

Abstract
The Tuberous Sclerosis Complex 2 (TSC2) gene product, tuberin, acts as a negative regulator of mTOR signaling, and loss of tuberin function leads to tumors of the brain, skin, kidney, heart and lungs. Previous studies have shown that loss of tuberin function affects the stability and subcellular localization of the cyclin-dependent kinase inhibitor (CKI) p27, although the mechanism(s) by which tuberin modulates p27 stability have not been elucidated. Previous studies have also shown that AMPK, which functions in an energy-sensing pathway in the cell, becomes activated in the absence of tuberin. Here we show that in Tsc2-null tumors and cell lines, AMPK activation correlates with an increase in p27 levels, and inhibition of AMPK signaling decreases p27 levels in these cells. In addition, activation of AMPK led to phosphorylation of p27 at the conserved terminal threonine residue of murine p27 (T197) in both in vitro kinase assays and in cells. Phosphorylation of p27 at T197 led to increased interaction between p27 and 14-3-3 proteins and increased the protein stability of p27. Furthermore, activation of AMPK signaling promoted the interaction between p27 and 14-3-3 proteins and increased the stability of the p27 protein in a manner that was dependent on T197. These data identify a conserved mechanism for regulation of p27 stability via phosphorylation at the terminal threonine (mT197/hT198), which when AMPK is activated, results in stabilization of the p27 protein.
Keywords: Tuberous sclerosis complex, TSC2, energy sensing
INTRODUCTION
Loss of the Tsc2 gene product, tuberin, or the Tsc1 gene product, hamartin leads to Tuberous Sclerosis Complex (TSC), an autosomal dominant tumor suppressor gene syndrome associated with tumors of the brain, skin, kidney, heart and lungs [1,2]. At the molecular level, tuberin interacts with hamartin [3,4] and functions as a GTPase activating protein (GAP) for the small GTPase Rheb (Ras homologue enriched in brain), decreasing Rheb-induced activation of mTOR signaling [5-10]. Tuberin is phosphorylated by AKT, repressing tuberin-mediated inhibition of Rheb and activating mTOR signaling [11-14]. Tuberin is also phosphorylated by the AMP-activated protein kinase (AMPK), which in contrast to AKT, activates tuberin suppression of Rheb and inhibits mTOR signaling [15].
AMPK is a heterotrimeric protein complex consisting of AMPK-α, AMPK-β and AMPK-γ subunits. AMPK functions downstream of LKB1 in a signaling pathway that regulates energy consuming (anabolic) and energy generating (catabolic) processes [16,17]. Changes in cellular AMP/ATP ratios promote allosteric interaction between AMP and the AMPK-γ subunit, which promotes phosphorylation of AMPK-α subunit at T172 and activation of AMPK signaling [16,18,19]. Recently, increased cellular AMP/ATP ratios and AMPK signaling have been observed in response to decreased AKT signaling [20]. AKT activity is repressed in Tsc2-null cells due to a negative feedback loop from mTOR/S6K to IRS1/PI3K [21-23], and this loss of AKT activity in Tsc2-null cells has been shown to result in elevated AMP levels and increased AMPK signaling [20].
In addition to aberrant AKT and AMPK signaling, loss of tuberin is associated with altered subcellular localization and expression of p27 [24], a member of the CIP/KIP family of cyclin-dependent kinase inhibitors (CKIs) [25]. We have recently shown that Tsc2-null cells derived from Eker rats (Tsc2Ek/+), a rat model of susceptibility to TSC-associated lesions caused by a retroviral insertion in one Tsc2 allele [26], have constitutively active AMPK signaling [27]. In these cells, AMPK signaling and phosphorylation of p27 at T170, which is adjacent to the p27 NLS, results in cytoplasmic sequestration of this CKI [27].
Here we show that elevated AMPK signaling characteristic of Tsc2-null tumors and tumor-derived cell lines from Eker rats increases the stability of p27 and results in elevated p27 levels in these cells, which can be reversed by inhibiting AMPK activity. We also show that AMPK phosphorylates murine p27 at T197 in vitro, and increasing AMPK signaling in cells promotes phosphorylation of murine p27 at T197. AMPK signaling enhances the interaction between p27 and 14-3-3 and enhances stabilization of p27 in a manner that is dependent on phosphorylation at T197. Therefore, we have identified a conserved mechanism for regulation of p27 stability via phosphorylation at the terminal threonine (mT197/hT198) in response to AMPK signaling.
MATERIALS AND METHODS
Cell Lines
NIH3T3 and HEK293 cells were maintained in DMEM supplemented with 10% FBS, and ELT-3 cells [28] were maintained in DF8 media [28]. NIH3T3 cells grown overnight in media lacking serum and glucose were treated with Compound C (Calbiochem, San Diego, CA). HEK293 cells were treated with AICAR for 1 h (Cell Signaling Technologies) with or without Compound C for 3 h.
