

Abstract
Clinical outcome remains poor in high-risk neuroblastoma patients, where chemoresistant relapse is common following high-intensity conventional multimodal therapy. Novel treatment approaches are required. Although recent genomic profiling initiatives have not revealed a high frequency of mutations in any significant number of therapeutically targeted genes two exceptions, amplification of the MYCN oncogene and somatically acquired tyrosine-kinase domain point mutations in ALK, present exciting possibilities for targeted therapy. In contrast to the situation with ALK, where a robust pipeline of pharmacologic agents is available from early clinical use in adult malignancy, therapeutic targeting of MYCN (and MYC oncoproteins in general) represents a significant medicinal chemistry challenge that has remained unsolved for two decades. We review the latest approaches envisioned for blockade of ALK activity in neuroblastoma, present a classification of potential approaches for therapeutic targeting of MYCN, and discuss how recent developments in targeting of MYC proteins appear to make therapeutic inhibition of MYCN a reality in the clinic.
Background
The need for novel therapies
Neuroblastoma is the most frequently occurring solid extra-cranial tumor of childhood, with 1200 new cases per year diagnosed in the USA and Europe. Of these, nearly half are classified as INRG (International Neuroblastoma Risk Group) high-risk, and these patients contribute approximately 15% of all childhood cancer-related mortality (1). In the last decade, the addition to 13-cis-retinoic acid (isotretinoin) of disialoganglioside (GD2) targeted ch14.18 chimeric antibody in combination with the cytokines GM-CSF and IL2 during continuation therapy has improved two-year progression-free survival (from time of randomization) by 20% (2). Nevertheless, outcome remains poor in this patient cohort despite intensified multimodal treatment. A considerable proportion of patients experience disease relapse and are refractory to conventional treatment approaches. With a long-term survival of approximately 8% (3), these children need improved treatment options.
Gene mutations in neuroblastoma
Genome sequencing studies of neuroblastoma tumor tissue from diagnostic biopsies have revealed a low mutation rate in a small number of individual genes. In a recent study of 240 tumors the median frequency of mutation was 0.60 mutations per Mb, which is markedly lower than that found in adult solid tumors (4). However, recurrent changes in five genes were identified with statistical and biological significance for neuroblastoma: PTPN11, ATRX, NRA, MYCN and ALK. Among hereditary neuroblastomas, which account for only 2% of all cases and are mostly associated with ALK germline mutations, 6% exhibited germline mutation of PHOX2B (5), and somatic mutations were detected even more rarely (6). Other known genetic alterations include loss-of-function mutations and deletions in TIAM1, ARID1A and ARID1B (7, 8). We will discuss altered expression of MYCN and ALK, as potential targets for novel therapy approaches, and assess whether existing and planned treatment strategies could improve treatment outcomes for neuroblastoma.
Aberrant expression of MYCN in neuroblastoma
Amplification of the MYCN gene defines approximately 20% of all neuroblastomas and 45% of high-risk cases (9). MYCN amplification is strongly associated with poor outcome and until recently was the only genetic factor integrated into risk stratification and treatment planning (10). MYC family members are bHLH-leucine zipper transcription factors that bind to DNA at conserved elements within the promoters of an extensive network of genes, and appear to act as direct amplifiers of transcriptionally active genes, neither directly inducing de novo gene transcription nor silencing of expressed genes (11). Therefore, the degree to which the function of MYCN can be targeted through the selective inhibition of specific genes is unclear. Further complicating matters, the non-linear relationship between MYCN gene copy number, mRNA expression, oncoprotein levels and clinical outcome (12) has called into question whether MYCN gene-copy number should be replaced as a clinical classifier with a measurement more indicative of MYCN function. Several MYCN mRNA expression signatures have been developed(13), including a 157 gene-set defining a class of high-risk tumors both amplified and diploid for MYCN (14). Patients included in this study also displayed prominent dysregulation of the phosphoinositide-3-kinase/Akt (protein kinase B, PKB)/mammalian target of rapamycin (PI3K/Akt/mTOR), a pathway known to drive oncogenic stabilization of MYCN protein (15). Thus a significant majority of high-risk patients are defined by altered expression or stabilization of MYCN and could potentially be targeted using clinically available PI3K/mTOR inhibitors already in early phase trials (16). Finally, it is worth noting that expression of MYCN is confined to maturing neural crest (17), making this oncoprotein one of few bona fide, tumor-specific targets in pediatric cancer, and a major priority for direct therapeutic targeting.
