

Abstract
Analogues of mitoQ and idebenone were synthesized to define the structural elements that support oxygen consumption in the mitochondrial respiratory chain. Eight analogues were prepared and fully characterized, then evaluated for their ability to support oxygen consumption in the mitochondrial respiratory chain. While oxygen consumption was strongly inhibited by mitoQ analogues 2–4 in a chain length-dependent manner, modification of idebenone by replacement of the quinone methoxy groups by methyl groups (analogues 6 – 8) reduced, but did not eliminate, oxygen consumption. Idebenone analogues 6 – 8 also displayed significant cytoprotective properties toward cultured mammalian cells in which glutathione had been depleted by treatment with diethyl maleate.
Keywords: Mitochondrial respiration, Oxygen consumption, Idebenone, Ubiquinones
1. Introduction
Mitochondrial dysfunction is thought to contribute to a number of human degenerative diseases, many of them lacking specific treatments.1 Because of their central role in energy production mitochondria, and particularly the mitochondrial respiratory chain, are important potential drug targets.2 The study and remediation of impaired electron flow through the complexes of the mitochondrial respiratory chain has been hampered by the difficulty in delivering redox-active molecules selectively to mitochondria in vivo. Coenzyme Q10 (Fig. 1) is one of the electron carriers used in the respiratory chain, but its bioavailability as an exogenous agent might be expected to be low due to its extreme hydrophobicity. In addition to transporting electrons between complexes I/II and III of the respiratory chain, the hydroquinone moiety of coenzyme Q has been shown to possess antioxidant properties. The multiple functions associated with coenzyme Q highlights the need for quinones that can be used to provide better insight into the fate of electrons circulating within the mitochondrial inner membrane and potentially facilitate the design of therapeutic agents. Such compounds should ideally accumulate within mitochondria, and mediate electron transport through all or defined parts of the respiratory chain.
Figure 1.
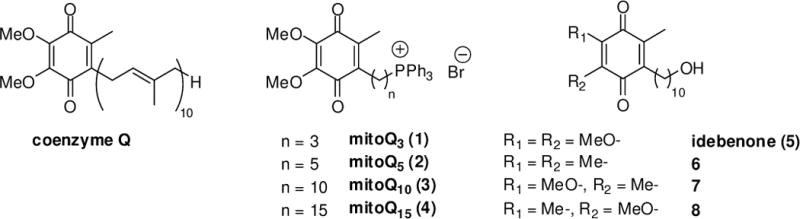
Coenzyme Q, and structurally related ubiquinones.
A family of mitochondria targeted ubiquinones denoted mitoQs has been developed recently;3,4 they feature a ubiquinone moiety attached to alkyl chains of varying length, which terminate with a triphenylphosphonium cation. This latter functional group enables such lipophilic cations to accumulate within the mitochondria as a consequence of the large mitochondrial membrane potential. A second family of ubiquinones was developed in the present study based on the idebenone scaffold as a coenzyme Q surrogate. Although its side chain is less lipophilic than that of coenzyme Q, it partitions non-specifically into phospholipid bilayers, but does not accumulate efficiently in mitochondria. As part of an effort to define the structural elements in coenzyme Q10 essential to support mitochondrial respiration, eight ubiquinones (1–8) were synthesized (Fig. 1), namely mitoQ3 (1), mitoQ5 (2), mitoQ10 (3), mitoQ15 (4), idebenone (5), and threeanalogues of idebenone (6–8). Compounds 6–8 include methyl groups and methoxy groups at varying positions on the quinone ring. The syntheses and characterization of these compounds is described herein. Analogues 1–8 were then evaluated for their ability to support oxygen consumption in the mitochondrial respiratory chain, and also for their ability to confer protection from oxidative stress to cultured CEM cells treated with diethyl maleate.
2. Results and discussion
2.1. Synthesis of mitoQ analogues 1–4
While all four of the mitoQ derivatives described here have been reported previously,5,6 a variety of alkylation chemistries were employed for introduction of side chain precursors, and for their subsequent conversion to triphenylphosphonium derivatives, the latter of which were also prepared as several different salt derivatives. Variations of this approach were employed in the present study for the syntheses of mitoQs 3 and 4 to obtain the products more conveniently, in an improved state of purity, and with a uniform bromide counterion to facilitate direct comparison of biochemical properties. One important variation involved reduction of the intermediate quinones to the hydroquinones prior to introduction of the triphenylphosphonium moiety; this afforded the (reoxidized) final products in an improved state of purity.
For mitoQs 1 and 2, new synthetic strategies were employed to facilitate side chain introduction and to enhance the potential flexibility of the introduced substituent as a precursor to a variety of functionalized side chains. The syntheses of mitoQ3 (1) and mitoQ5 (2) involved the use of allyl arene 13 (Scheme 1) as a common synthetic intermediate.
Scheme 1.
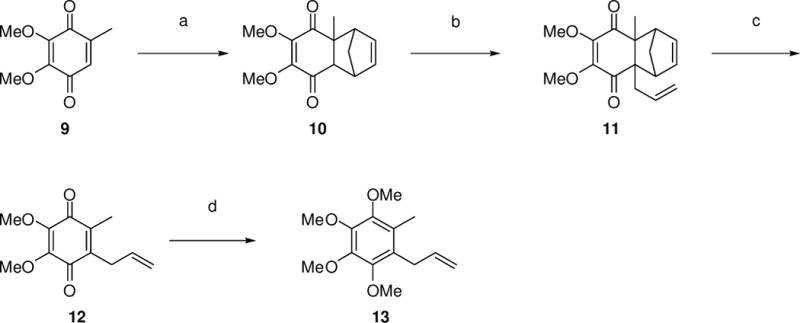
Synthesis of intermediate 13. Reagents and conditions: (a) cyclopentadiene, AcOH, 23 °C, 94%; (b) t-BuOK, allyl bromide, THF, 0 °C, 80%; (c) toluene, 110 °C, 98%; (d) (i) Na2S2O4, n-Bu4NBr, 1:1 THF/H2O, (ii) (MeO)2SO2, NaOH, 1:1 THF/H2O, 81%.
Treatment of coenzyme Q0 (9) with freshly distilled cyclopentadiene in acetic acid at 23 °C led to Diels-Alder cycloadduct 10 in 94% yield.7 The angular proton in position 7 was removed chemoselectively using potassium tert-butoxide in THF at 0 °C, and the resulting anion was treated with allyl bromide to provide 11 in 80% yield. Compound 11 was isolated as a racemate, but having exclusively a cis-orientation of methyl and allyl groups. Heating 11 in toluene at reflux led to cleavage of the cyclopentadiene moiety, providing quinone 12 in quantitative yield. This synthetic sequence was carried out on a gram scale without purifying any of the intermediates by column chromatography. This synthetic strategy allowed facile regioselective functionalization with the allyl substituent, and the mild conditions employed in this step should allow introduction of substituents bearing different functional groups. This should permit the convenient syntheses of coenzyme Q analogues. Finally, reactive quinone 12 was reduced using sodium dithionite in 1:1 THF–water, in the presence of a catalytic amount of n-BuN4Br, and maintained as the hydroquinone by the use of a reducing agent through phase transfer catalysis. These conditions were compatible with permethylation of the hydroquinone using dimethyl sulfate and sodium hydroxide, and yielded 13 in 81% yield over two steps. This one-pot procedure afforded 13 in much higher yield, as compared to the reduction of quinone 12 with NaBH4 or LiAlH4, followed by methylation of the obtained crude hydroquinone, even when strictly anaerobic conditions were employed in the latter case.
Hydroboration of 13 using 9-borabicyclo[3.3.1]nonane (9-BBN) in THF (Scheme 2), followed by oxidation using hydrogen peroxide/aqueous sodium hydroxide led to primary alcohol 14 in 79% yield. Alcohol 14 was converted to the corresponding mesylate 15, using MsCl and triethylamine in CH2Cl2, followed by treatment with sodium bromide in DMF at 23 °C to afford bromide 16. Finally, treatment of 12 with CAN in 1:1 acetonitrile/water led to the chemoselective oxidative cleavage of the two para-methoxy groups to afford quinone 17 in 95% yield.8 Slow addition of 4-bromo-1-butene to a dilute (0.01 M) solution containing 13 and Grubb’s 2nd generation catalyst9 (1,3-bis-(2,4,6-trimethylphenyl)-2-(imidazolidene)(dichlorophenylmethylene)(tricyclohexylphosphine)ruthenium) in CH2Cl2 at reflux led to the product of olefin cross-metathesis (18) in 64% yield. While ring closing metathesis and polymeric olefin metathesis have been studied extensively, olefin cross-metathesis has been relatively underrepresented in the area of olefin metathesis.9 The present report is to date the only example of olefin cross-metathesis employed in the synthesis of redox active molecules. Treatment of 18 with CAN in 1:1 acetonitrile/water led to oxidative cleavage of the two para-methoxy groups to afford quinone 19 in 71% yield. The quinone and the double bond were then reduced concomitantly by catalytic hydrogenation at atmospheric pressure (Pd/C in MeOH) to yield a hydroquinone, which air-oxidized spontaneously10 to quinone bromide 20 in 70% yield over two steps.
Scheme 2.
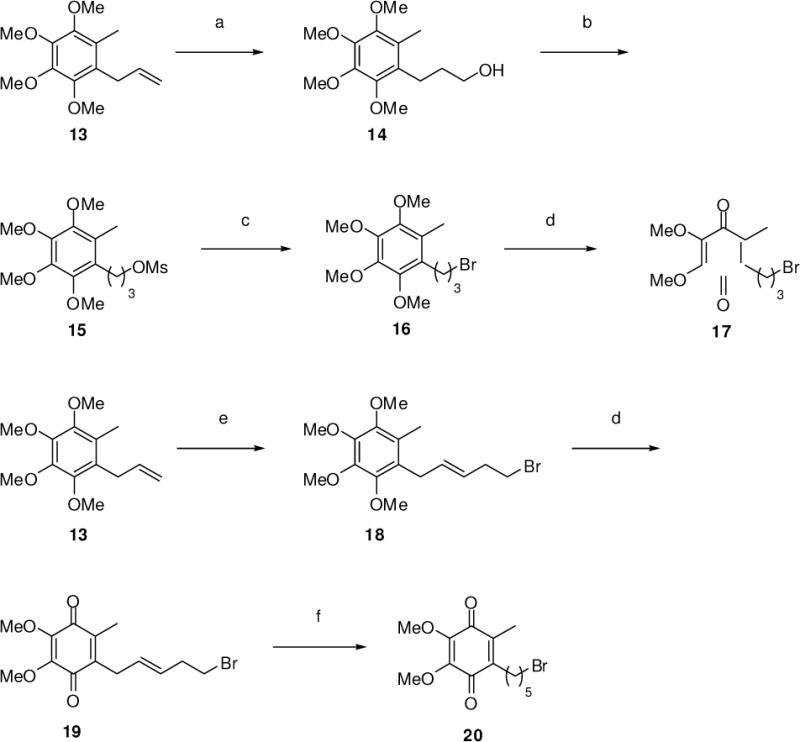
Syntheses of quinones 17 and 20. Reagents and conditions: (a) (i) 9-BBN, THF, 0 °C, then 60 °C, (ii) aq. NaOH, aq. H2O2, 0 °C, 79%; (b) MsCl, Et3N, DMAP, CH2Cl2, 23 °C, 97%; (c) NaBr, DMF, 23 °C, 65%; (d) CAN, 1:1 CH3CN-H2O, 0 °C, then 23 °C, 95% (17), 71% (19); (e) 4-bromo-1-butene, Grubbs’ 2nd generation catalyst, CH2Cl2, 40 °C, 64%; (f) H2 (1 atm), Pd/C, MeOH, 23 °C, 99%.