Plasmid Preparation and Transfection
The FLAG-p27-WT fusion protein was generated as described previously [27], and mutagenesis was performed using the QuickChange XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol to generate mutant Flag-p27 constructs. The AMPK-α constructs [7] were a gift of Dr. K. Guan. Expression constructs were transiently transfected into NIH3T3 and HEK293 cells using Lipofectamine 2000 (Invitrogen) or into ELT-3 cells using Effectene (Qiagen, Valencia, CA) according to the manufacturer's instructions.
In Vitro AMPK Kinase Assays
The GST-p27 fusion proteins were generated as described previously [27]. AMPK (10mU; Upstate) was incubated with 1 mg of the indicated recombinant p27 protein for 15 min at 30°C. The reactions were then separated by SDS-PAGE and dried. Dried gels were then visualized and quantitated using a Typhoon 9410 Variable Mode Imager (Amersham Pharmacia Biotech).
Cell Lysates
Cell lines were lysed in cold lysis buffer (20mM Tris (pH 7.5), 150mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton X-100, 2.5mM sodium pyrophosphate) containing 1X Complete protease inhibitor (Roche, Mannheim, Germany) and 1mM Na3VO4. Lysates were used for western blotting with the following antibodies: p27 (K5020) (BD Transduction Laboratories, San Diego, CA); 14-3-3β (K-19), cyclin D1 and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA); AMPK, phospho-AMPK-α (T172) (Cell Signaling Technology, Beverly, MA). Anti-rabbit or anti-mouse IgG secondary antibodies conjugated to HRP were from Santa Cruz Biotechnology. Western blots were visualized using LumiGLO™ (KPL, Gaithersburg, MD) substrate.
For two-dimensional western blotting, 150-mg of total protein from each lysate, either untreated or treated with 330 U of Calf Intestinal Alkaline Phosphatase (CIAP) (CalBiochem) at 30°C for two hours, was separated using IPG strips of pH 3-10 (BioRad). Gels were blotted onto PVDF membrane and immunoblotted using an anti-p27 antibody (K5020) (BD Transduction Laboratories, San Diego, CA).
Protein Microarray
A FLEXYS® Robotic workstation (Genomic Solution, Ann Arbor, MI) was used to spot the indicated proteins onto a glass slide that had been pre-coated with a nitrocellulose polymer (FAST™ PAK; Schleicher & Schuell, Keene, NH). p27 peptides and phospho-peptides were synthesized by the W. M. Keck Biotechnology Resource Center (New Haven, CT). Biotinylated peptides (10-μg) were prebound to 5 μL of Cy3-Streptavidin (Fluorolink; Amersham Pharmacia Biotech, Piscataway, NJ) in 500 μL of PBST and incubated with 20 μL of biotin agarose beads (Sigma) to remove the free Streptavidin label. Arrayed slides were blocked in PBST containing 3% fat-free milk, followed by the addition of 400 μL of fluorophore-tagged peptide for 1 h at room temperature. Unbound peptide was washed with PBST, slides were centrifuged dry, and fluorescent signal was detected using a Gene Pix 4200A scanner (Axon Instruments, Sunnyvale, CA). A 532 nm filter was used to detect FITC-conjugated secondary antibodies and Cy3-labeled probes. Bound GST fusion proteins were then detected by probing for 1 h with an anti-GST antibody (Amersham Pharmacia Biotech). The array was washed three times for 10 min in PBST, and the primary antibody was recognized with an appropriate FITC-conjugated secondary antibody.
GST Pulldown Assays
pGEX-GST 14-3-3 constructs were obtained from Dr. Michael B. Yaffe (g,e, and s) and Dr. Ari Elson (b,z,h, and t). Overexpression of GST fusion proteins in Escherichia coli DH5α cells (Invitrogen) was induced with 0.2 mM isopropyl–b-D-thiogalactopyranoside (IPTG), and cells were broken by sonication. GST fusion proteins were then batch-purified from extracts by binding to glutathione Sepharose 4B beads (Amersham Pharmacia Biotech) according to the manufacturer's instructions. Purified fusion proteins (5-10 mg) bound to agarose beads were mixed with 200-mg protein from pre-cleared lysates of cultured cells and incubated overnight at 4°C. Beads were washed in PBS with protease inhibitors, eluted in 2x SDS sample buffer (125mM Tris-HCl, 20% glycerol, 4% SDS, and 0.005% bromphenol blue), heated to 95°C, and analyzed by western blotting.
p27 Stability
Cells transfected with p27 plasmids were cultured overnight and subsequently grown for 1 h in media lacking cysteine and methionine (Invitrogen), and then pulsed for 1 h in media containing 50- to 75-mCi/mL 35S-labeled cysteine and methionine (Amersham Pharmacia Biotech). Cell lysates were generated as described above, quantitated, and immunoprecipitated using M2-Agarose beads (Sigma) according to the manufacturer's instructions. After immunoprecipitation, proteins were eluted from beads using 2x SDS sample buffer, resolved by SDS-PAGE and visualized/quantitated using a Typhoon 9410 Variable Mode Imager (Amersham Pharmacia Biotech). Quantified p27 levels were then compared to the level of p27 present at the 0-hour time-point and expressed as % p27 remaining. The 1/2-life for p27 for each experiment (n=2) was determined using at least two different time-points compared to the 0-hour time-point, and the mean 1/2-life for each experiment was then determined.