ALK mutations in neuroblastoma
Approximately 2% of neuroblastoma patients have familial predisposition, and in the majority of cases, germline mutations occur within the tyrosine kinase (TK) domain of the ALK (Anaplastic Lymphoma Kinase) gene, implying a putative role for this orphan receptor kinase in the genesis of neuroblastoma (18-21). A restricted set of TK domain mutations are present in the germline, but a wider array, with varying ability to activate ALK kinase activity is present in 8–14% of sporadic neuroblastomas. Targeted therapeutics with excellent selectivity and potency against ALK are in current clinical trials and are in development. Preliminary response data indicates that ALK is a therapeutic target of great interest (discussed below).
On the Horizon
Recent developments in biologic understanding of MYCN and ALK have made therapeutic inhibition of both targets a practical matter in the clinic. Here we discuss a mechanistically based classification system (Table 1) ordering five classes of existing direct and indirect inhibitors of MYCN and clinical strategies to target ALK using either small-molecule or immunotherapeutic approaches, all of which are in late development or existing clinical trials.
Table 1.
Targeted therapies against MYCN and ALK currently in development
| Drugs targeting MYCN | Drug | Development phase | Pediatric trial identifier | |
|---|---|---|---|---|
| Class I – Drugs targeting DNA binding functions of MYCN | ||||
| MYCN/MAX heterodimerization | 10058-F4 | Pre-clinical | ||
| Mycro3 | Pre-clinical | |||
|
| ||||
| Class II – Drugs targeting transcription of MYCN | ||||
| BET bromodomain | OTX015 | Phase I | – Adult | |
| JQ1 | Pre-clinical | |||
| I-BET762 | Pre-clinical | |||
| Pfi1 | Pre-clinical | |||
| CPI203 | Pre-clinical | |||
|
| ||||
| Class III – Drugs targeting synthetic lethal interactions of MYCN | ||||
| CHK1, CDK2, and CHK2 | SCH900776 | Phase I | – Adult | |
| CHK1 | LY2606368 | Phase I | – Adult | |
| GDC-0425 | Phase I | – Adult | ||
| GDC-0575 | Phase I | – Adult | ||
| CDK2 | SCH727965 | Phase I/II | – Adult | |
|
| ||||
| Class IV – Drugs targeting oncogenic stabilization of MYCN | ||||
| mTOR – rapalogs | Rapamycin | Phase I/II | NCT01331135, NCT01625351, NCT01804634 | |
| Temsirolimus | Phase I/II | NCT01204450, NCT00808899 | ||
| Everolimus | Phase I | NCT00106353, NCT01049841, NCT01637194 | ||
| Ridaforolimus | Phase I | NCT00704054, NCT01431547, NCT01431534 | ||
| mTOR – ATP competitive | AZD2014 | Phase I | – Adult | |
| OSI027 | Phase I | – Adult | ||
| MLN0128 | Phase I | – Adult | ||
| PI3K/mTOR dual specificity | BKM120 | Phase I/II/III | – Adult | |
| GDC-0980 | Phase I/II | – Adult | ||
| NVP-BEZ235 | Phase I/II | – Adult | ||
| Akt | Perifosine | Phase I | NCT00776867, NCT01049841 | |
| MK2206 | Phase I | NCT01231919 | ||
| Aurora A | AT9283 | Phase I | NCT01767194 | |
| MLN8237 | Phase I/II | NCT01601535 | ||
|
| ||||
| Class V – Drugs targeting the expression or function of MYCN | ||||
| MYCN expression | Isotretinoin | Current therapy | ||
| Drugs targeting ALK | Drug | Development phase | Pediatric trial identifier | |
|
| ||||
| Inhibition of ALK kinase activity | ||||
| ALK/c-Met dual specificity | Crizotinib | Phase I/II | NCT01644773, NCT00939770 | |
| ALK/EGFR dual specificity | AP26113 | Phase I/II | – Adult | |
| ALK | LDK378 | Phase I | NCT01742286 | |
| ALK | CH5424802 | Phase I/II | – Adult | |
|
| ||||
| Immunotherapy of ALK | ||||
| ALK | PF-03446962 | Phase I/II | – Adult | |
Class I - Targeting DNA binding functions of MYCN