Treatment of 11-bromoundecanoic acid (Scheme 3) with thionyl chloride at reflux led to the corresponding acyl chloride, which was then immediately treated with aqueous hydrogen peroxide in the presence of pyridine in diethyl ether to afford 11-bromoundecanoic peroxo anhydride.11
Scheme 3.
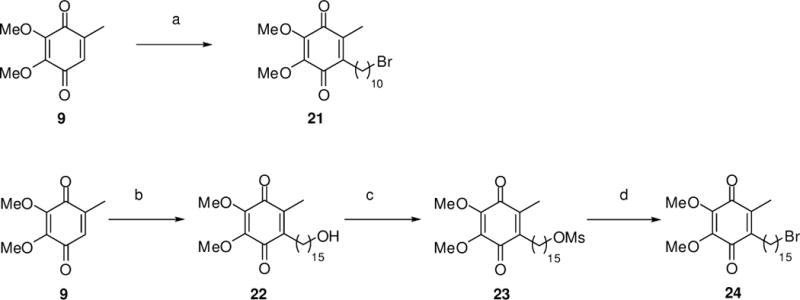
Syntheses of quinones 21 and 24. Reagents and conditions: (a) (i) 11-bromoundecanoic acid, SOCl2, 79 °C, then pyridine, aq. H2O2, Et2O, 23 °C, (ii) 5, AcOH, 118 °C, 22%; (b) 16-hydroxyhexadecanoic acid, AgNO3, K2S2O8, 1:1 CH3CN/H2O, 75 °C, 21%; (c) MsCl, Et3N, DMAP, CH2Cl2, 23 °C, 97%; (d) NaBr, acetone, 56 °C, 66%.
The crude solid so obtained was immediately stirred with coenzyme Q0 (9) in acetic acid at reflux to afford quinone bromide 21 in a modest 22% yield. Similarly, treatment of 15-hydroxyhexadecanoic acid and of 9 with silver nitrate and potassium peroxosulfate in 1:1 acetonitrile–water12 at 75 °C led to quinone 22 in 21% yield. Conversion of 22 to the corresponding mesylate 23 using MsCl and triethylamine in CH2Cl2, followed by treatment with sodium bromide in acetone at reflux led to quinone 24 in 64% yield over two steps.
Quinones 17, 20, 21, and 24 (Scheme 4) were reduced using aqueous sodium dithionite. The crude hydroquinones so obtained were then treated immediately with a stoichiometric amount of triphenylphosphine in ethanol in a sealed tube in the dark at 85 °C to afford, after purification by silica gel column chromatography, mitoQ3 (1), mitoQ5 (2), mitoQ10 (3) and mitoQ15 (4) in 55–92% yields. While mitoQ5 (2) was obtained as the quinone, 1 was obtained as a mixture containing the quinone (20%) and the corresponding hydroquinone (80%); mitoQ10 (3) and mitoQ15 (4) were obtained as a mixture of quinone (40%) and hydroquinone (60%). Stirring these mixtures in methanol in the presence of a catalytic amount palladium-on-carbon under aerobic conditions led to the pure quinones in each case. This last step proceeded in higher yields when the quinone was first reduced to the hydroquinones with Na2S2O4. In any case, the small scale at which the reaction was run prevented the use of solventless conditions.6 The use of non-polar aprotic solvents such as toluene did not favor the formation of the final mitoQ, nor did polar aprotic solvents such as acetonitrile. The conversion worked best using ethanol, a solvent in which both reagents and products were soluble. Contrary to the reported procedure,6 purification of the reaction mixture and removal of the excess PPh3, and of the Ph3PO generated during the reaction, upon washing with ether–CH2Cl2, did not lead to the isolation of any product having a satisfactory level of purity. In comparison, purification by column chromatography using CH2Cl2-EtOH enabled efficient separation of the product from unreacted starting material. Consistent with their structures, the mitoQs do not readily crystallize; under anhydrous conditions, they form foams that collapse into gums when exposed to the air.
Scheme 4.
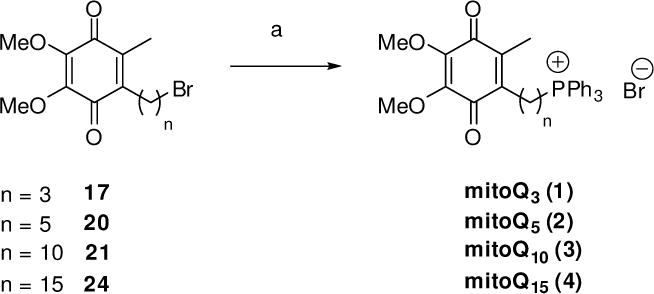
Syntheses of mitoQs 1–4. Reagents and conditions: (a) (i) Na2S2O4, 1:1 EtOAc/H2O, (ii) PPh3, EtOH, sealed tube, 85 °C, (iii) air, Pd/C, MeOH, 23 °C, 55–92%.
2.2. Synthesis of idebenone (5) and idebenone analogues 6–8
The syntheses of analogues 6–8 commenced with the preparation of their corresponding quinone precursors 26, 30, and 35 (Scheme 5). Thus oxidation of 2,3,5-trimethylphenol (25) using a mixture of iodine, sulfuric acid and aqueous hydrogen peroxide13 at 23 °C afforded 2,3,5-trimethyl-p-benzoquinone (26) in 90% yield. Treatment of 2,5-dimethyl-p-benzoquinone with acetic anhydride and boron trifluoride-etherate14 at 40 °C afforded 1,2,4-triacetoxy-3,6-dimethylbenzene (28) in 92% yield; 28 then treated with sodium hydroxide and dimethyl sulfate in methanol14 at 23 °C to provide 3,6-dimethyl-1,2,4-trimethoxybenzene (29) in 82% yield. Finally, 29 was oxidized using phenyliodine diacetate (PIDA)15 to obtain 3,6-trimethyl-2-methoxy-p-benzoquinone (30) in 65% yield. Jones oxidation of 2,6-dimethylphenol (31) provided 2,6-dimethyl-p-benzoquinone (32) in 94% yield1.16 The quinone so obtained was then subjected to the same synthetic procedures used for the preparation of 30 from 27, in order to provide quinone 35.
Scheme 5.
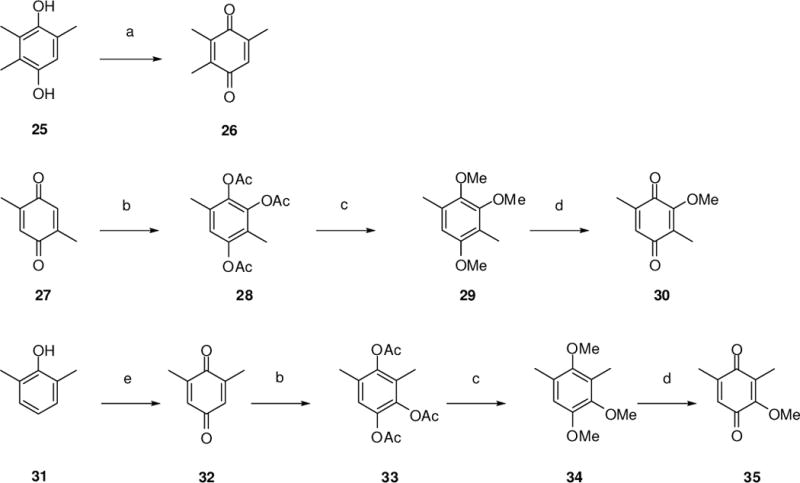
Syntheses of quinones 26, 30, and 35. Reagents and conditions: (a) I2, H2O2, H2SO4, MeOH, 23 °C, 94%; (b) Ac2O, BF3•OEt2, 40 °C, 92%; (c) Me2SO4, aq. NaOH, MeOH, 23 °C, 82%; (d) PIDA, 9:1 H2O/MeOH, 40 °C, 65%; (e) Jones reagent, 1:1 Et2O/H2O, 23 °C, 94%.
Commercially available 11-bromoundecanoic acid (Scheme 6) was treated with the sodium salt of benzyl alcohol in DMF at 75 °C to afford the O-benzylated derivative 36 in 60% yield, or with aqueous potassium hydroxide at reflux to afford 37 in 96% yield. Treatment of 9 and of 26 with 36, silver nitrate, and potassium persulfate, in acetonitrile–water12 in the dark at 75 °C afforded 38 and 39 in 17% and 37% yields, respectively. Cleavage of the benzyl ether protecting group by catalytic hydrogenation using palladium-on-carbon in methanol led to idebenone (5) and to idebenone analogue 6 in 82% and 51% yields, respectively. Similarly, treatment of 30 and of 35 with 37, silver nitrate, and potassium persulfate, in acetonitrile–water12 in the dark at 75 °C afforded 7 and 8 in 15% and 10% yields, respectively.
Scheme 6.
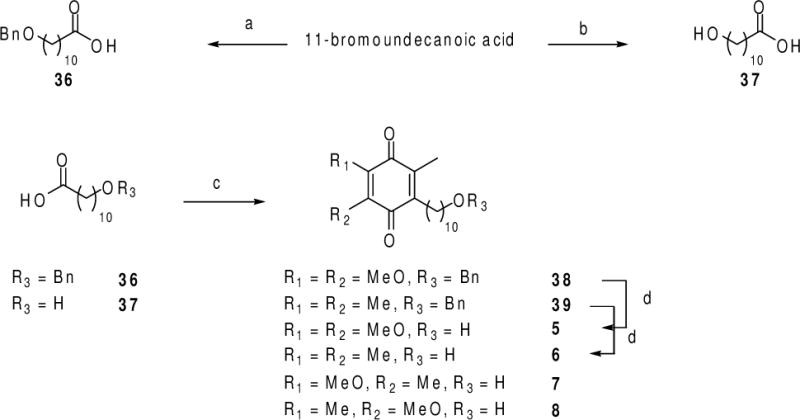
Syntheses of idebenone (analogues) 5–8. Reagents and conditions: (a) BnBr, NaH, DMF, 75 °C, 60%; (b) aq. KOH, 100 °C, 96%; (c) 9, 26, 30, or 35, AgNO3, K2S2O8, 1:1 CH3CN/H2O, 75 °C, 10–37%; (d) H2, Pd/C, MeOH, 23 °C, then air, Pd/C, MeOH, 23 °C, 53–82%.
2.3. Oxygen consumption assay
C2C12 cells pretreated with 25nM rotenone were further treated with 10 μM compounds 1–8, either in the presence or absence of 5 μM FCCP. Oxygen consumption was monitored for 2 h and quantified. As shown in Figure 2, FCCP activated mitochondrial oxygen consumption under basal conditions, and mitoQ3 did not inhibit O2 consumption, but mitoQ5, mitoQ10 and mitoQ15 each inhibited O2 consumption; this inhibition increased with increasing side chain length. Thus coenzyme Q analogues having the triphenylphosphonium functionality within their side chain function poorly in supporting mitochondrial O2 consumption. In contrast, rotenone + idebenone stimulated mitochondrial O2 consumption about 50% above the basal level. This was consistent for each of the idebenone analogues as well, and all four compounds stimulated maximal O2 consumption about 3-fold. While the errors associated with this measurement do not permit a more detailed analysis of those structural elements within the quinone moiety conducive to mitochondrial oxygen consumption, they do suggest that some variation in substituents on the quinone moiety can be tolerated, and thus support the concept that exogenously supplied coenzyme Q analogues may envisioned as mechanistically and therapeutically relevant probes.