RESULTS
Modulation of AMPK signaling alters p27 expression levels
To determine whether AMPK signaling regulated p27 expression, we first examined both AMPK signaling and p27 levels in Tsc2-null tumor-derived cells from Eker rats [28]. In accordance with a previous study from our lab, ELT-3 cells expressed both p27 and an active AMPK signaling pathway as indicated by phosphorylation of AMPK-α at Thr172 (Figure 1A) [27]. However, treatment of ELT-3 cells with Compound C, an inhibitor of AMPK [29], reduced AMPK-α phosphorylation and p27 expression in these cells (Figure 1A). In addition, AMPK signaling and p27 levels were examined in NIH3T3 cells that were deprived of glucose to activate AMPK signaling. Both AMPK signaling and p27 expression was increased upon removal of glucose and serum (Figure 1B and data not shown). In both ELT-3 and NIH3T3 cells p27 levels were reduced by treatment with AMPK inhibitor Compound C (Figures 1A and 1B).
Figure 1. AMPK signaling correlates with p27 expression.

A, Lysates from Tsc2-null ELT-3 cells that were treated overnight with Compound C were immunoblotted using anti-p27, anti-phospho-AMPK-α (T172) and anti-AMPK-α antibodies. B, Lysates from NIH3T3 cells grown overnight in the absence of serum and glucose, treated with Compound C for 3h were immunoblotted using anti-p27 and anti-GAPDH antibodies. C, Protein lysates from neonatal and adult kidney and five independent kidney tumors from Eker rats (Tsc2Ek/+) were immunoblotted using anti-p27, anti-phospho-AMPK-α (T172), anti-AMPK-α, anti-AMPK-α1, anti-AMPK-α2, anti-phospho-S6 and anti-S6 antibodies.
To determine whether elevated AMPK signaling also correlated with increased p27 expression in primary tumors, renal cell carcinomas (RCC) from Tsc2Ek/+ rats that had lost tuberin were examined for AMPK activation and p27 expression. AMPK activation, as assessed by phosphorylation of AMPK-α at T172, and p27 levels were both dramatically increased in tumors relative to normal adult kidney of Tsc2Ek/+ rats (Figure 1C). Both neonatal and adult kidney expressed AMPK α (both α -1 and α -2 isoforms), but activated AMPK (phosphorylated at T172) was only detected in RCCs. As anticipated, phosphorylation of S6 was increased in RCCs, indicative of loss of tuberin function. Thus, both primary tumors and cell lines that had lost tuberin had high levels of p27, and elevated p27 levels correlated with activation of AMPK.
T197 of p27 is phosphorylated by AMPK
Previous studies have indicated that human p27 is phosphorylated at the terminal threonine, T198, by kinases in the PI3K, MAPK, and AMPK pathways [30,31], and the carboxy-terminal amino acid sequence of p27 is conserved between human and murine p27 (Figure 2A). A Scansite search (scansite.mit.edu) confirmed that T197 of murine p27 was also a putative AMPK phopshorylation site [32]. In addition to T197, p27 contains other potential phosphorylation sites for AMPK (Scansite.mit.edu [32]), including T170, and our group has previously shown that T170 phosphorylation directs p27 to the cytoplasm in response to AMPK signaling [27]. To demonstrate that T197 was a conserved phosphorylation target for AMPK signaling, we first performed a series of in vitro kinase assays using recombinant murine wild-type p27 (p27-WT), mutant p27 which has an alanine substitution at T197 (T197A), mutant p27 with an alanine substitution at T170 adjacent to the NLS (T170A) (positive control) and a double mutant murine p27 construct (T170A/T197A) that cannot be phosphorylated at these sites. AMPK phosphorylated recombinant murine p27-WT in vitro in an AMP-dependent manner, with an approximately 5-fold increase in p27 phosphorylation in the presence of AMP compared to the absence of AMP (Figure 2B). Mutation of T197 led to a 20% reduction in phosphorylation of p27 when compared to p27-WT, similar to what was observed with the T170A mutant (35% reduction), and mutation of T170 and T197 in combination was additive, reducing p27 phosphorylation approximately 50% when compared to p27-WT (Figure 2B).
Figure 2. AMPK phosphorylates p27 at T197 in vitro.

A, Schematic of human and rat p27 proteins showing a conserved sequence at the C-terminus of the protein, including the terminal threonine residue, which is a 14-3-3 binding site and predicted to be a phosphorylation site for AMPK. B, Recombinant rat p27proteins, wild-type or mutated at the indicated potential AMPK target sites, were utilized as substrates for an in vitro kinase assay with purified AMPK in the absence or presence of 300-mM AMP. Kinase reactions were then separated by SDS-PAGE and visualized by autoradiography (a representative experiment is shown on the left). Autoradiographs were quantified, and the amount of 32P incorporated into each protein was normalized to p27-WT (right).
