

Abstract
The reaction of low energy electrons (LEEs; 10 eV) with 5′-TpXpT-3′ (TXT), where X is uracil (U), thymine (T) and 5-bromouracil (5BrU), was examined by HPLC-UV analysis. The presence of 5BrU increased total damage by > 50%. The radiation products of T5BrUT included TUT (40%), free U, T, 5BrU (23%) and fragments (13%). These products may be explained by initial capture of LEEs by the nucleobase to form a transient anion, followed by transfer of the electron within the molecule, and cleavage of susceptible bonds by dissociative electron attachment (C-Br, C-N, or C-O bonds). In addition, these products may arise from the uracilyl-5-yl (U-5-yl) radicals that undergo H-atom abstraction from the sugar moiety. Interestingly, several products contained two sites of cleavage (U, pUT and TUp). The formation of these products was linear with dose, and thus, they arise from the single electron reactions. To explain these products, we propose that the reaction of LEEs (10 eV) involves the coupling of two dissociative processes in the same molecule (for example, dissociative excitation and dissociative electron attachment). The latter reactions may contribute to the formation of clustered damage, which is the most deleterious damage induced by ionizing radiation.
Keywords: 5-Bromouracil, radiosensitizer, clustered damage, secondary electrons
Introduction
The absorption of 1 MeV of ionizing radiation leads to the generation of ~ 3 × 104 of secondary low energy electrons (LEEs).1 Although LEEs are largely responsible for the ionization of water and DNA components upon exposure to ionizing radiation, they may also react directly with DNA components below the ionization threshold of DNA, and thereby, lead to DNA damage via a transient molecular ion.2 The reaction of LEE with DNA has been investigated using simple DNA components (bases, nucleosides, nucleotides), oligonucleotides, and plasmids).3–8 The majority of these studies have been performed under ultra high vacuum in which volatile and small fragments (<100 a.m.u.) of electron induced desorption from the films were detected. These studies have been extended to the analysis of products remaining in the film of irradiated targets. Once initial radicals leading to products are formed by LEE, they will either remain within the solid film, until the film is dissolved in solution, or they will form products directly within the film. The results of the latter studies indicate that LEEs induce DNA damage by base release and strand breaks, which likely involve C-N cleavage of the N-glycosidic bond and C-O cleavage of the phosphodiester bond, respectively.9–12 In addition, another reaction was recently reported involving the formation of 5,6-dihydrothymine within TTT as a major base modification induced by irradiation with LEEs.13 Numerous studies with plasmids under high vacuum demonstrate the formation of single and double strand breaks using gel electrophoresis to separate DNA fragments following exposure to LEEs.14–17 Together, the above studies support a general mechanism of LEE-induced DNA damage in which LEE forms a transient molecular ion that dissociates into an anion and a radical through a resonance process, referred to as dissociative electron attachment (DEA). This mechanism is supported by theoretical studies involving quantum theory simulation, in particular, density functional theory (DFT).14 Theoretical studies, however, are restricted to the interaction of small DNA components, for example, dinucleotide monophosphate model systems are among the largest structures that can be studied using reliable theoretical methods.15 On the basis of theoretical studies, an electron transfer model has been proposed in which LEEs are initially captured by the nucleobase and then transferred to either the N-glycosidic bond or phosphodiester-sugar bond leading to cleavage.16–19 Despite much experimental and theoretical efforts, the intermediates, i.e., radicals and excited states, and other chemical steps leading to the formation of LEE-induced DNA damage remain poorly understood.
The study of 5-bromouracil (5BrU) is important because of its potential as a radiosensitizer in medicine.20 The van der Waals radius of a bromine atom is very similar to that of a methyl group, and thus, 5BrU replaces thymine (T) in DNA without any significantly change the DNA structure or base coding properties. Indeed, the 2′-deoxyribonucleoside derivative of 5BrU is phosphorylated by cells and incorporated into cellular DNA, and the presence of 5BrU in cellular DNA sensitizes cells to radiation-induced DNA damage and cell death.24–28 Although it is generally thought that solvated electrons (i.e., thermalized) are involved in the mechanism of radiosensitization by 5BrU,21,22 it is reasonable to propose that LEEs are also important.