Figure 2.
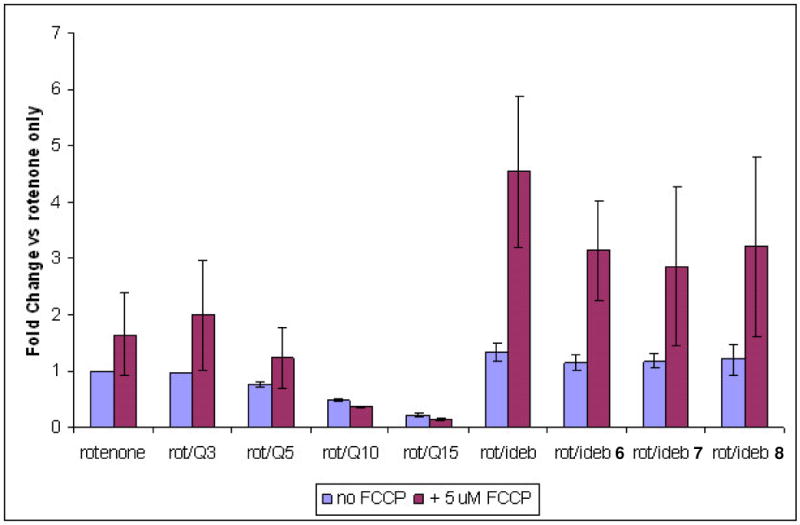
Effect of compounds 1–8 in supporting mitochondrial O2 consumption. C2C12 cells were pretreated with 25nM rotenone to simulate a partial blockage of mitochondrial Complex I, after which 10 μM compounds 1–8, either without or with 5 μM FCCP (to maximally stimulate mitochondrial oxygen consumption), were added. Oxygen consumption was monitored for 2 h and quantified by calculating M/T (see section 4.2.1). Compounds were tested in triplicate. Error bars represent median absolute deviation. In the left part of the figure, one sees that under basal conditions, FCCP activated mitochondrial oxygen consumption, and that mitoQ3 did not inhibit O2 consumption, but that mitoQ5, mitoQ10 and mitoQ15 each inhibited O2, with inhibition increasing with increased side chain length. In the right part of the figure, one sees that rotenone + idebenone stimulated mitochondrial O2 consumption about 50% above the basal level This was consistent for each of the idebenone analogues (6–8) as well, and all of the idebenone analogues stimulated maximal O2 consumption by approximately 3-fold.
2.4 Cytoprotective effects of idebenone analogues
The promising effects noted with idebenone (5) and its analogues (6 – 8) in supporting mitochondrial oxygen consumption prompted us to assay these compounds for their ability to protect cultured mammalian cells from the effects of oxidative stress. Accordingly, cultured CEM leukemia cells were treated with 5 mM diethyl maleate to deplete cellular glutathione, thus exposing the cells to the effects of oxidative stress. Pretreatment of the cells with compounds 5 – 8 prior to diethyl maleate treatment actually did confer protection to the cells, as summarized in Table 1. As shown in the Table, compound 6, containing two methyl groups in place of the methoxyl groups in idebenone, was actually the most effective, conferring virtually complete protection against depletion of glutathione when employed at a concentration of 2.5 μM.
Table 1.
Cytoprotective effects of compounds 5 – 8 on cultured CEM cells treated with diethyl maleatea
| Compound | Viable cells
|
||
|---|---|---|---|
| 0.1 μM | 0.5 μM | 2.5 μM | |
| 5 | 24.6 ± 1.2 | 35.8 ± 3.3 | 86.2 ± 4.1 |
| 6 | 34.8 ± 3.4 | 77.3 ± 2.8 | 96.3 ± 3.7 |
| 7 | 22.9 ± 0.8 | 74.7 ± 7.7 | 82.3 ± 3.7 |
| 8 | 8.27 ± 0.7 | 21.2 ± 1.0 | 77.1 ± 1.4 |
CEM leukemia cells were pretreated with compounds 5 – 8 at the concentrations indicated for 18 hours. The cells were then treated with 5 mM diethyl maleate for 4 hours and subsequently assayed for viability using trypan blue.
3. Conclusion
The syntheses of four mitoQs (1–4), as well as idebenone (5) and three idebenone analogues (6–8) have been accomplished. While the side chains of mitoQs 3–4 and of idebenone analogues 5–8 were obtained upon radical alkylation of their quinone precursors, Diels-Alder cycloadduct 10 allowed the facile functionalization of 9, leading to the precursors of mitoQ3 (1) and mitoQ5 (2). This strategy should be readily applicable to modification of the redox core of coenzyme Q and the introduction of a variety of side chains. Olefin cross-metathesis was used successfully to elongate the side chain of intermediate 13, en route to mitoQ5 (2). While mitoQ analogue 1 did not materially increase or decrease basal or maximal mitochondrial O2 consumption in the presence of rotenone, 2, 3 and 4 decreased basal and maximal mitochondrial O2 consumption in order of increasing side chain length. By contrast, idebenone (5) and each of its analogues 6–8 promoted basal mitochondrial O2 consumption by about 50%, and maximal mitochondrial O2 consumption by about 300%, i.e. much more than in the presence of rotenone itself. The method by which idebenone and its analogues increase mitochondrial O2 consumption could be either through uncoupling mitochondria, or through supporting maximal electron transport activity. Since rotenone + FCCP only produced a 50% increase in O2 over rotenone alone, whereas idebenone and analogues + FCCP produced a 300% increase, the latter hypothesis (i.e. facilitation of maximal electron transport activity) is better supported. However, further studies of absolute mitochondrial productivity (i.e. ATP synthesis) are necessary to confirm this idea. The ability of the idebenone analogues to protect diethyl maleate-treated CEM cells from oxidative stress is also quite encouraging, especially in view of the enhanced protection relative to idebenone obtained with compound 6.
4. Experimental
4.1. Chemistry
Reactions were carried out under an atmosphere of argon unless specified otherwise. The glassware was dried in an oven at 110 °C prior to use. Tetrahydrofuran was distilled from sodium/benzophenone ketyl, dichloromethane was distilled from calcium hydride, benzene and toluene were distilled from sodium, and triethylamine and diisopropylamine were distilled from potassium hydroxide. All other solvents were of analytical grade and were used without any further purification. Flash column chromatography was carried out using silica gel (Silicycle R10030B, 60 Å particle size, 230–400 mesh), applying a low-pressure stream of nitrogen. Analytical thin layer chromatographic separations were carried on glass plates coated with silica gel (60 Å particle size, 250 μm thickness, F-254, Silicycle). The TLC chromatograms were developed using iodine vapor, or by immersing the plates either in 2.5% phosphomolybdic acid in ethanol, or in 2.0% anisaldehyde in ethanol/sulfuric acid/acetic acid, followed by heating (heat gun). 1H NMR chemical shifts were reported relative to residual CHCl3 at 7.26 ppm or DMSO at 3.31ppm; 13C NMR chemical shifts were reported relative to the central line of CDCl3 at 77.0 ppm or of DMSO-d6 at 39.5 ppm, and 31P NMR shifts were reported relative to the central line of H3PO4 at 0.00 ppm.
4.1.1. 4,5-Dimethoxy-2-methyltricyclo[6.2.1.02,7]undeca-4,9-diene-3,6-dione (10)7
A stirred solution containing 2.00 g (11.0 mmol) of 2,3-dimethoxy-5-methyl-1,4-benzoquinone (9) and 1.24 mL (16.5 mmol) of freshly distilled cyclopentadiene in 6.6 mL of glacial acetic acid was stirred at 23 °C for 18 h. The reaction solution was chilled by the mean of an ice–water bath and was then treated with saturated aq NaOH until a basic pH was reached. The product was extracted with five 20-mL portions of ethyl acetate. The combined organic layer was washed successively with 20 mL of distilled water, and 20 mL of brine, and was then dried (MgSO4), and concentrated under diminished pressure to provide 4,5-dimethoxy-2-methyltricyclo[6.2.1.02,7]undeca-4,9-diene-3,6-dione (10) as a dark yellow oil: yield 2.56 g (94%); silica gel TLC Rf 0.20 (3:2 hexanes/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 1.45 (s, 3H), 1.45–1.65 (m, 2H, AB system), 2.80 (d, 1H, J = 3.85 Hz),3.06 (br m, 1H), 3.40 (br m, 1H),3.93 (s, 3H), 3.95 (s, 3H), 6.00 (dd, 1H, J = 5.5, 3.8 Hz) and 6.14 (dd, 1H, J = 5.5, 2.8 Hz); 13C NMR (125 MHz, CDCl3) δ 26.8,44.5,46.6,49.1, 53.7, 57.3,58.8, 60.9, 134.6, 137.3, 138.27, 138.48, 150.63 and 150.69.
4.1.2. 2-Allyl-4,5-dimethoxy-7-methyltricyclo[6.2.1.02,7]-undeca-4,9-diene-3,6-dione (11)
To a stirred solution containing 1.00 g (4.04 mmol) of 4,5-dimethoxy-2-methyltricyclo[6.2.1.02,7]-undeca-4,9-diene-3,6-dione (10) in 6 mL of tetrahydrofuran at 0 °C, was added portionwise 0.72 g (6.06 mmol) of potassium tert-butoxide. The reaction mixture turned from yellow to dark red and was stirred at 0 °C for 30 min. A solution containing 0.53 mL (6.12 mmol) of allyl bromide in 6 mL of tetrahydrofuran was added dropwise to the reaction suspension at 0 °C. The reaction mixture was stirred at 0 °C for 1.5 h, and was then treated with 8 mL of distilled water. The product was extracted with five 10-mL portions of ether. The combined organic layer was washed with 10 mL of distilled water, then with 10 mL of brine, and was then dried (MgSO4) and concentrated under diminished pressure to provide 2-allyl-4,5-dimethoxy-7-methyltricyclo[6.2.1.02,7]-undeca-4,9-diene-3,6-dione (11) as a yellow oil: yield 0.94 g (80%); silica gel TLC Rf 0.33 (2:1 hexanes/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 1.46 (m, 1H),1.49 (s, 3H), 1.75 (d, 1H, J = 9.77 Hz), 2.54 (m, 1H, AB system), 2.66 (m, 1H, AB system),3.01 (s, 1H), 3.10 (s, 1H), 3.88 (s, 3H), 3.89 (s, 3H), 5.04 (m, 2H),5.77 (m, 1H) and 6.04 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 23.8,41.8,43.4,52.7, 54.4, 56.5, 59.3,60.4, 60.5,95.0, 118.9, 134.2, 137.7, 138.1, 149.5, 150.6 and 177.0; mass spectrum (LCT electrospray), m/z 311.1260 (M+Na)+(C17H20O4Na requires 311.1259).
4.1.3. 2-Allyl-3-methyl-5,6-dimethoxy-1,4-benzoquinone (12)
A solution containing 0.91 g (3.17 mmol) of 2-allyl-4,5-dimethoxy-7-methyltricyclo[6.2.1.02,7]-undeca-4,9-diene-3,6-dione (11) in 16 mL of toluene was stirred at reflux for 5 h, during which time the reaction solution turned from yellow to dark orange. The solvent was concentrated under diminished pressure to provide 2-allyl-3-methyl-5,6-dimethoxy-1,4-benzoquinone (12) as a red oil: yield 0.69 g (98%); silica gel TLC Rf 0.38 (2:1 hexanes/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 2.02 (s, 3H), 3.23 (d, 2H, J = 7.1 Hz), 3.99 (s, 6H),5.04 (m, 2H) and 5.75 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 12.2, 15.7, 18.2, 30.0, 30.5, 61.4, 116.8, 133.2, 140.0,144.5, 183.7 and 184.6; mass spectrum (LCT electrospray), m/z 223.0962 (M+H)+ (C12H15O4 requires 223.0970).