To determine whether AMPK signaling could phosphorylate p27 at T197 in cells, we performed two-dimensional western blot analysis using lysates from NIH3T3 cells transfected with a wild-type murine p27 (Flag-p27-WT) construct or a mutant murine p27 (Flag-p27-T197A) construct grown in the presence or absence of glucose to activate AMPK. Consistent with a previous report [33], wild-type and mutant p27 were detected at three different isoelectric points (pIs) when cells were grown in the presence of glucose (Figure 3, top and middle panels). When comparing wild-type p27 and mutant p27 (T197A) in cells grown in the absence of glucose, which activated AMPK signaling (Figure 3, bottom panel), wild-type but not mutant p27 was detected at additional isoelectric points with lower pI values, indicative of phosphorylation (Figure 3, top and middle panels). Furthermore, migration of wild-type p27 protein at isoelectric points with lower pI values (indicative of phosphorylation), was ablated by phosphatase treatment (Figure 3, top panel). These data indicate that rodent p27 is phosphorylated in vitro and in cells at T197 when AMPK signaling is activated.
Figure 3. Activation of AMPK signaling increases p27 phosphorylation at T197.

NIH3T3 cells were transfected with Flag-p27-WT or Flag-p27-T197A constructs, grown in the presence or absence of glucose, and then utilized to generate whole-cell lysates that were untreated or treated with CIAP. Lysates were then separated on 2-D gels and immunoblotted for p27 or separated by SDS-PAGE and immunoblotted for AMPK-α and phospho-AMPK-α (T172). Arrows A, B, and C indicate p27 isoforms detectable under all conditions, and arrows D and E indicate additional p27 isoforms with lower pIs (indicative of phosphorylation) that are detectable only upon glucose deprivation, which activates AMPK signaling.
T197 of murine p27 promotes interaction with 14-3-3
Phosphorylation of murine p27 at T197 in response to activation of AMPK signaling would be predicted to create a 14-3-3 binding site (Scansite.mit.edu [32]). Therefore, protein microarrays were utilized to determine whether peptides corresponding to phosphorylated or nonphosphorylated T197 (rat) or T198 (human), as well as peptides corresponding to several other potential phosphorylation sites (S10, S118, and S140 see Supplemental Table 1) were able to bind to 14-3-3 proteins. In this assay, phosphorylated T197 and T198 peptides (but not S10, S118, or S140 peptides) bound to all seven 14-3-3 isoforms (beta, gamma, epsilon, sigma, tau, zeta, and eta), while the non-phosphorylated peptides did not interact with 14-3-3 proteins (Figure 4A).
Figure 4. Phosphorylation of p27 at T197 promotes interaction with 14-3-3.

A, A protein array composed of 14-3-3 isoforms or other protein domains indicated in protein-protein interactions were fused to GST and spotted in duplicate onto a glass chip. Protein arrays were then incubated with phosphorylated and non-phosphorylated peptides corresponding to S10, S118, S140 and T197/T198 phosphorylation sites of p27 (predicted to be 14-3-3 interaction sites) that were labeled with Cy3. Interaction between Cy3-labeled peptides and the protein array was then visualized. A key indicating the area on the chip corresponding to each 14-3-3 isoform is shown below. B, ELT-3 cells were transfected with Flag-p27-WT or Flag-p27-T197A and used to generate whole-cell lysates. Lysates were equalized for p27-WT or p27-T197A expression and affinity purified using GST-14-3-3 or GST alone. Lysates and affinity-purified lysates were immunoblotted using an anti-p27 antibody. C, ELT-3 cells were transfected with Flag-p27-WT, Flag-p27-T197A or Flag-p27-T197E and used to generate whole-cell lysates. Lysates were equalized for exogenous p27 expression and affinity purified using GST-14-3-3. Lysates and affinity-purified lysates were immunoblotted using an anti-p27 antibody.
To further determine whether phosphorylation of p27 at T197 generated a 14-3-3 recognition site, wild-type p27 (Flag-p27-WT) and mutant p27 (Flag-p27-T197A) constructs were expressed in Tsc2-null ELT-3 cells and affinity purified using GST-14-3-3. Both Flag-p27-WT and endogenous p27 were capable of interacting with 14-3-3 (Figure 4B). However, there was no increase in binding of Flag-p27-T197A (or endogenous p27) observed when comparing affinity purifications using GST-14-3-3 with GST alone, indicating no specific interaction between T197A mutant p27 and 14-3-3 (Figure 4B). Phosphomimetic p27 (Flag-p27-T197E) was also more efficiently affinity purified with GST-14-3-3 than Flag-p27-T197A (Figure 4C). Interestingly, the amount of both exogenous and endogenous p27 affinity purified with GST-14-3-3 increased when Flag-p27-WT or Flag-p27-T197E was overexpressed (Figures 4B and 4C). In addition, increased amounts of endogenous p27 could be affinity purified by GST-14-3-3 in the presence of phosphorylated T197 peptide but not the non-phosphorylated T197 peptide (data not shown). These data confirm a conserved role for the terminal threonine of p27, specifically phosphorylated T197, in 14-3-3 binding and suggests a potential cooperativity in binding of p27 by the 14-3-3 dimer.