Attempts to develop small molecules that directly target MYC family members have focused on blocking the interaction of MYC with MAX, an approach that has been technically challenging(22). Studies with a dominant-negative MYC mutant, Omomyc, have highlighted the clinical potential of this approach. Omomyc, which binds to all MYC family members and prevents dimerization with MAX, exerts a dramatic therapeutic impact in MYC-addicted cancers (23, 24). Recently, a compound (10058-F4) that inhibits MYC:MAX interactions in vitro demonstrated a modest survival benefit in vivo (25) in a genetically modified, MYCN-dependent mouse model (TH-MYCN) of neuroblastoma (26).
Class II – Targeting transcription of MYCN
Much recent interest has been generated in the MYC targeting field following the recognition that bromodomain and extra terminal (BET) family adaptor proteins (BRD2, BRD3, BRD4) localize to MYC promoters. BET proteins contain acetyl-lysine recognition motifs, or bromodomains, that bind acetylated lysine residues in histone tails usually associated with an open chromatin state and transcriptional activation (27). BRD4 was identified as a key therapeutic target in AML, via a focused RNAi screen targeting 243 genes of chromatin regulators (28), and small molecule inhibitors (such as the prototypic BET inhibitor JQ1) that bind the bromodomain and disrupt BET recruitment to chromatin, downregulating expression of MYC (28). More recently, a screen of 673 genetically characterized cancer cell lines for sensitivity to the JQ1 identified MYCN amplification in neuroblastoma cells as a major predictor of response (29). This study found that treatment with JQ1 downregulated the MYC/MYCN transcriptional program, as well as suppressing transcription of MYCN itself. This was accompanied by displacement of BRD4 from the MYCN promoter and was phenocopied by RNAi knockdown of BRD4. JQ1 treatment conferred a significant survival advantage in subcutaneous neuroblastoma cell line xenografts, primary human neuroblastoma orthotopic xenografts, and in TH-MYCN transgenic mice (29). Currently, OTX015 (OncoEthix), an orally bioavailable BRD2/3/4-selective inhibitor is the only BET inhibitor undergoing early phase clinical testing (Table 1). In preclinical studies, OTX015 caused transient downregulation of MYC mRNA in anaplastic large cell lymphoma (ALCL) (30).
Class III - Targeting synthetic lethal interactions of MYCN
Expression of MYC proteins unleashes a powerful oncogenic stimulus that necessitates remodeling of critical cellular control pathways and exposes synthetic-lethal gene interactions that can be therapeutically targeted. Genes that are synthetic lethal for MYCN expression have been identified through shRNA library screens in cancer cells and include AURKA, CDK1, CDK2 and CHK1 (31-33). In some cases the mechanisms underlying these synthetic lethal interactions are understood. CHK1 is an essential kinase involved in DNA repair, which is significantly modulated by expression of MYC or MYCN through induction of replicative stress and in response to this both DNA repair and cell cycle checkpoint pathways are activated (34). CHK1 mRNA expression is significantly elevated in patients with high-risk disease and MYCN amplified neuroblastomas (31). CCT244747, a highly selective, orally active CHK1 inhibitor has recently been shown to have therapeutic activity in TH-MYCN mice (35). Several additional CHK1 inhibitors are in early phase trials in adults but none are being clinically evaluated in children. Sensitivity to CDK inhibition may relate to the role of CDK proteins in maintenance of MYC protein stability (discussed below) (36, 37). Several CDK inhibitors with excellent selectivity and potency are under development and may prove to be effective inhibitors of MYCN (38, 39).