4.1.4. 1-Allyl-2,3,4,5-tetramethoxy-6-methylbenzene (13)
To a vigorously stirred solution at 23 °C containing 0.474 g (2.13 mmol) of 2-allyl-3-methyl-5,6-dimethoxy-1,4-benzoquinone (12) and 0.069 g (0.214 mmol) of n-tetrabutylammonium bromide in 42 mL of 1:1 tetrahydrofuran/water was added portionwise 4.37 g (21.3 mmol) of sodium dithionite. The reaction mixture was stirred vigorously at 23 °C for 0.5 h, time during which the reaction solution turned from orange to light yellow. The reaction mixture was then chilled by means of an ice–water bath, then 1.28 g (32.0 mmol) of solid sodium hydroxide was added portion-wise, followed immediately by 4.00 mL (42.3 mmol) of dimethyl sulfate. The cooling bath was removed and the reaction mixture was stirred at 23 °C for 2.5 h. The product was extracted with five 30-mL portions of ether. The combined organic layer was washed successively with 30 mL of distilled water, then with 30 mL of brine and was dried (MgSO4) and concentrated under diminished pressure. The crude product was applied to a silica gel column (12 × 4 cm); elution with 7:1 hexanes/ethyl acetate provided 1-allyl-2,3,4,5-tetramethoxy-6-methylbenzene (13) as a colorless oil: yield 0.438 g (81%); silica gel TLC Rf 0.63 (2:1 hexane/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 2.12 (s, 3H), 3.39 (d, 2H, J = 5.9 Hz), 3.76 (s, 3H), 3.78 (s, 3H), 3.88 (s, 3H), 3.89 (s, 3H), 4.92 (d, 1H, J = 16.6 Hz), 5.00 (d, 1H, J = 9.8 Hz) and 5.92 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 11.9,31.2, 61.0, 61.3, 61.4, 61.5, 115.0, 125.8, 127.0,136.7, 144.8, 145.4, 148.0 and 148.0; mass spectrum (LCT electrospray), m/z 275.1249 (M+Na)+ (C14H20O4Na requires 275.1259).
4.1.5. 1-(3-Hydroxypropyl)-2,3,4,5-tetramethoxy-6-methylbenzene (14)17
To a stirred solution containing 307 mg (1.22 mmol) of 1-allyl-2,3,4,5-tetramethoxy-6-methylbenzene (13) in 1.8 mL of tetrahydrofuran at 0 °C was added dropwise 3.00 mL of a 1.0 M 9-BBN solution in tetrahydrofuran. The reaction mixture was stirred at 23 °C for 16 h, then at reflux for 2 h. The reaction solution was then treated successively at 0 °C with 2.0 mL (6.0 mmol) of 3.0 M aq sodium hydroxide and then with 2.0 mL (5.83 mmol) of 30% aq hydrogen peroxide. The reaction mixture was stirred at 0 – 2 °C for 0.5 h. The reaction mixture was diluted with 20 mL of distilled water and the product was extracted with four 20-mL portions of ether. The combined organic layer was washed with 20 mL of brine, dried (Na2SO4) and then concentrated under diminished pressure. The crude oil was applied to a silica gel column (12 × 3 cm); step-gradient elution with 4:1 → 2:1 hexanes/ethyl acetate provided 1-(3-hydroxypropyl)-2,3,4,5-tetramethoxy-6-methylbenzene (14) as a colorless oil: yield 261 mg (79%); silica gel TLC Rf 0.08 (2:1 hexanes/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 1.71 (quint, 2H, J = 5.9 Hz),2.15 (s, 3H), 2.69 (t, 2H, J = 7.3 Hz), 3.52 (t, 2H, J = 5.9 Hz),3.76 (s, 3H), 3.82 (s, 3H), 3.87 (s, 3H) and 3.89 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 11.8, 22.7, 30.0,32.3, 60.9, 61.3, 61.5, 61.7,125.3, 128.4, 144.5, 145.2, 147.9 and 148.3; mass spectrum (LCT electrospray), m/z 271.1539 (M+H)+ (C14H23O5 requires 271.1545).
4.1.6. 1-(3-Methanesulfoxypropyl)-2,3,4,5-tetramethoxy-6-methylbenzene (15)
To a solution containing 221 mg (0.817 mmol) of 1-(3-hydroxypropyl)-2,3,4,5-tetramethoxy-6-methylbenzene (14), 500 μL (3.27 mmol) of triethylamine, and 2.0 mg (0.016 mmol) of dimethylaminopyridine in 5.5 mL of dichloromethane was added 200 μL (1.72 mmol) of methanesulfonyl chloride. The reaction solution was stirred at 23 °C for 4.5 h. The reaction mixture was transferred into a separatory funnel containing 20 mL of distilled water. The layers were separated and the aqueous layer was extracted with three 15-mL portions of ether. The combined organic layer was washed with 15 mL of brine, and then dried (MgSO4) and concentrated under diminished pressure. The crude product was applied to a column of silica gel (13 × 3 cm); elution with 2:1 hexanes/ethyl acetate afforded 1-(3-methanesulfoxy)-2,3,4,5-tetramethoxy-6-methylbenzene (15) as a clear colorless oil: yield 275 mg (97%); silica gel TLC Rf 0.15 (2:1 hexanes/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 1.93 (dt, 2H, J = 15.9, 6.3 Hz), 2.17 (s, 3H),2.70 (t, 2H, J = 7.7 Hz), 3.03 (s, 3H),3.78 (s, 3H),3.82 (s, 3H),3.89 (s, 3H), 3.91 (s, 3H) and 4.27 (t, 2H, J = 6.3 Hz); 13C NMR (125 MHz, CDCl3) δ 11.6,22.9,29.4, 37.4, 60.1, 60.6, 61.0, 70.0, 125.0, 127.7, 144.7, 145.3,147.7 and 147.9; mass spectrum (LCT electrospray), m/z 371.1147 (M+Na)+ (C15H24O7SNa requires 371.1140).
4.1.7. 1-(3-Bromopropyl)-2,3,4,5-tetramethoxy-6-methylbenzene (16)
A solution containing 230 mg (0.660 mmol) of 1-(3-methanesulfoxypropyl)-2,3,4,5-tetramethoxy-6-methylbenzene (15) in 3 mL of dimethylformamide was treated with 682 mg (6.60 mmol) of sodium bromide at 23 °C for 24 h. The reaction mixture was partitioned between distilled water (5 mL) and ethyl acetate (5 mL). The aqueous layer was further extracted with six 5-mL portions of ethyl acetate. The combined organic layer was washed with one 10-mL portion of 3% aq HCl, then with two 10-mL portions of distilled water and 10 mL of brine. The organic phase was dried (MgSO4) and concentrated under diminished pressure. The resulting oil was applied to a silica gel column (13 × 3 cm); elution with 4:1 hexanes/ethyl acetate afforded 1-(3-bromopropyl)-2,3,4,5-tetramethoxy-6-methylbenzene (16) as a colorless oil: yield 199 mg (90%); silica gel TLC Rf 0.32 (1:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 2.02 (m, 2H), 2.18 (s, 3H), 2.72 (dd, 2H, J = 8.8, 6.8 Hz),3.47 (t, 2H, J = 6.8 Hz),3.78 (s, 3H), 3.83 (s, 3H), 3.89 (s, 3H) and 3.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 11.7,25.7,33.2, 33.9, 60.6, 60.98, 61.01, 61.1, 125.0, 128.1, 144.6, 145.1, 145.2 and 147.8; mass spectrum (LCT electrospray), m/z 355.0525 (M+Na)+ (C14H21O4BrNa requires 355.0521).
4.1.8. 2-(5-Bromopropyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (17)
To a stirred solution containing 199 mg (0.597 mmol) of 1-(3-bromopropyl)-2,3,4,5-tetramethoxy-6-methylbenzene (16) in 6 mL of 1:1 acetonitrile/water was added a solution containing 833 mg (1.49 mmol) of cerium (IV) ammonium nitrate in 6 mL of 1:1 acetonitrile/water. The cooling bath was removed, and the reaction solution was stirred at 23 °C for 1 h. The reaction mixture was partitioned between 10 mL of distilled water and 10 mL of ethyl acetate. The aqueous phase was extracted with five 10-mL portions of ethyl acetate. The combined organic layer was washed with 5 mL of brine, then dried (NaSO4) and concentrated under diminished pressure. The resulting oil was applied to a silica gel column (13 × 3 cm); elution with 2:1 hexanes/ethyl acetate afforded 2-(5-bromopropyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (17) as a yellow oil: yield 164 mg (95%); silica gel TLC Rf 0.36 (2:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.96 (m, 2H), 2.05 (s, 3H), 2.62 (t, 2H, J = 7.6 Hz), 3.43 (t, 2H, J = 6.8 Hz) and 3.98 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 12.0, 25.2,31.5, 33.1, 61.2, 139.7, 141.1, 144.3, 144.4, 184.0 and 184.4; mass spectrum (APCI), m/z 303.0233 (M+H)+ (C12H16O4Br requires 303.0232).
4.1.9. 1-(5-Bromo-2-pentenyl)-2,3,4,5-tetramethoxy-6-methylbenzene (18)
A solution containing 115 mg (0.453 mmol) of 1-allyl-2,3,4,5-tetramethoxy-6-methylbenzene (13) and 230 μL (2.27 mmol) of 4-bromo-1-butene was stirred in 45 mL of dichloromethane in the presence of 19.4 mg (0.023 mmol) of benzylidene [1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene] dichloro (tricyclohexylphosphine) ruthenium (Grubbs’ second generation catalyst11) at reflux for 6 h. The solvent was concentrated under diminished pressure and the crude residue was applied to a silica gel column (14 × 3 cm); elution with 6:1 hexanes/ethyl acetate afforded 1-(5-bromo-2-pentenyl)-2,3,4,5-tetramethoxy-6-methylbenzene (18) as a light amber oil: yield 105 mg (64%); silica gel TLC Rf 0.51 (2:1 hexanes/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 2.12 (s, 3H), 2.52 (q, 2H, J = 6.8 Hz), 3.32 (m, 4H), 3.76 (s, 3H), 3.78 (s, 3H), 3.88 (s, 3H),3.89 (s, 3H), 5.30 (m, 1H) and 5.59 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 11.9, 30.0, 33.0,36.1,60.9, 61.27, 61.33, 61.5, 125.7, 127.19, 127.22, 131.6, 144.8, 145.4, 147.85 and 147.96; mass spectrum (LCT electrospray), m/z 381.0688 (M+Na)+ (C16H23O4BrNa requires 381.0677).
4.1.10. 2-(5-Bromo-2-penten)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (19)
To a stirred solution at 0 °C containing 133 mg (0.369 mmol) of 1-(5-bromo-pentenyl)-2,3,4,5-tetramethoxy-6-methylbenzene (18) in 3.7 mL of 1:1 acetonitrile/water was added a solution containing 514 mg (0.923 mmol) of cerium (IV) ammonium nitrate in 3.7 mL of 1:1 acetonitrile/water. The reaction mixture was stirred at 23 °C for 2.5 h. The reaction mixture was partitioned between 10 mL of distilled water and 10 mL of ethyl acetate. The aqueous layer was extracted with three 5-mL portions of ethyl acetate. The combined organic layer was washed with 5 mL of brine, then dried (Na2SO4) and concentrated under diminished pressure. The resulting crude oil was applied to a silica gel column (14 × 3 cm); elution with 2:1 hexanes/ethyl acetate afforded 2-(5-bromo-2-pentenyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (19)as a red oil: yield 86.5 mg (71%); silica gel TLC Rf 0.09 (4:1 hexanes/ether; 1H NMR (300 MHz, CDCl3) δ 1.99 (s, 3H),2.50 (q, 2H, J = 5.5 Hz), 3.16 (d, 2H, J = 3.3 Hz), 3.31 (t, 2H, J = 6.8 Hz),3.95 (s, 6H) and 5.42 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 12.0,29.2,29.8, 32.4, 35.2, 53.4, 61.2, 127.8, 129.1, 184.4 and 185.0; mass spectrum (LCT electrospray), m/z 351.0208 (M+Na)+ (C14H17O4BrNa requires 351.0214).