Experimental Methods
Sample Irradiation
Experimental details of the LEE irradiator and the procedure to irradiate thin films of oligonucleotides have been reported elsewhere.17 Briefly, approximately 80 μg of HPLC-UV purified compound was dissolved in 5 mL of nanopure grade H2O (Milli-Q water system, 18 MΩ·cm, 25°C) and the solution was deposited by spin-coating onto the inner surface of seven chemically clean tantalum cylinders (3.2 cm × 2.5 cm diameter). The average thickness of the film on the cylinder was 2.5 ± 0.1 nm (4 to 5 monolayers (ML)), assuming that the molecules are uniformly distributed on the inner surface of cylinders and that the average density of DNA is 1.7 g cm−3.23 All manipulations of samples, before and immediately after irradiation, were carried out in a sealed glove box under an atmosphere of dry nitrogen. After spin-coating, the samples were transferred to the LEE irradiation chamber, which was subsequently evacuated for ~24 h to reach a pressure of about 10−9 Torr at ambient temperature. The irradiator generated a uniform electron beam over the entire sample surface of the cylinder with an energy resolution of 0.5 eV full width at half-maximum (FWHM). Each cylinder containing sample was irradiated individually with constant irradiation time, beam current, and incident electron energy. Under present conditions, the time of irradiation was 2.5 min giving a total exposure of approximately 1016 electrons per cylinder. The current and irradiation time were adjusted to give an exposure well within the linear regime of the dose response curve.9 The average thickness of the film (2.5 nm) was considerably smaller than the penetration depth (5–20nm) of 10 eV electrons in either liquid water or amorphous ice.24 Because the penetration depth is in fact smaller than the inelastic mean free path for electronic excitation of biological solids (9–28 nm), electrons impinging on the film will be transmitted to the metal substrate and will, at most, lead to single inelastic scattering events with target molecules.25
HPLC-UV analysis
After irradiation with LEEs, samples were removed from ultra high vacuum and placed into a dry nitrogen-purged glove box. The compounds and their radiation products were recovered from the surface of the tantalum cylinders by the addition of 12 mL of nanopure grade H2O (Milli-Q water system, 18 MΩ·cm, 25°C). The nonirradiated samples (three cylinders) and the irradiated samples (four cylinders) were pooled, frozen and lyophilized to dryness. Nonirradiated and irradiated samples were dissolved in 150 μL and 200 μL of nanopure grade H2O, respectively, in order to have an equal amount of product per volume in each sample. Half of the sample was analyzed by HPLC while the other half was first treated with alkaline phosphatase (1 unit, Roche Applied Science) for 1 h at 37°C to remove the terminal phosphate group of nucleotides, and then analyzed by HPLC-UV under the same conditions as the non-treated sample. In the chromatogram, the identity of DNA fragments containing a terminal phosphate group was supported by their conversion to derivatives without a terminal phosphate group upon treating with alkaline phosphatase (see Figure 1). The HPLC-UV system consisted of a Waters alliance HT system equipped with a refrigerated autosampler, a 2795 separations module and a 2487 dual wavelength absorbance detector. The separation of products was achieved using an analytical YMC-Pack ODS-A column (250 × 6 mm), maintained at 30°C, using a linear gradient from 1% to 10% acetonitrile in buffer containing 25 mM NaH2PO4 (pH 5.7) over an interval of 60 min and at a flow rate of 1.0 mL/min. All products were detected at 210 and 260 nm. The total yield of damage in terms of the number of damaged molecules per 1000 target molecules was estimated by the amount of nonmodified trimer in irradiated and nonirradiated samples. The yield of LEE-induced products was determined by calibration with authentic reference compounds.
Figure 1.
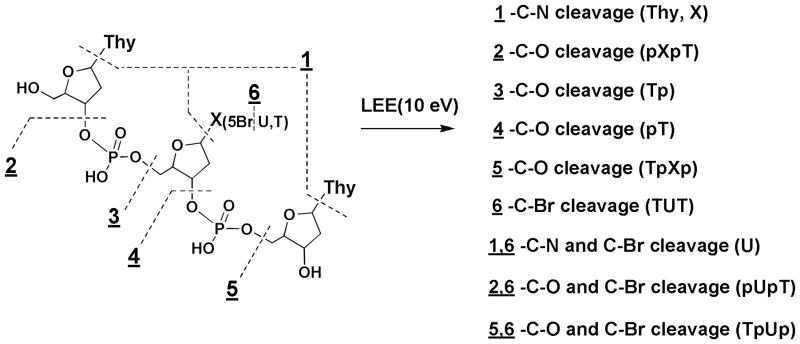
Structure of TTT and positions of cleavage for the N-glycosidic bond (1), phosphodiester bond (2–5), and C-Br bond cleavage (6).
Reference Compounds
The identity of LEE-induced products was confirmed by co-elution with reference compounds using HPLC-UV. TTT and TUT were purchased from Alpha DNA (Montreal, QC) and T5BrUT was purchased from UCDNA Services (Calgary, AB). 5-Bromouracil (5BrU), uracil (U), thymine (T), thymidine (dThd), and mononucleotides (pT and Tp) were purchased from Sigma-Aldrich (St. Louis, MO). In addition, several standard dinucleotide fragments of TpXpT (X represents T, U, 5BrU respectively) containing either a 3′ or 5′ terminal phosphate group were prepared by enzymatic digestion of the corresponding trimers with micrococcal nuclease (Roche Applied Science, giving TXp) and with P1 nuclease (MP Biomedical, giving pXT).21 All samples were purified by HPLC-UV using the same methods as described above for irradiated samples except that a volatile buffer solution, i.e., triethylamine acetate (20 mM, pH 7) was used. The purified solution was lyophilized to dryness and redissolved in nanopure grade H2O before spin-coating and irradiation. The yield of LEE-induced DNA fragments was determined by comparison of the peak area at 260 nm with the peak area of standard solutions prepared from commercially available compounds (nucleobases, 2′-deoxynucleosides, and mononucleotides (pT, Tp)). In addition, other fragments containing a terminal phosphate (pXpT and TpXp) were obtained by the partial digestion of trinucleotides with nucleases together with HPLC purification of the fragments. The concentration of standard solutions of trinucleotides and their fragments was estimated by their optical absorption at 260 nm taking the given molar absorptivity of DNA bases (http://www.basic.northwestern.edu/biotools/oligocalc.html). The optical density was measured using a spectrophotometer (Hitachi U-2000).