Class IV - Targeting oncogenic stabilization of MYCN
Degradation of MYCN is required for terminal differentiation of neuronal precursors, so that control of MYCN protein is tightly controlled in a cell-cycle-specific manner. MYC proteins bind to a proteasomal degradation complex that includes Aurora A kinase (AURKA), E3 ubiquitin ligases (FBXW7 and HUWE1), and undefined additional proteins (33). The interaction of “F-box” oncoproteins with FBXW7 is specified by phosphorylation at threonine 58 (T58) and serine 62 (S62) residues within a conserved phosphodegron domain (CPD) (36, 40, 41). The phosphorylation status of T58 is critical to the oncogenic activity of MYC proteins and is regulated by GSK3β, a direct target of the PI3K/mTOR pathway (36). Aberrant PI3K/mTOR activity in neuroblastoma correlates with poor outcome (42), drives oncogenic stabilization of MYCN (15) and can be targeted using clinical PI3K/mTOR inhibitors for which recommended phase 2 doses have been established in children (43, 44). Early phase trials of compounds active against either PI3K or mTOR are underway using late-generation rapalog inhibitors such as temsirolimus and ridaforolimus, which have improved bioavailability and inhibition of mTORC1 and mTORC2 (Table 1). Several ATP-competitive inhibitors of mTOR, including MLN0128 (INK-128) (45) and AZD2014 (46) are in early clinical development. MK2206, an Akt inhibitor effective against neuroblastoma alone or in combination with etoposide or rapamycin(47) is also undergoing phase I testing in patients under 16 years of age (Table 1).
Another strategy to target oncogenic stabilization of MYCN is to promote dissociation of the Aurora A Kinase:MYCN complex, which results in rapid proteasomal degradation of MYCN (33). Certain Aurora A inhibitors such as MLN8237 induce a particular conformational change in the kinase that actively re-initiates MYCN degradation through this mechanism, independent of any requirement for enzymatic inhibition of the kinase itself (48). MLN8237 was identified as a promising agent for neuroblastoma (49), but has not displayed robust antitumor activity in early-phase pediatric studies (50). This recent data on the mechanism of Aurora A kinase inhibition indicates that MLN8327 is not a structurally optimal inhibitor of MYCN:Aurora A interactions, making the development of improved inhibitors a priority.
Class V – Targeting the expression or function of MYCN
Novel targets that either regulate the expression of MYC proteins or modify MYC function have long been known and are currently being identified. The recognition that retinoids modulate the ability of MYCN to regulate neuronal differentiation led to the use of 13-cis-retinoic acid (CRA) in neuroblastoma, and is one of few therapeutic interventions in recent years that has extended long-term survival in high-risk patients (51). CRA, which is used in the continuation phase of neuroblastoma treatment (2), down-regulates MYCN expression, induces cell cycle arrest and stimulates neuronal differentiation (52). Finally, a novel mechanism regulating MYCN expression involves modulation of the let-7 family of microRNAs, which negatively regulate MYCN expression (53). Let-7 expression is suppressed by LIN28B, which is amplified and overexpressed in high-risk neuroblastoma. Overexpression of LIN28B has been elegantly modeled in genetically-engineered mice as a primary driver of neuroblastoma tumorigenesis (53), leads to generation of neuroblastoma with elevated levels of MYCN expression and will likely provide data for novel strategies to target this mechanism.