Phosphorylation at T197 stabilizes p27
We routinely observed that Flag-p27-T197A expression levels were lower than Flag-p27-WT in several cell types including Tsc2-null ELT-3 cells (Figure 5A), HEK293 human embryonic kidney cells (Figure 5B) and NIH3T3 mouse cells (data not shown). In addition, Flag-p27-T197E, with a phosphomimetic glutamic acid residue instead of threonine, was expressed at much higher levels than Flag-p27-T197A and at slightly higher levels than Flag-p27-WT in HEK293 cells (Figure 5B).
Figure 5. Phosphorylation of T197 alters p27 stability.

A, ELT-3 cells were transfected with Flag-p27-WT or Flag-p27-T197A constructs and analyzed by immunoblotting using an anti-p27 antibody (top) or utilized in pulse-chase experiments as described in Materials and Methods. Flag-p27 was immunoprecipitated from lysates, resuspended, separated by SDS-PAGE and analyzed by autoradiography. A graph from a representative experiment depicting the amount of p27 remaining at the indicated time-points is shown and the half-life of p27-WT and p27-T197A is given below (bottom). B, HEK293 cells were transfected with Flag-p27-WT, Flag-p27-T197A or Flag-p27-T197E constructs and analyzed by immunoblotting using an anti-p27 antibody (top) or utilized in pulse-chase experiments as described in Materials and Methods. Flag-p27 was then immunoprecipitated from cell lysates, resuspended, separated by SDS-PAGE and analyzed by autoradiography. A graph from a representative experiment depicting the amount of p27 remaining at the indicated time-points is shown and the half-life of exogenous p27 is given below (bottom).
To determine whether phosphorylation of murine p27 at T197 regulated its stability, the half-life of wild-type (Flag-p27-WT) and mutant p27 (Flag-p27-T197A) in Tsc2-null ELT-3 cells with activated AMPK was determined. Pulse-chase experiments revealed that Flag-p27-T197A had a shorter half-life (4.4 hrs) than Flag-p27-WT (16.7 hrs) in ELT-3 cells (Figure 5A). Similar to what was observed in ELT-3 cells, Flag-p27-T197A had a shorter half-life (3.1 hrs) than Flag-p27-WT (7.2 hrs) in HEK293 cells, and Flag-p27-T197E had a longer half-life (17 hrs) than Flag-p27-WT or Flag-p27-T197A in HEK293 cells (Figure 5B), demonstrating that the terminal threonine of murine p27 regulates its stability.
AMPK signaling regulates the interaction between p27 and 14-3-3
Since AMPK signaling increases phosphorylation of p27 at T197 and phosphorylation at this site promotes interaction between p27 and 14-3-3, we sought to determine whether AMPK signaling regulated p27 interaction with 14-3-3. Initially, endogenous rat p27 was immunoprecipitated from ELT-3 cell lysates (which have activated AMPK signaling), and analyzed for 14-3-3 binding. 14-3-3 was readily detected in anti-p27 immunoprecipitates from ELT-3 lysates (Figure 6A). p27 could also be detected by western blotting anti-14-3-3 immunoprecipitates from ELT-3 cell lysates and immunoprecipitates from tuberin-null primary tumors (data not shown).
Figure 6. AMPK signaling alters interaction between p27 and 14-3-3.

A, Whole-cell lysates from ELT-3 cells were immunoprecipitated using a control antibody or an anti-p27 antibody. The immunoprecipitated pellets were then resuspended and immunoblotted with anti-p27 and anti-14-3-3 antibodies. B, HEK293 cells, which were transfected with Flag-p27-WT, were treated as indicated and used to generate whole-cell lysates. Lysates were analyzed for exogenous p27 expression or utilized for affinity purification using GST-14-3-3 or GST alone and analyzed by immunoblotting with an anti-p27 antibody. In addition, lysates were used for immunoprecipitation with an anti-p27 antibody, and immunoprecipitates were analyzed by immunoblotting using an anti-14-3-3 antibody.
In addition, we analyzed the interaction between 14-3-3 and p27 upon modulation of AMPK signaling in HEK293 cells transfected with a Flag-p27-WT construct treated with AICAR to activate and/or with Compound C to inhibit AMPK signaling. AICAR treatment increased the amount of wild-type rat p27 that was affinity purified using GST-14-3-3 when compared to non-treated cells (Figure 6B), and pre-treatment with Compound C inhibited the AICAR-mediated increase in p27 biding to GST-14-3-3 (Figure 6B). Similarly, AICAR treatment of HEK293 cells increased the amount of 14-3-3 detected in anti-p27 immunoprecipitates, and pre-treatment with Compound C could block this increased amount of 14-3-3 detected by western blotting p27 immunoprecipitates (Figure 6B). These data indicate that AMPK signaling regulates the interaction between p27 and 14-3-3.
