]
- 29.Li W, Ghosh M, Eftekhari S, Yuan XM. Lipid accumulation and lysosomal pathways contribute to dysfunction and apoptosis of human endothelial cells caused by 7-oxysterols. Biochemical and biophysical research communications. 2011;409:711–716. doi: 10.1016/j.bbrc.2011.05.071. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Mehta JL, Haider N, Zhang X, Narula J, Li D. Role of caspases in ox-ldl-induced apoptotic cascade in human coronary artery endothelial cells. Circulation research. 2004;94:370–376. doi: 10.1161/01.RES.0000113782.07824.BE. [DOI] [PubMed] [Google Scholar]
- 31.Yang R, Muller C, Huynh V, Fung YK, Yee AS, Koeffler HP. Functions of cyclin a1 in the cell cycle and its interactions with transcription factor e2f-1 and the rb family of proteins. Molecular and cellular biology. 1999;19:2400–2407. doi: 10.1128/mcb.19.3.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong D, Ferrell JE., Jr The roles of cyclin a2, b1, and b2 in early and late mitotic events. Molecular biology of the cell. 2010;21:3149–3161. doi: 10.1091/mbc.E10-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chun HK, Chung KS, Kim HC, Kang JE, Kang MA, Kim JT, Choi EH, Jung KE, Kim MH, Song EY, Kim SY, Won M, Lee HG. Oip5 is a highly expressed potential therapeutic target for colorectal and gastric cancers. BMB reports. 2010;43:349–354. doi: 10.5483/bmbrep.2010.43.5.349. [DOI] [PubMed] [Google Scholar]
- 34.Koinuma J, Akiyama H, Fujita M, Hosokawa M, Tsuchiya E, Kondo S, Nakamura Y, Daigo Y. Characterization of an opa interacting protein 5 involved in lung and esophageal carcinogenesis. Cancer science. 2012;103:577–586. doi: 10.1111/j.1349-7006.2011.02167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crane R, Gadea B, Littlepage L, Wu H, Ruderman JV. Aurora a, meiosis and mitosis. Biology of the cell / under the auspices of the European Cell Biology Organization. 2004;96:215–229. doi: 10.1016/j.biolcel.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 36.Martinsson-Ahlzen HS, Liberal V, Grunenfelder B, Chaves SR, Spruck CH, Reed SI. Cyclin-dependent kinase-associated proteins cks1 and cks2 are essential during early embryogenesis and for cell cycle progression in somatic cells. Molecular and cellular biology. 2008;28:5698–5709. doi: 10.1128/MCB.01833-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto K, Ohishi M, Ho C, Kurtz TW, Rakugi H. Telmisartan-induced inhibition of vascular cell proliferation beyond angiotensin receptor blockade and peroxisome proliferator-activated receptor-gamma activation. Hypertension. 2009;54:1353–1359. doi: 10.1161/HYPERTENSIONAHA.109.138750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benson SC, Iguchi R, Ho CI, Yamamoto K, Kurtz TW. Inhibition of cardiovascular cell proliferation by angiotensin receptor blockers: Are all molecules the same? Journal of hypertension. 2008;26:973–980. doi: 10.1097/HJH.0b013e3282f56ba5. [DOI] [PubMed] [Google Scholar]
- 39.Jang JY, Choi Y, Jeon YK, Aung KC, Kim CW. Over-expression of adenine nucleotide translocase 1 (ant1) induces apoptosis and tumor regression in vivo. BMC cancer. 2008;8:160. doi: 10.1186/1471-2407-8-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel vdac. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 41.Vucic D, Deshayes K, Ackerly H, Pisabarro MT, Kadkhodayan S, Fairbrother WJ, Dixit VM. Smac negatively regulates the anti-apoptotic activity of melanoma inhibitor of apoptosis (ml-iap) The Journal of biological chemistry. 2002;277:12275–12279. doi: 10.1074/jbc.M112045200. [DOI] [PubMed] [Google Scholar]
- 42.Davies PF. Spatial hemodynamics, the endothelium, and focal atherogenesis: A cell cycle link? Circulation research. 2000;86:114–116. doi: 10.1161/01.res.86.2.114. [DOI] [PubMed] [Google Scholar]
- 43.Slevin M, Krupinski J, Badimon L. Controlling the angiogenic switch in developing atherosclerotic plaques: Possible targets for therapeutic intervention. Journal of angiogenesis research. 2009;1:4. doi: 10.1186/2040-2384-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choy JC, Granville DJ, Hunt DW, McManus BM. Endothelial cell apoptosis: Biochemical characteristics and potential implications for atherosclerosis. Journal of molecular and cellular cardiology. 2001;33:1673–1690. doi: 10.1006/jmcc.2001.1419. [DOI] [PubMed] [Google Scholar]