Inhibition of ALK kinase activity
The recognition that germline and somatic mutations occur in the TK domain of ALK and are targetable using the existing clinical therapeutic crizotinib (PF-02341066, an ATP-competitive dual-specific inhibitor of ALK/c-Met) (54), has generated significant interest. ALK mutations drive constitutive phosphorylation of ALK and are critical for growth of neuroblasts (54). In common with MYCN, normal ALK expression is confined to developing neural tissues. Thus ALK represents a bona fide target in neuroblastoma and its inhibition is not predicted to result in undesirable systemic side effects. Crizotinib has been clinically evaluated in adult ALCL (55), non-small cell lung cancer (56) and ALK-rearranged inflammatory myofibroblastic tumor (IMFT)(57). In these early phase trials, excellent clinical responses have been achieved in ALK-rearranged patients in which oncogenesis is driven by the resultant fusion proteins displaying constitutive ALK TK activity. Analogous to BCR/ABL-positive chronic myelogenous leukemia (CML) in which clonal evolution of gatekeeper mutations drives resistance to targeted TK inhibition, crizotinib treatment is complicated by the development of ALK TK domain point mutations, which reduce the effectiveness of crizotinib through an increased affinity for ATP(54). Results suggest that crizotinib inhibits proliferation of neuroblastoma cells harboring R1275Q-mutated ALK or amplified wild-type ALK, but not those possessing the F1174L mutation (54). Crizotinib was evaluated in a phase I/II trial and exhibited activity against IMT, ALK-translocated ALCL and neuroblastoma (58). In 11 of 34 neuroblastoma cases, with known ALK status, one complete response and two stable disease responses were observed. The limited number of patients with defined ALK status makes interpretation of crizotinib activity in this setting premature (58). A combination trial of crizotinib with chemotherapy is planned (NCT01606878). Several structurally distinct, second-generation ALK inhibitors with enhanced potency and specificity are currently in early phase clinical trials. These include LDK378 (59), CH5424802 (RO5424802) (60), and AP26113 (a dual-specificity ALK/EGFR inhibitor) (61) (Table 1). Questions surrounding the significance of ALK as a target in neuroblastoma, as well as therapeutic resistance to crizotinib, will likely be addressed in the near future using these available therapeutic tools.
Immunotherapy of ALK
Immunodominant peptide epitopes of ALK with restriction for common MHC types have been described for both class I and class II MHC and high circulating levels of ALK specific T cells recognizing these peptide/MHC combinations have been reported specifically in patients with ALCL, although this has not yet been identified in neuroblastoma patients (62). T-cells with specificity for the ALK peptide/MHC specificities are effective at lysing ALK-positive cancer cells. This evidence for natural immunity against ALK in cancer is supportive for the development of peptide vaccine based immunotherapy approaches for neuroblastoma. However the low class I MHC expression in neuroblastoma will assist in immune evasion.
An alternative approach to target ALK makes use of its cell surface localization, which allows for targeting by antibody-based strategies. Monoclonal antibodies (mAb) directed against the ALK ectodomain have been generated (63), and these have been shown to induce antibody-dependent cell-mediated cytotoxicity (ADCC) against ALK-positive neuroblastoma cells (64). Interestingly, ADCC was increased when anti-ALK mAb was used in combination with crizotinib, which upregulated expression of ALK in this experimental system. This is supportive of the concept of combinatorial small molecule inhibition of ALK TK activity and anti-ALK immunotherapy. Other potential antibody-based strategies are to develop neutralizing antibodies specific for mutated ALK, or to use non-lytic antibodies to deliver payloads such as radionuclides or immunotoxins. The bright cell surface expression of ALK makes it an attractive target for immunotherapy using chimeric antigen receptor-transduced T cells, which combine MHC-unrestricted antibody specificity with potent T cells activation. Using this approach against a similar cancer cell specific target antigen in leukemia (CD19) has resulted in dramatic clinical responses in chemotherapy-resistant patients and the development of long-lived memory responses (65).