AMPK signaling regulates p27 stability via phosphorylation at T197
In addition to regulating interaction between p27 and 14-3-3, phosphorylation of p27 at T197 also regulated p27 protein stability (Figure 4). Interestingly, the half-life of Flag-p27-WT was almost twice as long in Tsc2-null ELT-3 cells (that had elevated AMPK signaling) as it was in HEK293 cells. The phosphomimetic Flag-p27-T197E showed a similar half-life (i.e. 17 hours) to Flag-p27-WT in ELT-3 cells, consistent with AMPK phosphorylation stabilizing the p27 protein (Figure 4).
Therefore, we sought to directly determine whether AMPK signaling could regulate the stability of p27 via phosphorylation at T197. Wild-type p27 (Flag-p27-WT) was expressed in HEK293 cells with or without constitutively active AMPK (HA-CA-AMPK) and pulse-chase experiments were performed to examine p27 degradation. Co-expression of constitutively active AMPK increased the stability of p27 in HEK293 cells when compared to the stability of p27 expressed in the absence of constitutively active AMPK (Figure 7A). To determine whether the increased stability of p27 upon co-expression of constitutively active AMPK was dependent on the T197 residue, pulse-chase experiments were performed in HEK293 cells that had been cotransfected with HA-CA-AMPK and Flag-p27-WT or Flag-p27-T197A. Again, co-expression of constitutively active AMPK with wild-type p27 increased the stability of p27; however, the stability of T197A mutant p27 was not increased when co-expressed with constitutively active AMPK (Figure 7B). These data indicate that AMPK signaling increases p27 protein stability in a manner that is dependent on phosphorylation of T197.
Figure 7. AMPK signaling alters p27 stability via phosphorylation at T197.
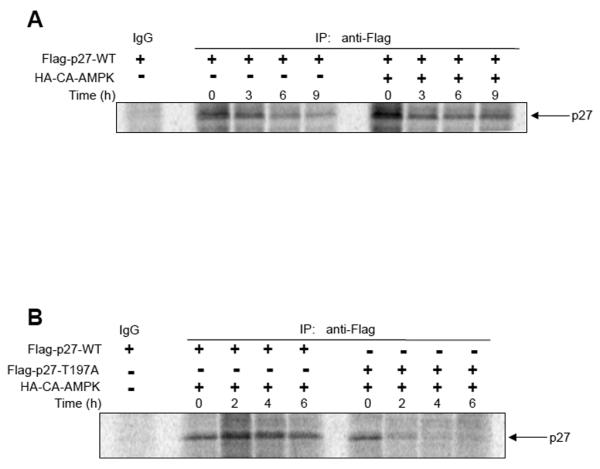
A, HEK293 cells were transfected either with Flag-p27-WT alone or Flag-p27-WT and HA-CA-AMPK constructs, and cells were utilized in pulse-chase experiments for the indicated times as described in Materials and Methods. Flag-p27 was immunoprecipitated from lysates, resuspended, separated by SDS-PAGE and analyzed by autoradiography. B, HEK293 cells were co-transfected with Flag-p27-WT or Flag-p27-T197A and HA-CA-AMPK constructs, and cells were utilized in pulse-chase experiments for the indicated times as described in Materials and Methods. Flag-p27 was immunoprecipitated from lysates, resuspended, separated by SDS-PAGE and analyzed by autoradiography.
DISCUSSION
We report here that activated AMPK signaling and increased levels of p27 are observed in Tsc2-null cells and tumors from Eker rats, and inhibition of AMPK signaling in these cells can decrease p27 levels. We also demonstrated that activation of AMPK signaling leads to phosphorylation of the conserved terminal threonine, T197, of murine p27 both in vitro and in cells, a mechanism for stabilizing p27 that is preserved across species. Moreover, we showed that AMPK signaling induces an interaction between p27 and 14-3-3 proteins via phosphorylation of the terminal threonine of murine p27.
Loss of tuberin has previously been reported to result in altered sub-cellular localization and expression of p27 [24], and our group has previously reported that human p27 is phosphorylated on its terminal residue, T198, which increases the stability of p27 [30]. Similar to our data, previous studies by Rattan et al. showed activation of AMPK signaling in several types of cancer cells results in elevated expression of p21 and p27 [34]. However, other studies have reported a decrease in p27 expression as well as a primarily cytosolic localization pattern in certain tuberin-deficient cells, and over expression of Tsc2 can increase p27 expression levels [35-37]. In those studies, tuberin was proposed to interact with p27 and increase nuclear p27 levels by regulating the interaction between AKT-phosphorylated p27 and 14-3-3 [35]. However, the biological plausibility of this mechanism is questionable as both Tsc2-null cell lines and tumors have been shown to exhibit decreased AKT activity due to a negative feedback from S6K to IRS1 [data not shown and [21,22,38]], suggesting that other kinase(s) may be regulating p27 in tuberin-null cells. Our study provides a novel mechanism for regulation of p27 stability in Tsc2-null cells via AMPK signaling.