4.1.11. 2-(5-Bromopentyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (20)17
A solution containing 85 mg (0.26 mmol) of 2-(5-bromo-2-pentenyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (19) in 2.6 mL of methanol was stirred in the presence of 9 mg of 10% palladium-on-carbon under hydrogen (1 atm) at 23 °C for 3 h, during which time the reaction mixture turned from yellow to colorless. The catalyst was removed by filtration through a short pad of silica gel and the filtrate was stirred at 23 °C under aerobic conditions for 0.5 h, and was then concentrated under diminished pressure to afford a mixture containing ~ 80% of 2-(5-bromopentyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (20), and ~ 20% of the corresponding hydroquinone as an orange oil: yield 84 mg (99%); treatment of an aerated methanolic solution of the hydroquinone/quinone mixture with 10% palladium-on-carbon provided the quinone quantitatively; silica gel TLC Rf 0.35 (1:1 hexanes/ethyl acetate); 1H NMR (300 MHz, CDCl3) δ 0.89 (m, 2H),1.34 (m, 6H), 2.01 (s, 3H), 2.45 (m, 2H, J = 7.4 Hz) and 3.98 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 12.1, 14.2, 22.7, 26.6, 28.6, 32.2, 61.4, 138.9, 143.3, 144.5, 144.6, 184.4 and 185.0; mass spectrum (APCI), m/z 331.0548 (M+H)+ (C14H20O4Br requires 331.0545).
4.1.12. 2-(10-Bromodecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (21)6
A solution containing 3.00 g (11.3 mmol) of 11-bromoundecanoic acid in 1.50 mL (20.6 mmol) of thionyl chloride was stirred at reflux for 20 min. Excess thionyl chloride was removed by distillation under diminished pressure; the residue was diluted with 10 mL of ether and the resulting light yellow clear solution was stirred at 0 °C. It was treated with 1.40 mL of 30% aq hydrogen peroxide at 0 °C, followed by the slow addition at 0 °C (over the course of 15 min) of a solution containing 1 mL of pyridine in 1 mL of ether. As the pyridine was added, a white precipitate formed. Once the addition was complete, the resulting suspension was stirred at room temperature for 1.25 h. The entire reaction mixture was then transferred into a separatory funnel containing 150 mL of ether and 50 mL of distilled water. The layers were separated and the organic layer was washed successively with two 50-mL portions of distilled water, 50 mL of 10% aq HCl, 50 mL of distilled water, two 50-mL portions of 0.5 M aq sodium bicarbonate, 50 mL of distilled water, and then with 50 mL of brine. The organic phase was dried (MgSO4) and concentrated under diminished pressure to afford the crude peroxo-anhydride as a white solid which was used immediately for the coupling step with 2,3-dimethoxy-5-methyl-1,4-benzoquinone (9): yield 2.98 g (94%).
A solution containing 1.29 g (7.08 mmol) of 2,3-dimethoxy-5-methyl-1,4-benzoquinone (9) and 2.98 g (10.6 mmol) of the freshly prepared peroxo-anhydride in 42 mL of glacial acetic acid was stirred at reflux for 20 h. The resulting red solution was allowed to cool to room temperature and was then transferred into a separatory funnel containing 300 mL of ether. The solution was washed successively with two 100-mL portions of distilled water, two 100-mL portions of 10% aq HCl, 100 mL of distilled water, two 100-mL portions of 0.5 M aq sodium bicarbonate, 100 mL of distilled water, and then with two 100-mL portions of brine. The dried organic layer (MgSO4) was concentrated under diminished pressure and the resulting red solid was applied to a silca gel column (15 × 4 cm); elution with 20:1 dichloromethane/diethyl ether afforded 3-(10-bromodecyl)-2-methyl-5,6-dimethoxy-1,4-benzoquinone (21) as a red oil: yield 0.62 g (22%); silica gel TLC Rf 0.72 (2:1 hexanes/ethyl acetate); 1H NMR (500 MHz, CDCl3) δ 1.25–1.40 (m, 14H),1.82 (dt, 2H, J = 14.7, 6.8 Hz), 1.98 (s, 3H), 2.42 (t, 2H, J = 6.8 Hz), 3.37 (t, 2H, J = 6.8 Hz), 3.95 (s, 3H) and 3.96 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 12.2, 26.7, 28.4, 28.9, 29.0, 29.5, 29.6, 29.7, 30.1, 33.1, 34.3, 61.4, 138.8, 143.2, 144.4, 184.2 and 184.7.
4.1.13. 2-(15-Hydroxypentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (22)6
To a stirred suspension containing 546 mg (3.00 mmol) of 2,3-dimethoxy-5-methyl-1,4-benzoquinone (9), 816 mg (3.00 mmol) of 16-hydroxyhexadecanoic acid, and 464 mg (2.73 mmol) of silver nitrate in 60 mL of 1:1 acetonitrile/water at 75 °C, was added slowly over the course of 2 h a solution containing 900 mg (3.33 mmol) of potassium persulfate in 35 mL of distilled water. When the addition was complete, the reaction mixture was stirred at 75 °C for 1 h, then was allowed to cool to room temperature, and the precipitated solid was filtered under vacuum. The filtrate was transferred to a separatory funnel and the product was extracted with five 50-mL portions of ether. The combined organic layer was washed successively with two 100-mL portions of distilled water, two 100-mL portions of 1.0 M aq sodium bicarbonate, 100 mL of distilled water and 50 mL portion of brine. The dried (Na2SO4) organic phase was then concentrated under diminished pressure. The crude solid was applied to a silica gel column (12 × 3 cm); elution with 10:1 dichloromethane/ether afforded 2-(15-hydroxylpentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (22) as a dark yellow solid: yield 260 mg (21%); silica gel TLC Rf 0.10 (20:1 dichloromethane/ether); mp: 63 – 64 °C; 1H NMR (500 MHz, CDCl3) δ 1.23–1.36 (m, 24H),1.54 (quint, 2H, J = 6.8 Hz),1.99 (s, 3H), 2.42 (t, 2H, J = 7.8 Hz), 3.62 (t, 2H, J = 6.8 Hz), 3.96 (s, 3H) and 3.97 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 12.2, 26.0,26.7, 29.0, 29.68, 29.73, 29.80,29.88, 29.90, 29.93, 30.1, 33.1, 61.4, 63.4, 138.8,143.3, 144.4, 184.2 and 184.8; mass spectrum (LCT electrospray), m/z 431.2772 (M+Na)+ (C24H40O5Na requires 431.2773).
4.1.14. 2-(15-Methanesulfoxypentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (23)
To a stirred solution at 23 °C containing 91.8 mg (0.225 mmol) of 2-(15-hydroxypentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (22), 70.0 μL (0.502 mmol) of triethylamine, and 3.1 mg (0.025 mmol) of dimethylaminopyridine in 0.5 mL of dichloromethane, was added 21.0 μL (0.271 mmol) of methanesulfonyl chloride. After stirring at 23 °C for 1 h, the reaction mixture was diluted with 5 mL of ether and was poured into a separatory funnel containing 5 mL of distilled water. The layers were separated and the aqueous layer was extracted with two 3-mL portions of ether. The combined organic layer was washed with two 3-mL portions 0.5 M aq sodium bicarbonate, then with 3 mL of brine. The dried (MgSO4) organic layer was then concentrated under diminished pressure to afford 2-(15-methanesulfoxypentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (23) as a yellow solid: yield 107 mg (97%); silica gel TLC Rf 0.40 (20:1 dichloromethane/ether); mp 42 – 44 °C; 1H NMR (500 MHz, CDCl3) δ 1.23–1.38 (m, 24H),1.72 (quint, 2H, J = 6.8 Hz),1.98 (s, 3H), 2.42 (t, 2H, J = 6.8 Hz), 2.98 (s, 3H), 3.96 (s, 6H) and 4.20 (t, 2H, J = 6.8 Hz); 13C NMR (125 MHz, CDCl3) δ 12.2, 25.7, 26.7, 29.0, 29.3, 29.4,29.66,29.70, 29.79,29.88, 29.90,30.1, 37.6,61.4,70.4, 138.8, 143.2, 144.0,144.4, 184.2 and 184.8; mass spectrum (LCT electrospray), m/z 509.2555 (M+Na)+ (C25H42O7SNa requires 509.2549).
4.1.15. 2-(15-Bromopentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (24)
A solution containing 58.0 mg (0.119 mmol) of 2-(15-methanesulfoxypentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (23) and 123 mg (1.19 mmol) of finely ground sodium bromide in 1.2 mL of acetone was stirred at reflux for 6.5 h. The acetone was concentrated under diminished pressure; the crude residue was suspended in a minimum amount of dichloromethane and the supernatant was applied to a silica gel column (13 × 2 cm). Elution with 4:1 hexanes/ethyl acetate afforded 2-(15-bromopentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (24) as a yellow solid: yield 37.0 mg (66%) as well as essentially all unreacted starting material: yield 18.7 mg (32%); silica gel TLC Rf 0.53 (4:1 hexanes/ethyl acetate); mp: 43 – 44 °C; 1H NMR (500 MHz, CDCl3) δ 1.23–1.42 (m, 24H), 1.83 (quint, 2H, J = 6.8 Hz), 1.98 (s, 3H), 2.42 (t, 2H, J = 6.8 Hz), 3.38 (t, 2H, J = 6.8 Hz) and 3.96 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 12.0, 26.5, 28.2, 28.8,29.45,29.50, 29.58, 29.59, 29.7, 29.8, 29.9, 32.9,34.1, 61.2, 138.6, 143.0, 183.9 and 184.6; mass spectrum (LCT electrospray), m/z 493.1924 (M+Na)+ (C24H39O4BrNa requires 493.1929).
4.1.16. 2-(5-Triphenylphosphoniumpropyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone bromide (MitoQ3, 1)
A solution containing 81.8 mg (0.266 mmol) of 2-(5-bromopropyl)-3-methyl-5,6-dimethoxy-1,4-dihydroxybenzene (17) in 2.7 mL of ethyl acetate was thoroughly shaken in a separatory formed in the presence of a solution containing 1.10 g (5.37 mmol) of sodium dithionite in 5.4 mL of distilled water. The layers were separated and the aqueous layer was extracted with 2.7 mL portion of ethyl acetate. The combined organic layer was dried (MgSO4), and was then concentrated under diminished pressure. The crude hydroquinone so obtained was quickly dissolved in 2.7 mL of 95% ethanol and the resulting light yellow solution was stirred in the presence of 72.4 mg (0.276 mmol) of triphenylphosphine in a sealed tube in the dark at 85 °C for 72 h. The solvent was concentrated under diminished pressure and the resulting yellow foam was applied to a silica gel column (12 × 3 cm); elution with 9:1 dichloromethane/ethanol afforded 2-(5-triphenylphosphoniumpropyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone bromide (1) as a yellow foam and as a mixture of quinone and hydroquinone (Q:QH2 ~ 20:80): yield 126 mg (82%); treatment of an aerated methanolic solution of the hydroquinone/quinone mixture with 10% palladium-on-carbon provided the quinone quantitatively; silica gel TLC Rf 0.22 (9:1 dichloromethane/ethanol); 1H NMR (400 MHz, CDCl3) δ 1.87 (m, 2H), 2.16 (s, 3H), 2.94 (dd, 2H, J = 7.6, 7.4 Hz), 3.76 (m, 2H), 3.94 (s, 3H), 3.95 (s, 3H) and 7.66–7.89 (m, 15H); 13C NMR (100 MHz, CDCl3) δ 11.5,21.8,22.1, 26.7,60.9, 61.0,117.3, 118.0,118.7, 120.6, 130.5,133.7, 135.0, 137.9, 184.2 and 184.4; 31P NMR (162 MHz, CDCl3) δ 24.3; mass spectrum (LCT electrospray), m/z 485.1883 (M-Br) (C30H30O4P requires 485.1882).