Conclusions
A number of the drugs described in this review are already in early phase clinical testing in adult and pediatric settings (see Table 1) and others are at an advanced stage of pre-clinical evaluation. A rational approach and alternatives to mechanistic targeting of MYC proteins in general, and MYCN in particular, is emerging using clinically available therapeutics (Figure 1). Strategies based on both enzymatic and immunotherapeutic targeting of ALK are advancing rapidly. Our own work and that of others implies that MYCN and ALK are functionally synergistic and that MYCN gene amplification (and/or oncogenic protein stabilization) and ALK mutation may in fact co-associate in a proportion of neuroblastoma patients with high-risk disease (66-70). It is anticipated that many therapeutic options will become available: for example, the oncogenic activity of ALK appears to proceed primarily via aberrant PI3K/mTOR pathway activity. Therefore, therapeutic strategies targeting both oncoproteins can easily be envisioned (such as combinations of ALK and mTOR inhibitors) and may have enhanced efficacy. In summary, the likelihood is that therapeutic suppression of the activities of MYCN and mutated ALK, arising from the two most common and potentially significant genetic alterations in neuroblastoma, will become a clinical reality.
Fig 1.
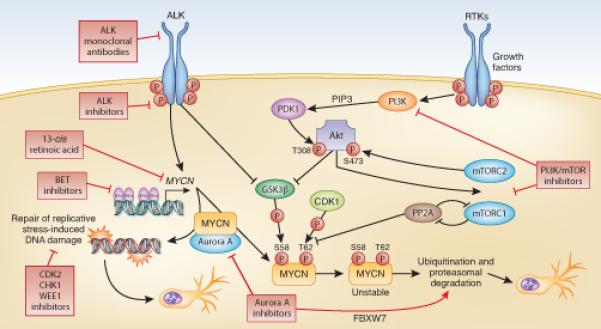
Schematic representation of therapeutic strategies targeting MYCN and ALK in neuroblastoma.
Acknowledgments
Grant support The Neuroblastoma Society (A1100139), Cancer Research UK (C34648/A140) (L.C.), Cancer Research UK (C347/A15403); Oak Foundation (OCAY-12-287) (A.D.J.P). NIHR Biomedical Research Centre (G.B., A.D.J.P., K.P. and L.C.). Wellcome Trust clinical research fellowship (G.B.).
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed by the authors.
References
- 1.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Pediatr Clin North Am. 2008;55:97–120. doi: 10.1016/j.pcl.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 2.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–34. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.London WB, Castel V, Monclair T, Ambros PF, Pearson AD, Cohn SL, et al. Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J Clin Oncol. 2011;29:3286–92. doi: 10.1200/JCO.2010.34.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nature genetics. 2013;45:279–84. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosse YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, et al. Germline PHOX2B mutation in hereditary neuroblastoma. American journal of human genetics. 2004;75:727–30. doi: 10.1086/424530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serra A, Haberle B, Konig IR, Kappler R, Suttorp M, Schackert HK, et al. Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. Journal of pediatric hematology/oncology. 2008;30:728–32. doi: 10.1097/MPH.0b013e3181772141. [DOI] [PubMed] [Google Scholar]
- 7.Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483:589–93. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 8.Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nature genetics. 2013;45:12–7. doi: 10.1038/ng.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–4. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 10.Goto S, Umehara S, Gerbing RB, Stram DO, Brodeur GM, Seeger RC, et al. Histopathology (International Neuroblastoma Pathology Classification) and MYCN status in patients with peripheral neuroblastic tumors: a report from the Children’s Cancer Group. Cancer. 2001;92:2699–708. doi: 10.1002/1097-0142(20011115)92:10<2699::aid-cncr1624>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 11.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohn SL, London WB, Huang D, Katzenstein HM, Salwen HR, Reinhart T, et al. MYCN expression is not prognostic of adverse outcome in advanced-stage neuroblastoma with nonamplified MYCN. J Clin Oncol. 2000;18:3604–13. doi: 10.1200/JCO.2000.18.21.3604. [DOI] [PubMed] [Google Scholar]
- 13.Stutterheim J, Tytgat GM, Schoot CE. Pediatric Neuroblastoma: Molecular Detection of Minimal Residual Disease. In: Hayat MA, editor. Neuroblastoma. Springer; Netherlands: 2012. pp. 47–63. [Google Scholar]