Although AMPK stabilized p27 in Tsc2-null kidney cells, elevation of AMPK signaling does not lead to increased p27 expression in all Tsc2-null cell lines or tumors. For example, Tsc2-null uterine leiomyomas with elevated AMPK signaling express p27 at levels comparable to normal myometrium, although p27 is mislocalized to the cytoplasm in these leiomyomas [27]. Others have also reported p27 expression being unaffected by activation of AMPK signaling [39]. However, it should be noted that basal p27 expression levels were high in cells and tissues in which increased AMPK signaling did not alter p27 expression [data not shown and [27,39]]. Thus, it is likely that the effect of AMPK on p27 levels is context-dependent, and may depend on other pathways that are known to regulate p27 expression and stability.
Activation of AMPK signaling has also been postulated to increase p27 expression by increasing activity of the p27 5′-UTR [34]. Although the p27 constructs used in this study did not possess the p27 promoter region or 5′-UTR, it is plausible that AMPK signaling also contributes to elevated p27 levels by modulating p27 expression at the transcriptional or translational level in addition to modulating stability via post-translational modifications. Nonetheless, while the relationship between AMPK signaling and p27 is complex and likely cell-type or tissue-specific, our data clearly demonstrate that p27 is targeted in a conserved manner by this signaling pathway for phosphorylation of its terminal threonine residue, which stabilizes p27 in cells with activated AMPK signaling.
The terminal threonine of human p27 has previously been reported to be phosphorylated by several kinases including AKT, p90-RSK and AMPK [30,32,40,41], although a recent report calls into question whether T198 of human p27 is a bona fide target site for AKT [33]. In addition, T198 of human p27 was reported to lie within a motif capable of being phosphorylated by other kinases, including PKA and CHK1 [30], suggesting that the terminal threonine of p27 may serve as the target for multiple signaling pathways to stabilize p27. However, we did not observe differences in localization when comparing wild-type p27 and mutant p27 (T197A) under normal growth conditions or under conditions where AMPK signaling was elevated [27], and a recent report indicates that the nucleo-cytoplasmic shuttling and degradation of mutant human p27 (T198A) are independent processes [31]. While phosphorylation of T197 did not affect p27 localization, we have previously identified another site on p27 at T170, which lies adjacent to the p27 NLS, as a target for AMPK in vitro, and have found that this site determines localization of p27 in the cell [27]. Therefore, it appears that AMPK signaling can cause both mislocalization (via phosphorylation at T170) and stabilization (via phosphorylation of the terminal T197) of murine p27 through independent phosphorylation events.
Finally, activation of the AMPK signaling pathway has been proposed as a potential therapy for cancer [42]. In fact, clinical studies have shown that metformin, an anti-diabetic drug that activates AMPK signaling, protects against cancer in some diabetic patients [43,44], and others have shown that activation of AMPK signaling can inhibit proliferation of prostate cancer cells [45]. However, none of these prior studies analyzed the effect of AMPK activation on p27 expression levels. We have previously shown that AMPK-mediated phosphorylation and stabilization of p27 inhibits apoptosis while promoting cellular autophagy [30], promoting tumor cell survival. Therefore, it is possible that the effect of AMPK signaling on promotion or inhibition of tumor growth and survival could depend on whether p27 expression is retained in the specific tumor being treated. Further elucidation of the context-specific events that regulate AMPK-mediated phosphorylation of p27 and its cellular consequences will help determine whether activation or inhibition of AMPK signaling is more appropriate for cancer therapy, alone, or in combination with other treatments.
Supplementary Material
{kind=link}
ACKNOWLEDGEMENTS
We thank Dr. K. Guan for providing AMPK-α constructs and Drs. M.B. Yaffe and A. Elson for providing GST-14-3-3 constructs. Also, we thank T. Berry, A. Espejo, S. Hensley and C. Johnson for technical assistance.
Grant Support: This work was supported in part by grants as follows: NIEHS (ES007784), NIH (CA041996 and CA032737) to FT; NIH (CA064602 and CA099031) and DAMD (17-02-01-0694) to GBM; and NCI (CA063613), NIEHS (ES008263) and NICHD (HD046282) to CLW.