4.1.17. 2-(5-Triphenylphosphoniumpentyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone bromide (MitoQ5, 2)
A solution containing 47 mg (0.14 mmol) of 2-(5-bromopentyl)-3-methyl-5,6-dimethoxy-1,4-dihydroxybenzene (20) in 2.8 mL of ethyl acetate was shaken thoroughly in a separatory funnel in the presence of a solution containing 582 mg (2.84 mmol) of sodium dithionite in 5.6 mL of distilled water. The layers were separated and the aqueous layer was extracted with 2.8 mL of ethyl acetate. The combined organic layer was dried (MgSO4) and was then concentrated under diminished pressure. The crude hydroquinone was quickly dissolved in 1.4 mL of 95% ethanol and the resulting light yellow solution was stirred in the presence of 38 mg (0.14 mmol) of triphenylphosphine in a sealed tube in the dark at 85 °C for 40 h. The solvent was concentrated under diminished pressure and the resulting yellow foam was applied to a silica gel column (12 × 3 cm); elution with 9:1 dichloromethane/ethanol afforded 2-(5-triphenylphosphoniumpentyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (2) as a yellow foam: yield 46 mg (55%); silica gel TLC Rf 0.22 (9:1 dichloromethane/ethanol); 1H NMR (400 MHz, CDCl3) δ 1.40 (m, 2H),1.65 (m, 2H), 1.71 (m, 2H), 1.96 (s, 3H), 2.36 (dd, 2H, J = 8.2, 8.0), 3.80 (m, 2H), 3.95 (s, 6H) and 7.66–7.85 (m, 15H); 13C NMR (100 MHz, CDCl3) δ 12.0, 22.1,22.6, 25.8, 27.8, 29.9, 61.11, 61.13, 117.9,118.6, 130.4, 133.7, 134.9, 139.3,142.1, 144.2, 184.2 and 184.5; 31P NMR (162 MHz, CDCl3) δ 24.56; mass spectrum (LCT electrospray), m/z 513.2198 (M-Br)+ (C32H34O4P requires 513.2195).
4.1.18. 2-(10-Triphenylphosphoniumdecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone bromide (MitoQ10, 3)
A solution containing 97.7 mg (0.243 mmol) of 2-(10-bromodecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone (21) in 5 mL of ethyl acetate was thoroughly shaken in a separatory funnel in the presence of a solution containing 499 mg (2.44 mmol) of sodium dithionite in 5 mL of distilled water. The layers were separated and the organic layer was shaken a second time with a solution containing 499 mg (2.44 mmol) of sodium dithionite in 5 mL distilled water. The combined aqueous layer was extracted with 5 mL of ethyl acetate. The combined organic layer was dried (MgSO4) and concentrated under diminished pressure. The crude hydroquinone was quickly dissolved in 2.4 mL of 95% ethanol and the resulting light yellow solution was stirred in the presence of 64.0 mg (0.244 mmol) of triphenylphosphine in a sealed tube in the dark at 85 °C for 60 h. The solvent was concentrated under diminished pressure and the resulting crude yellow oil was applied to a silica gel column (13 × 2 cm); elution with 9:1 dichloromethane/ethanol afforded 2-(10-triphenylphosphoniumdecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone bromide (3) as a yellow foam and as a mixture of quinone and hydroquinone (Q:QH2 ~ 40:60): yield 149 mg (92%); treatment of an aerated methanolic solution of the hydroquinone/quinone mixture with 10% palladium-on-carbon provided the quinone quantitatively; 1H NMR (400 MHz, CDCl3) δ 1.21–1.62 (m, 16H), 2.13 (s, 3H), 2.56 (dd, 2H, J = 8.2, 8.0 Hz), 3.82 (m, 2H), 3.98 (s, 6H) and 7.68–7.88 (m, 15H); 13C NMR (100 MHz, CDCl3) δ 11.9, 15.3,22.7, 23.0, 26.4, 28.7, 29.2,29.4, 29.7,30.4, 31.6, 60.8, 61.2, 118.2,118.9, 130.5, 133.8, 134.9, 138.7, 143.0, 144.3, 184.2 and 184.7; 31P NMR (162 MHz, CDCl3) δ 24.7; mass spectrum (LCT electrospray), m/z 583.2975 (M-Br)+ (C37H44O4P requires 583.2977).
4.1.19. 2-(15-Triphenylphosphoniumpentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone bromide (MitoQ15, 4)
A solution containing 66.8 mg (0.142 mmol) of 24 in 2.80 mL of ethyl acetate was thoroughly shaken in a separatory funnel in the presence of a solution containing 581mg (2.83 mmol) of sodium dithionite in 2.8 mL of distilled water. The layers were separated and the aqueous layer was extracted with 3 mL of ethyl acetate. The combined organic layer was washed with 5 mL of brine and then dried (MgSO4) and concentrated under diminished pressure. The crude hydroquinone was quickly dissolved in 1.4 mL of 95% ethanol and the resulting yellow solution was treated with 37.2 mg (0.142 mmol) of triphenylphosphine in a sealed tube in the dark at 85 °C for 63 h. The solvent was concentrated under diminished pressure and the resulting crude yellow oil was applied to a silica gel column (13 × 2 cm); elution with 9:1 dichloromethane/ethanol afforded 2-(15-triphenylphosphoniumpentadecyl)-3-methyl-5,6-dimethoxy-1,4-benzoquinone bromide (4) as a yellow foam and as a mixture of quinone and hydroquinone (Q:QH2 ~ 40:60): yield 57.0 mg (55%); treatment of an aerated methanolic solution of the hydroquinone/quinone mixture with 10% palladium-on-carbon provided the quinone quantitatively; silica gel TLC Rf 0.30 (9:1 dichloromethane/ethanol); 1H NMR (400 MHz, CDCl3) δ 1.15–1.42 (m, 22H), 1.58 (m, 4H),2.10 (s, 3H), 2.54 (dd, 2H, J = 8.2, 8.0 Hz), 3.68 (m, 2H), 3.85 (s, 6H) and 7.66–7.82 (m, 15H); 13C NMR (100 MHz, CDCl3) δ 11.8,22.47,22.53,22.6, 22.9, 26.25, 26.30, 28.6, 29.1,29.27, 29.39, 29.46, 29.49, 29.53,29.8,30.4,60.7, 61.1, 117.9, 118.6, 130.5,133.6, 135.0, 136.6, 138.6, 139.9, 144.2,184.1 and 184.6; 31P NMR (162 MHz, CDCl3) δ 24.5; mass spectrum (LCT electrospray), m/z 653.3754 (M-Br)+ (C42H54O4P requires 653.3760).
4.1.20. Trimethyl-p-ben (26)13
To a stirred solution containing 1.00 g (6.57 mmol) of trimethyl-p-hydroquinone (25) in 20 mL of methanol at 23 °C were added 83.3 mg (0.33 mmol) of iodine followed by 329 μL (2.90 mmol) of 30% aq hydrogen peroxide and 329 μL (0.93 mmol) of sulfuric acid. The reaction mixture was stirred at 23 °C for 3 h and was then diluted with 150 mL of ether. The organic layer was washed with three 75-mL portions of water, then with one 75-mL portion of satd aq sodium thiosulfate and then with 75mL of brine. The organic layer was dried (MgSO4) and concentrated under diminished pressure to afford trimethyl-p-benzoquinone (26) as yellow crystals: yield 930 mg (94%); mp: 29–30°C; silica gel TLC Rf 0.65 (3:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 2.03 (m, 9H) and 6.56 (s, 1H); 13C NMR (CDCl3) δ 12.0,12.3, 15.9,133.0, 140.7, 140.9, 145.3,187.5, and 187.9.
4.1.21. 1,2,4-Acetoxy-3,6-dimethylbenzene (28)14
To stirred solution at 23 °C containing 1.00 g (7.34 mmol) of 2,5-dimethylparabenzoquinone (27) in 8.0 mL of acetic anhydride was added 400 μL (3.13 mmol) of boron trifluoride etherate. The reaction mixture was stirred at 40 °C for 48 h and was then poured into 100 mL of water. The formed precipitate was collected by filtration, and was then dried under diminished pressure to afford 1,2,4-acetoxy-3,6-dimethylbenzene (28) as a colorless solid: yield 1.89 g (92%); mp: 97–98 °C; silica gel TLC Rf 0.5 (1:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.95 (s, 3H),2.14 (s, 3H), 2.29 (s, 9H) and 6.84 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 10.1,16.0,20.2, 20.7, 20.8, 121.4, 122.5, 129.5, 139.2, 141.8, 146.7, 167.8, 167.9 and 168.8; mass spectrum (EI), m/z 280.0927 (M)+ (C14H16O6 requires 280.0947).
4.1.22. 3,6-Dimethyl-1,2,4-trimethoxybenzene (29)14
To a stirred solution at 23 °C containing 1.84 g (6.74 mmol) of 1,2,4-acetoxy-3,6-dimethylbenzene (28) in 6.0 mL of methanol were added 5.0 mL (52.3 mmol) of dimethyl sulfate followed by the slow addition of a solution containing 2.17 g (54.3 mmol) of sodium hydroxide in 2.5 mL of water. The reaction mixture was stirred at 23 °C for 16 h. The reaction mixture was poured into 60 mL of water and was then extracted with 80 mL of 1:1 diethyl ether/hexanes and then with 60 mL of hexanes. The combined organic layer was washed with 80mL of water and then with 80mL of brine. The solution was dried (MgSO4) and was then concentrated under diminished pressure. The residue was applied to a silica gel column (10 × 6 cm); elution with 3:1 hexanes/ethyl acetate afforded 3,6-dimethyl-1,2,4-trimethoxybenzene (29) as a colorless oil; yield 1.08 g (82%); silica gel TLC Rf 0.62 (4:1 hexanes/diethyl ether); 1H NMR (400 MHz, CDCl3)δ 2.12 (s, 3H), 2.27 (s, 3H), 3.78 (s, 3H),3.79 (s, 3H), 3.83 (s, 3H) and 6.43 (s, 1H); 13C NMR (100 MHz, CDCl3) δ8.7,16.0, 55.7, 60.3, 60.3,107.5, 118.0, 128.5, 145.3, 151.8 and 153.8; mass spectrum (EI), m/z 196.1094 (M)+ (C11H16O3 requires 196.1100).