- 14.Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans ME, et al. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19190–5. doi: 10.1073/pnas.1208215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, et al. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006;66:8139–46. doi: 10.1158/0008-5472.CAN-05-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shuttleworth SJ, Silva FA, Cecil AR, Tomassi CD, Hill TJ, Raynaud FI, et al. Progress in the preclinical discovery and clinical development of class I and dual class I/IV phosphoinositide 3-kinase (PI3K) inhibitors. Curr Med Chem. 2011;18:2686–714. doi: 10.2174/092986711796011229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by N-myc: promotion of ventral migration and neuronal differentiation. Development. 1997;124:1953–62. doi: 10.1242/dev.124.10.1953. [DOI] [PubMed] [Google Scholar]
- 18.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–5. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–4. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 20.George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–8. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–70. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 22.Prochownik EV, Vogt PK. Therapeutic Targeting of Myc. Genes & cancer. 2010;1:650–9. doi: 10.1177/1947601910377494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–83. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes & development. 2013;27:504–13. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zirath H, Frenzel A, Oliynyk G, Segerstrom L, Westermark UK, Larsson K, et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2013 doi: 10.1073/pnas.1222404110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–95. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–7. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- 28.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer discovery. 2013;3:308–23. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boi M, Bonetti P, Ponzoni M, Tibiletti MG, Stathis A, Cvitkovic E, et al. The Brd-Inhibitor OTX015 Shows Pre-Clinical Activity in Anaplastic Large T-Cell Lymphoma (ALCL) ASH Annual Meeting Abstracts. 2012;120:4872. [Google Scholar]
- 31.Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3336–41. doi: 10.1073/pnas.1012351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molenaar JJ, Ebus ME, Geerts D, Koster J, Lamers F, Valentijn LJ, et al. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12968–73. doi: 10.1073/pnas.0901418106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–51. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- 35.Walton MI, Eve PD, Hayes A, Valenti MR, De Haven Brandon AK, Box G, et al. CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:5650–61. doi: 10.1158/1078-0432.CCR-12-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev Cell. 2005;9:327–38. doi: 10.1016/j.devcel.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 37.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nature cell biology. 2004;6:308–18. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, Tsai YH, Tseng SH. Inhibition of cyclin-dependent kinase 1-induced cell death in neuroblastoma cells through the microRNA-34a-MYCN-survivin pathway. Surgery. 2013;153:4–16. doi: 10.1016/j.surg.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 39.Gogolin S, Ehemann V, Becker G, Brueckner LM, Dreidax D, Bannert S, et al. CDK4 inhibition restores G(1)-S arrest in MYCN-amplified neuroblastoma cells in the context of doxorubicin-induced DNA damage. Cell cycle. 2013;12:1091–104. doi: 10.4161/cc.24091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X, Heng JI, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, et al. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nature cell biology. 2008;10:643–53. doi: 10.1038/ncb1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007;67:735–45. doi: 10.1158/0008-5472.CAN-06-2201. [DOI] [PubMed] [Google Scholar]
- 43.Gore L, Trippett TM, Katzenstein H, Boklan J, Narendran A, Smith A, et al. A Multicenter, First-in-Pediatrics, Phase 1, Pharmacokinetic and Pharmacodynamic Study of Ridaforolimus in Patients with Refractory Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013 doi: 10.1158/1078-0432.CCR-12-3166. [DOI] [PubMed] [Google Scholar]
- 44.Schenone S, Brullo C, Musumeci F, Radi M, Botta M. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011;18:2995–3014. doi: 10.2174/092986711796391651. [DOI] [PubMed] [Google Scholar]