Abbreviations used are
- TSC
Tuberous Sclerosis Complex
- TSC2
Tuberous Sclerosis Complex 2
- CKI
Cyclin-dependent Kinase Inhibitor
- AMPK
AMP-Activated Protein Kinase
REFERENCES
- 1.Astrinidis A, Henske EP. Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease. Oncogene. 2005;24(50):7475–7481. doi: 10.1038/sj.onc.1209090. [DOI] [PubMed] [Google Scholar]
- 2.Lendvay TS, Marshall FF. The tuberous sclerosis complex and its highly variable manifestations. J Urol. 2003;169(5):1635–1642. doi: 10.1097/01.ju.0000058253.40352.60. [DOI] [PubMed] [Google Scholar]
- 3.Plank TL, Yeung RS, Henske EP. Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer Res. 1998;58(21):4766–4770. [PubMed] [Google Scholar]
- 4.van Slegtenhorst M, Nellist M, Nagelkerken B, et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet. 1998;7(6):1053–1057. doi: 10.1093/hmg/7.6.1053. [DOI] [PubMed] [Google Scholar]
- 5.Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278(35):32493–32496. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- 6.Garami A, Zwartkruis FJ, Nobukuni T, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11(6):1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 7.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5(6):566–571. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 9.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13(15):1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5(6):578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 11.Dan HC, Sun M, Yang L, et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem. 2002;277(38):35364–35370. doi: 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- 12.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 13.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 14.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4(9):658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 15.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 16.Hardie DG. The AMP-activated protein kinase pathway--new players upstream and downstream. J Cell Sci. 2004;117(Pt 23):5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 17.Witters LA, Kemp BE, Means AR. Chutes and Ladders: the search for protein kinases that act on AMPK. Trends Biochem Sci. 2006;31(1):13–16. doi: 10.1016/j.tibs.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 18.Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004;29(1):18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 19.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403(1):139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280(37):32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 21.Harrington LS, Findlay GM, Gray A, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19(15):1773–1778. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14(18):1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 24.Rosner M, Freilinger A, Hengstschlager M. The tuberous sclerosis genes and regulation of the cyclin-dependent kinase inhibitor p27. Mutat Res. 2006;613(1):10–16. doi: 10.1016/j.mrrev.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Borriello A, Cucciolla V, Oliva A, Zappia V, Della Ragione F. p27Kip1 metabolism: a fascinating labyrinth. Cell Cycle. 2007;6(9):1053–1061. doi: 10.4161/cc.6.9.4142. [DOI] [PubMed] [Google Scholar]
- 26.Walker C, Goldsworthy TL, Wolf DC, Everitt J. Predisposition to renal cell carcinoma due to alteration of a cancer susceptibility gene. Science. 1992;255(5052):1693–1695. doi: 10.1126/science.1553556. [DOI] [PubMed] [Google Scholar]
- 27.Short JD, Houston KD, Dere R, et al. AMP-activated protein kinase signaling results in cytoplasmic sequestration of p27. Cancer Res. 2008;68(16):6496–6506. doi: 10.1158/0008-5472.CAN-07-5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howe SR, Gottardis MM, Everitt JI, Goldsworthy TL, Wolf DC, Walker C. Rodent model of reproductive tract leiomyomata. Establishment and characterization of tumor-derived cell lines. Am J Pathol. 1995;146(6):1568–1579. [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang J, Shao SH, Xu ZX, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9(2):218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 31.Kossatz U, Vervoorts J, Nickeleit I, et al. C-terminal phosphorylation controls the stability and function of p27kip1. Embo J. 2006;25(21):5159–5170. doi: 10.1038/sj.emboj.7601388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31(13):3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nacusi LP, Sheaff RJ. Akt1 sequentially phosphorylates p27kip1 within a conserved but non-canonical region. Cell Div. 2006;1:11. doi: 10.1186/1747-1028-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eto I. Nutritional and chemopreventive anti-cancer agents up-regulate expression of p27Kip1, a cyclin-dependent kinase inhibitor, in mouse JB6 epidermal and human MCF7, MDA-MB-321 and AU565 breast cancer cells. Cancer Cell Int. 2006;6:20. doi: 10.1186/1475-2867-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosner M, Freilinger A, Hanneder M, et al. p27Kip1 localization depends on the tumor suppressor protein tuberin. Hum Mol Genet. 2007;16(13):1541–1556. doi: 10.1093/hmg/ddm103. [DOI] [PubMed] [Google Scholar]
- 36.Soucek T, Rosner M, Miloloza A, et al. Tuberous sclerosis causing mutants of the TSC2 gene product affect proliferation and p27 expression. Oncogene. 2001;20(35):4904–4909. doi: 10.1038/sj.onc.1204627. [DOI] [PubMed] [Google Scholar]
- 37.Soucek T, Yeung RS, Hengstschlager M. Inactivation of the cyclin-dependent kinase inhibitor p27 upon loss of the tuberous sclerosis complex gene-2. Proc Natl Acad Sci U S A. 1998;95(26):15653–15658. doi: 10.1073/pnas.95.26.15653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6(1):91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 39.Igata M, Motoshima H, Tsuruzoe K, et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res. 2005;97(8):837–844. doi: 10.1161/01.RES.0000185823.73556.06. [DOI] [PubMed] [Google Scholar]
- 40.Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002;277(32):28706–28713. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- 41.Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003;278(49):49254–49260. doi: 10.1074/jbc.M306614200. [DOI] [PubMed] [Google Scholar]
- 42.Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation--AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574(Pt 1):63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29(2):254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 44.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Bmj. 2005;330(7503):1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun. 2004;321(1):161–167. doi: 10.1016/j.bbrc.2004.06.133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.