4.1.23. 3,6-Dimethyl-2-methoxy-p-benzoquinone (30)
To a stirred solution containing 900 mg (4.85 mmol) of 3,6-dimethyl-1,2,4-trimethoxybenzene (29) in 30 mL of 9:1 water/methanolat 23 °C was added 2.21 g (6.86 mmol) of phenyliodine diacetate (PIDA). The reaction mixture was stirred at 40°C for 16 h, and then poured into 100 mL of water. The product was extracted with 150 mL of ether. The organic layer was washed with 100mL of water, then with 100mL of satd aq sodium bicarbonate solution and 100mL of brine, and was then dried (MgSO4) and concentrated under diminished pressure. The residue was applied to a silica gel column (10 × 6 cm); elution with 3:1 hexanes/ethyl acetate afforded 3,6-dimethyl-2-methoxy-p-benzoquinone (30) as a yellow solid: yield: 543 mg (65%); mp 58–59 °C; silica gel TLC Rf 0.46 (4:1 hexanes/ether); 1H NMR (400 MHz, CDCl3) δ 1.93 (s, 3H), 2.03 (s, 3H), 3.98 (s, 3H) and 6.51 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 8.8,15.7, 60.8, 128.8,133.1,143.7, 155.5, 183.6 and 188.5; mass spectrum (EI), m/z 166.0632 (M)+ (C9H10O3 requires 166.0630).
4.1.24. 2,6-Dimethyl-p-benzoquinone (32)16
To a stirred solution at 23 °C containing 2.00 g (16.36 mmol) of 2,6-dimethylphenol (31) in 20mL of diethyl ether was added dropwise a solution containing 11.0 g (36.9 mmol) of sodium dichromate-dihydrate and 7 mL of sulfuric acid in 20 mL of water (Jones reagent). The reaction mixture was stirred at 23 °C for 16 h, and was then poured into 150 mL of water. The product was extracted using three 150-mL portions of ether. The combined organic layer was washed with 150mL of water, then with 150mL of brine, and was then dried (MgSO4) and concentrated under diminished pressure to afford 2,6-dimethyl-p-benzoquinone (32) as a yellow solid; yield 2.10 g (94%); mp: 65–66 °C; silica gel TLC Rf 0.60 (5:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 2.05 (s, 6H) and 6.55 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 16.0, 17.0, 133.3, 133.3, 145.8, 145.8,187.6 and 188.2.
4.1.25. 1,2,4-Acetoxy-3,5-dimethylbenzene (33)14
To a stirred solution containing 1.10 g (8.08 mmol) of 2,6-dimethyl-p-benzoquinone (32)in 8.0 mL of acetic anhydrideat 23 °C was added 360 μL (3.23 mmol) of boron trifluoride-etherate. The reaction mixture was stirred at 40°C for 48 h. The reaction mixture was poured into 100 mL of water and the product was extracted with two 75-mL portions of ethyl acetate. The combined organic layer was washed with 60mL of water, then with 60mL of satd aq sodium bicarbonate and 60mL of brine, and was dried (MgSO4) and concentrated under diminished pressure. The residue was applied to a silica gel column (12 × 3 cm); elution with 3:1 hexanes/ethyl acetate afforded 1,2,4-acetoxy-3,5-dimethylbenzene (33) as a colorless solid: yield 1.86 g (82%); mp 94–95 °C; silica gel TLC Rf 0.25 (3:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.97 (s, 3H), 2.13 (s, 3H),2.25 (s, 3H),2.33 (s, 3H), 2.99 (s, 3H) and 6.92 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 10.48, 16.3, 20.2,20.4, 20.6, 121.9, 125.1,128.7, 139.2, 139.9, 145.8,167.8,168.3 and 168.3; mass spectrum (APCI), m/z 281.1031 (M+H)+ (C14H17O6 requires 281.1025).
4.1.26. 3,5-Dimethyl-1,2,4-trimethoxybenzene (34)14
To a stirred solution containing 1.77 g (6.31 mmol) of 1,2,4-acetoxy-3,5-dimethylbenzene (33) in 5.0 mL of methanol at 23 °C was added 5.0 mL (52.3 mmol) of dimethyl sulfate followed by the slow addition of a solution containing 5.25 g (56.3 mmol) of sodium hydroxide in 6 mL of water. The reaction mixture was stirred at 23 °C for 16 h, and was then poured into 60 mL of water. The product was extracted with 80mL of 1:1 ethyl acetate/hexanes and then with 60mL of hexanes. The combined organic layer was washed with 80mL of water, then with 80mL of brine, and was dried (MgSO4) and concentrated under diminished pressure. The residue was applied to a silica gel column (12 × 3 cm); elution with 3:1 hexanes/ethyl ether afforded 3,5-dimethyl-1,2,4-trimethoxybenzene (34) as a colorless oil: yield 292 mg (24%); silica gel TLC Rf 0.5 (9:1 hexanes/ether); 1H NMR (400 MHz, CDCl3) δ 2.21 (s, 3H),2.52 (s, 3H), 3.66 (s, 3H),3.77 (s, 3H), 3.81 (s, 3H) and 6.56 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 9.4,16.0, 55.9, 59.9, 60.3, 111.5, 125.2, 125.7,145.8, 148.9 and 150.6; mass spectrum (EI), m/z 196.1094 (M)+ (C11H16O3 requires 196.1100).
4.1.27. 3,5-Dimethyl-2-methoxy-p-benzoquinone (35)
To a stirred solution containing 500 mg (2.55 mmol) of 3,5-dimethyl-1,2,4-trimethoxybenzene (34) in 15 mL of 9:1 water/methanolat 23 °C was added 1.22 g (3.78 mmol) of PIDA. The reaction mixture was stirred at 40°C for 16 h, and was then poured into 50 mL of water. The product was extracted with 150mL of ether. The organic layer was washed with 100mL of water, then with 100mL of satd aq sodium bicarbonate, and with 100mL of brine, and was dried (MgSO4) and concentrated under diminished pressure. The residue was applied to a silica gel column (12 × 3 cm); elution with 3:1 hexanes/diethyl ether afforded 3,5-dimethyl-2-methoxy-p-benzoquinone (35) as a yellow solid: yield 282 mg (66%); mp 55–56 °C; silica gel TLC Rf 0.47 (4:1 hexanes/diethyl ether); 1H NMR (400 MHz, CDCl3) δ 1.95 (s, 3H),2.03 (s, 3H),4.01 (s, 3H) and 6.43 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 8.8, 15.7,60.8, 128.8,131.4, 145.7, 155.5, 183.6 and 188.5; mass spectrum (EI), m/z 166.0625 (M)+ (C9H10O3 requires 166.0630).
4.1.28. 1-Benzoxyundecanoic acid (36)
To a stirred solution containing 330 mg (8.25 mmol) of sodium hydride in 15 mL of anhydrous DMFat 23 °C was added dropwise 510 mg (4.71 mmol) of anhydrous benzyl alcohol followed by 1.00 g (3.77 mmol) of 11-bromoundecanoic acid. The reaction mixture was stirred at 75 °C for 3 h, and was then poured into 100 mL of ether. The formed precipitate was collected by filtration. The solid was then dissolved in 70 mL of water and the pH was adjusted to ~1. The product was extracted with 75mL of dichloromethane. The organic layer was washed with two 60-mL portions of water, then with 60mL of brine, and was then dried (MgSO4) and concentrated under diminished pressure to afford 11-benzyloxyundecanoic acid (36) as a wax: yield 660 mg (60%); silica gel TLC Rf 0.15 (4:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.28 (m, 12H),1.62 (m, 4H), 2.34 (t, 2H, J = 7.6 Hz), 3.46 (t, 2H, J = 6.8 Hz), 4.50 (s, 2H), 6.98 (m, 1H) and 7.03 (m, 4H); 13C NMR (100 MHz, CDCl3) δ24.7, 26.1, 29.0,29.2, 29.3,29.4, 29.5, 29.7, 34.0, 70.5,72.8, 127.4, 127.6, 127.6, 128.3, 128.3, 138.7 and 179.7; mass spectrum (APCI), m/z 293.2106 (M+H)+ (C18H29O3 requires 293.2117).
4.1.29. 11-Hydroxyundecanoic acid (37)
To 70 mL of 2 M aq potassium hydroxide solution was added 1.60 g (6.08 mmol) of 11-bromoundecanoic acid. The reaction mixture was stirred at 100 °C for 16 h, and was then cooled to 23 °C. The pH was adjusted to ~1 by addition of conc HCl. The formed precipitate was collected by filtration and was then dried under diminished pressure to afford 11-hydroxyundecanoic acid (37)as a colorless solid: yield 1.18 g (96%); mp 65–66 °C; 1H NMR (400 MHz, CDCl3) δ 1.28 (m, 12H), 1.62 (m, 4H),2.34 (t, 2H, J = 7.6) and 3.63 (t, 2H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ24.6, 25.6, 29.0,29.1, 29.3,29.3, 29.4, 32.7, 34.0, 63.0 and 179.3; mass spectrum (APCI), m/z 203.1651 (M+H)+ (C11H23O3 requires 203.1647).
4.1.30. 2,3-Dimethoxy-6-methyl-5-benzyloxydecyl-p-benzoquinone (38)
To a stirred solution containing 900 mg (3.07 mmol) of 11-benzyloxyundecanoic acid (36) and 500 mg (2.74 mmol) of coenzyme Q0 (9) in 50 mL of acetonitrileat 23 °C was added 464 mg (2.74 mmol) of silver nitrate. The reaction mixture was heated to 75 °C, then 900 mg (3.32 mmol) of potassium persulfate in 50 mL of water was added very slowly. The reaction mixture was stirred at 75 °C for 4 h and was then cooled to 23 °C, and poured into 25 mL of water. The product was extracted with 50mL of ethyl acetate. The organic layer was washed with 25mL of satd aq sodium bicarbonate, then with 25mL of brine, and was then dried (MgSO4), and concentrated under diminished pressure. The residue was applied to a silica gel column (12 × 4 cm); elution with 3:1 hexanes/ethyl acetate afforded 2,3-dimethoxy-6-methyl-5-benzyloxydecyl-p-benzoquinone (38) as an orange oil: yield 200 mg (17%); silica gel TLC Rf 0.75 (4:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.34 (m, 14H),1.60 (quint, 2H, J = 5.2 Hz), 2.04 (s, 3H), 2.44 (t, 2H, J = 8.4 Hz), 3.46 (t, 2H, J = 6.4 Hz), 3.98 (s, 6H),4.50 (s, 2H),7.27 (m, 1H), 7.34 (m, 4H); 13C NMR (100 MHz, CDCl3) δ11.9, 26.2, 26.4, 28.7, 29.3, 29.4, 29.4, 29.5, 29.8, 29.8, 61.1, 61.1,70.5,72.8, 127.44, 127.6, 127.6, 128.3, 128.3, 138.6, 138.7,143.1, 144.29, 144.30, 184.1 and 184.7; mass spectrum (APCI), m/z 429.2637 (M+H)+(C26H37O5 requires 429.2641).
4.1.31. Idebenone (5)18
To a stirred solution containing 200 mg (0.467 mmol) of 2,3,6-trimethyl-5-benzyloxydecyl-p-benzoquinone (38) in 5 mL of anhyd methanol at 23 °C was added 15.0 mg (0.014 mmol) of 10 % Pd/C in one portion. The reaction mixture was stirred at 23 °C under an atmosphere of hydrogen for 24 h. Air was then bubbled through the reaction mixture at 23 °C for 24 h. The suspension was filtered through Celite and the filtrate was concentrated under diminished pressure to afford idebenone (5) as an orange solid: yield 130 mg (82%); mp: 46–47 °C; 1H NMR (400 MHz, CDCl3) δ 1.34 (m, 14H), 1.60 (quint, 2H, J = 7.6 Hz), 2.04 (s, 3H), 2.44 (t, 2H, J = 8.0 Hz), 3.63 (t, 2H, J = 6.8 Hz) and 3.99 (s, 6H); 13CNMR (100 MHz, CDCl3) δ 11.9, 25.7, 26.4, 28.7, 29.3, 29.3, 29.4, 29.5, 29.8, 32.7,61.1, 61.1, 63.0, 138.6,143.1,144.3, 144.3, 184.1 and 184.7.