- 45.Jessen K, Wang S, Kessler L, Guo X, Kucharski J, Staunton, et al. INK128 is a potent and selective TORC1/2 inhibitor with broad oral antitumor activity. Mol Cancer Ther; Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; Boston, MA. Nov 15–19, 2009; AACR; 2009. Abstract B148. [Google Scholar]
- 46.Guichard SM, Howard Z, Heathcote D, Roth M, Hughes G, Curwen J, et al. AZD2014, a dual mTORC1 and mTORC2 inhibitor is differentiated from allosteric inhibitors of mTORC1 in ER+ breast cancer. Cancer Res; Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; Chicago, IL Philadelphia (PA). 2012 Mar 31-Apr 4; AACR; 2012. Abstract nr 917. [Google Scholar]
- 47.Li Z, Yan S, Attayan N, Ramalingam S, Thiele CJ. Combination of an allosteric Akt Inhibitor MK-2206 with etoposide or rapamycin enhances the antitumor growth effect in neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:3603–15. doi: 10.1158/1078-0432.CCR-11-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer cell. 2013;24:75–89. doi: 10.1016/j.ccr.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatric blood & cancer. 2010;55:26–34. doi: 10.1002/pbc.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mosse YP, Lipsitz E, Fox E, Teachey DT, Maris JM, Weigel B, et al. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children’s Oncology Group Phase I Consortium study. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:6058–64. doi: 10.1158/1078-0432.CCR-11-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J Clin Oncol. 2009;27:1007–13. doi: 10.1200/JCO.2007.13.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thiele CJ, Reynolds CP, Israel MA. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature. 1985;313:404–6. doi: 10.1038/313404a0. [DOI] [PubMed] [Google Scholar]
- 53.Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nature genetics. 2012;44:1199–206. doi: 10.1038/ng.2436. [DOI] [PubMed] [Google Scholar]
- 54.Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Science translational medicine. 2011;3:108ra14. doi: 10.1126/scitranslmed.3002950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med. 2011;364:775–6. doi: 10.1056/NEJMc1013224. [DOI] [PubMed] [Google Scholar]
- 56.Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011–9. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Butrynski JE, D’Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–33. doi: 10.1056/NEJMoa1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472–80. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mehra R, Camidge DR, Sharma S, Felip E, Tan DS, Vansteenkiste JF, et al. First-in-human phase 1 study of the ALK inhibitor LDK378 in ALK+ solid tumors. Journal of Clinical Oncology (Meeting Abstracts) 2012;30 Abstract nr 3007. [Google Scholar]
- 60.Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013 doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- 61.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ait-Tahar K, Barnardo MC, Pulford K. CD4 T-helper responses to the anaplastic lymphoma kinase (ALK) protein in patients with ALK-positive anaplastic large-cell lymphoma. Cancer Res. 2007;67:1898–901. doi: 10.1158/0008-5472.CAN-06-4427. [DOI] [PubMed] [Google Scholar]
- 63.Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Brunet-de Carvalho N, Bureau J, et al. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. The Journal of biological chemistry. 2005;280:26039–48. doi: 10.1074/jbc.M501972200. [DOI] [PubMed] [Google Scholar]
- 64.Carpenter EL, Haglund EA, Mace EM, Deng D, Martinez D, Wood AC, et al. Antibody targeting of anaplastic lymphoma kinase induces cytotoxicity of human neuroblastoma. Oncogene. 2012;31:4859–67. doi: 10.1038/onc.2011.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) Mutation Potentiates the Oncogenic Activity of MYCN in Neuroblastoma. Cancer cell. 2012;22:117–30. doi: 10.1016/j.ccr.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:4353–62. doi: 10.1158/1078-0432.CCR-09-2660. [DOI] [PubMed] [Google Scholar]
- 68.Heukamp LC, Thor T, Schramm A, De Preter K, Kumps C, De Wilde B, et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Science translational medicine. 2012;4:141ra91. doi: 10.1126/scitranslmed.3003967. [DOI] [PubMed] [Google Scholar]
- 69.Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, Kutok JL, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer cell. 2012;21:362–73. doi: 10.1016/j.ccr.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schonherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. 2012;31:5193–200. doi: 10.1038/onc.2012.12. [DOI] [PubMed] [Google Scholar]