4.1.32. 2,3,6-Trimethyl-5-benzyloxydecyl-p-benzoquinone (39)
To a stirred solution containing 600 mg (3.11 mmol) of 11-benzyloxyundecanoic acid (36) and 450 mg (3.00 mmol) of trimethyl-p-benzoquinone (26) in 50 mL of acetonitrileat 23 °C was added 509 mg (3.00 mmol) of silver nitrate. The reaction mixture was heated to 75 °C and then 981 mg (3.63 mmol) of potassium persulfate in 50 mL of water was added very slowly. The reaction mixture was stirred at 75 °C for 3 h, was then allowed to cool to 23 °C, and was poured into 25 mL of water. The product was extracted with 50mL of ethyl acetate. The organic layer was washed with 25mL of water, then with 25mL of satd aq sodium bicarbonate, and 25mL of brine, then dried (MgSO4) and concentrated under diminished pressure. The residue was purified by chromatography on silica gel column (13 × 4 cm); elution with 3:1 hexanes/ethyl acetate afforded 2,3,6-trimethyl-5-benzyloxydecyl-p-benzoquinone (39)as an orange oil: yield 330 mg (37%); silica gel TLC Rf 0.75 (3:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.26 (m, 14H), 1.59 (quint, 2H, J = 7.2 Hz), 2.00 (m, 9H), 2.43 (t, 2H, J = 8.0 Hz),3.44 (t, 2H, J = 6.4 Hz),4.50 (s, 2H), 7.25 (m, 1H) and 7.31 (m, 4H); 13CNMR (100 MHz, CDCl3) δ12.1,12.3, 12.3, 26.2, 26.6, 28.8, 29.34, 29.4,29.4,29.5, 29.8, 29.9, 70.5,72.8, 127.4,127.6, 127.6,128.3, 128.3, 138.7, 140.0, 140.3, 140.3, 144.5,187.1 and 187.8; mass spectrum (EI), m/z 296.2676 (M)+ (C26H36O3 requires 396.2665).
4.1.33. 2,3,6-Trimethyl-5-hydroxydecyl-p-benzoquinone (6)
To a stirred solution containing 330 mg (0.832 mmol) of 2,3,6-trimethyl-5-benzyloxydecylparabenzoquinone (39)in 6 mL of anhydrous methanol at 23 °C was added 20.0 mg (0.018 mmol) of 10% Pd/C. The reaction mixture was stirred at 23 °C under a hydrogen atmosphere for 24 h, then air was bubbled through the reaction mixture at 23 °C for 24 h. The suspension was filtered through Celite and the filtrate was concentrated under diminished pressure. The residue was applied to a silica gel column (13× 4 cm); elution with 3:1 hexanes/ethyl acetate afforded 2,3,6-trimethyl-5-hydroxydecyl-p-benzoquinone (6) as a yellow solid: yield 130 mg (51%); mp 61–62 °C; silica gel TLC Rf 0.4 (3:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.26 (m, 14H),1.53 (quint, 2H, J = 6.4 Hz),2.00 (m, 9H),2.43 (t, 2H, J = 8.0 Hz) and 3.61 (t, 2H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ12.1, 12.3, 12.3, 25.7, 26.6,28.8, 29.31,29.34,29.4, 29.5, 29.8,32.7, 63.0, 140.0,140.3, 140.4, 144.5, 187.2 and 187.9; mass spectrum (APCI), m/z 307.2270 (M+H)+(C19H31O3 requires 307.2273).
4.1.34. 3,6-Dimethyl-2-methoxy-5-hydroxydecyl-p-benzoquinone (7)
To a stirred solution containing 110 mg (0.54 mmol) of 11-hydroxyundecanoic acid (37) and 85.0 mg (0.51 mmol) of 2,5-dimethyl-3-methoxy-p-benzoquinone (30) in 2.5 mL of acetonitrileat 23 °C was added 87.0 mg (0.51 mmol) of silver nitrate. The reaction mixture was heated to 75 °C, then 167 mg (0.61 mmol) of potassium persulfate in 2.5 mL of water was added very slowly. The reaction mixture was stirred at 75 °C for 4 h, and was then allowed to cool to 23 °C, and poured into 50 mL of water. The product was extracted with 75mL of dichloromethane. The organic layer was washed with 75mL of water, then with 75mL of satd aq sodium bicarbonate, and 75mL of brine, and was then dried (MgSO4) and concentrated under diminished pressure. The residue was applied to a silica gel column (10 × 3 cm); elution with 19:1 dichloromethane/ethyl ether afforded 3,6-dimethyl-2-methoxy-5-hydroxydecyl-p-benzoquinone (7) as an orange solid: yield 25 mg (15%); mp 58–59°C; silica gel TLC Rf 0.32 (3:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.28 (m, 14H),1.59 (quint, 2H, J = 7.0 Hz),1.93 (s, 3H),2.01 (s, 3H), 2.44 (t, 2H, J = 7.0 Hz), 3.62 (t, 2H, J = 6.5 Hz) and 3.95 (s, 3H); 13CNMR (100 MHz, CDCl3) δ 8.8,12.2, 26.16, 26.22,28.7, 29.4, 29.4, 29.5, 29.8, 29.8,60.9, 63.0, 72.8, 128.6, 140.3, 143.1, 155.4, 183.4 and 188.7; mass spectrum (APCI), m/z 322.2217 (M+H) (C19H31O4 requires 322.2222).
4.1.35. 3,5-Dimethyl-2-methoxy-6-hydroxydecyl-p-benzoquinone (8)
To a stirred solution at 23 °C containing 364 mg (1.79 mmol) of 11-hydroxyundecanoic acid (37)and 282 mg (1.69 mmol) of 3,5-dimethyl-2-methoxy-p-benzoquinone (35) in 10 mL of acetonitrileat 23 °C was added 288 mg (1.69 mmol) of silver nitrate. The reaction mixture was heated to 75 °C, then 552 mg (2.04 mmol) of potassium persulfate in 10 mL of water was added dropwise. The reaction mixture was stirred at 75 °C for 4 h, then allowed to cool to 23 °C, and then poured into 50 mL of water. The mixture was extracted with 75mL of dichloromethane. The organic layer was washed with 75mL of water, then with 75mL satd aq sodium bicarbonate, and 75mL of brine, and was then dried (MgSO4) and concentrated under diminished pressure. The residue was applied to a silica gel column (12 × 3 cm); elution with 19:1 dichloromethane/ether afforded 3,5-dimethyl-2-methoxy-6-hydroxydecyl-p-benzoquinone (8) as an orange wax: yield 56 mg (10%); silica gel TLC Rf 0.33 (3:1 hexanes/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 1.28 (m, 14H),1.59 (quint, 2H, J = 7.2 Hz),1.93 (s, 3H), 2.01 (s, 3H), 2.43 (t, 2H, J = 8.0 Hz), 3.62 (t, 2H, J = 6.8 Hz) and 3.95 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 8.8, 12.2,26.2, 26.2, 28.7, 29.4, 29.4,29.5, 29.8, 29.8,60.9, 63.0,72.8, 128.6, 140.3, 143.1, 155.4, 183.4 and 188.7; mass spectrum (APCI), m/z 323.2229 (M+H)+(C19H31O4 requires 323.2222).
4.2 Biological experiments
4.2.1 Measurement of oxygen consumption
C2C12 cells were grown in DMEM supplemented with 10% fetal bovine serum and 2 mM glutamine. Mitochondrial O2 consumption was performed essentially as described previously19 with minor modifications. Briefly, C2C12 cells were trypsinized and resuspended in phenol red-free growth medium, treated with 25 nM rotenone and plated at 200,000 cells/well in a 96-well Bd-D Oxygen Biosensor plate. Ten μM compounds 1–8 were added and fluorescence was monitored for 2 h in a PerSeptive Biosystems Cytofluor Series 4000 plate reader (ex 485 nm; em 620 nm). Oxygen consumption was quantified by calculating M/T, i.e. the slope at 50% maximum divided by the time to reach 50% maximum.
4.2.2 Measurement of cytoprotective effects of idebenone analogues
CEM cells were grown in RPMI medium 1640 (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Hyclone, South Logan, Utah) and 1% penicillin-streptomycin solution (Cellgro, Manassas, VA). In a 12- well cell culture plate 5 × 105 per well were seeded and treated with the idebenone analogues for 18 hours. Cells were then treated with 5 mM DEM (diethyl maleate) at 37° C for 4 h in a humidified atmosphere of 5% CO2 in air. The viability of the cells was measured by staining with 0.4% trypan blue. Viable cells exclude the dye and non- viable cells take up the dye. Cell viability is calculated as percentage of positive control (cells not treated with DEM or the antioxidants). Data is expressed as mean ± SD (n= 3).
Acknowledgments
This work was supported at Arizona State University by a grant from the Friedreich’s Ataxia Research Alliance. R.A.S. was supported by NIH RO1 AG16719.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.DiMauro S, Schon EA. N Engl J Med. 2003;348:2656. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong JS. Br J Pharmacol. 2007;151:1154. doi: 10.1038/sj.bjp.0707288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith RAJ, Porteus CM, Gane AM, Murphy MP. Proc Natl Acad Sci USA. 2003;100:5407. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith RAJ, Kelso GF, James AM, Murphy MP. Methods Enzymol. 2004;382:45. doi: 10.1016/S0076-6879(04)82003-2. [DOI] [PubMed] [Google Scholar]
- 5.Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RAJ, Murphy MP. J Biol Chem. 2001;276:4588. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 6.Asin-Cayuela J, Manas AR, James AM, Smith RA, Murphy MP. FEBS Lett. 2004;571:9. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 7.Ruttimann A, Lorenz P. Helv Chim Acta. 1990;73:790. [Google Scholar]
- 8.Jacob P, Callery PS, Shulgin AT, Castagnoli N. J Org Chem. 1976;41:3627. doi: 10.1021/jo00884a035. [DOI] [PubMed] [Google Scholar]
- 9.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 10.Luly JR, Rapoport H. J Org Chem. 1984;49:1671. [Google Scholar]
- 11.Fieser LF, Oxford AE. J Am Chem Soc. 1942;64:2060. [Google Scholar]
- 12.Jacobsen N, Torssell K. Liebigs Ann Chem. 1972;763:135. [Google Scholar]
- 13.Minisci F, Citterio A, Vismara E, Fontana F, De Bernardinis S, Correale M. J Org Chem. 1989;54:728. [Google Scholar]
- 14.Iyer S, Liebeskind LS. J Am Chem Soc. 1987;109:2759. [Google Scholar]
- 15.Tohma H, Morioka H, Harayama Y, Hashizume M, Kita Y. Tetrahedron Lett. 2001;42:6899. [Google Scholar]
- 16.Liotta D, Arbiser J, Short JW, Saindane M. J Org Chem. 1983;48:2932. [Google Scholar]
- 17.Yu CA, Yu L. Biochemistry. 1982;21:4096. doi: 10.1021/bi00260a028. [DOI] [PubMed] [Google Scholar]
- 18.Jung Y-S, Joe B-Y, Seong C-M, Park N-S. Synth Commun. 2001;31:2735. [Google Scholar]
- 19.Rolo AP, Palmeira CM, Cortopassi GA. Anal Biochem. 2009;385:176. doi: 10.1016/j.ab.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]