Results and Discussion
In the present study, we explore the chemistry of LEEs with a trinucleotides containing 5BrU (T5BrUT) in comparison to other trinucleotides (TTT and TUT; Figure 1). The targets were irradiated with monoenergetic low energy electrons (LEEs) of 10 eV in condensed films under high vacuum. Following irradiation, the profile of LEE-induced damage to trinucleotides remaining in the film was examined by HPLC-UV (Figure 2). The chromatogram of nonirradiated samples, which was subjected to analysis in parallel with irradiated samples, indicated a minor amount of decomposition of the initial trinucleotide as a result of spin-coating, pumping to UHV, and other steps of the experiments. The total damage was calculated from the difference of initial target molecules between non-irradiated and irradiated samples (Table 1). This quantity represents the sum of all possible sources of parent molecule loss, which includes not only non-volatile products remaining in the film (as measured by HPLC-UV) but also products that result from either LEE-induced desorption of fragments from the surface or LEE-induced attachment of products to the metal surface (not detected by HPLC-UV)). Thereby, the total damage for TTT and TUT was 82 and 92 modifications per 1000 initial target molecules, respectively, and the damage considerable increased to 138 modifications when the central T was replaced with 5BrU (an increase of ~50%). Furthermore, the main product of the reaction of LEE with T5BrUT was dehalogenated trimer (TUT; 39% of the total damage).
Figure 2.

Analysis of Tp5BrUpT radiation products by HPLC/UV detected at 260 nm. The trimer was exposed to 1016 electrons with an energy of 10 eV. The lower chromatogram (2C; green) depicts the analysis of a nonirradiated sample. The upper and middle chromatograms show the corresponding irradiated sample, which was divided in two parts: one was treated with alkaline phosphatase (2B; red) and the other received no treatment (2C; black). Blue arrows illustrate the conversion of products with a terminal phosphate to those without.
Table 1.
Yield of Products from LEE Induced DNA Damage.
| Samplea | Damageb | Bromine release | Base release | Phosphodiester bond cleavage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||||
| TXT | TUT | %c | T | X(U) | Sum | %d | pT | Tp | pXT(pUT) | TXp(TUp) | Sum | %e | ratiof | |
| X=5BrU | 138 | 53.5 | 38.8 | 25.8 | 1.8(4.4) | 32.0 | 23.2 | 4.3 | 2.5 | 3.0(4.5) | 2.1(1.3) | 17.7 | 12.8 | 1.81 |
| X=T | 92 | 17.0 | 17.0 | 18.5 | 1.6 | 1.2 | 4.3 | 2.7 | 9.8 | 10.7 | 1.73 | |||
| X=U | 82 | 14.8 | 2.5 | 17.3 | 21.1 | 1.1 | 0.8 | 2.4 | 2.1 | 6.4 | 7.8 | 2.70 | ||
DNA samples and fragments written from 5′ to 3′ with p indicating the position of terminal phosphate groups.
Total damage includes all losses of initial targeted molecules based on HPLC-UV analysis. The values are expressed as a ratio of damaged molecules to 1000 initial target molecules and are the average of three independent experiments; SD= ± 20%.
Percentage of C-Br cleavage based on total damage.
Percentage of C-N cleavage based on total damage.
Percentage of C-O cleavage based on total damage.
Ratio of C-N to C-O cleavage.
An increase in the total damage with trinucleotides containing 5BrU might result from the high electron affinity of this compound compared to T and U, which could translate into a higher electron capture cross section by TBrUT, but also from the low activation energy of C-Br bond cleavage following electron attachment.32–34 From studies in the gas phase, the reaction of LEE and 5BrU induces mainly the desorption of Br ions and U-5-yl radicals (i.e., 5BrU + e− (~0 eV) → 5BrU− → (U-yl)• + Br−).26–28 This process shows a resonance at very low electron energies (< 2.0 eV) indicating that it occurs by DEA.35, 36 Similar results are obtained for 5BrU in the condensed phase.29,30 The loss of Br ions from T5BrUT may be explained by the initial formation of a transient molecular ion followed DEA in which the negative charge goes to Br. Analysis of the spin density on Br along the anion radical PES (potential energy surface) of 5BrU as a function of C-Br distance indicates that the negative charge shifts to Br as the C-Br distance increases and at C-Br = 5 Å, the charge on Br is −0.74 and the spin is 0.25. The tendencies of charge/spin distribution suggest that bond cleavage of the BrU anion is more likely to result in a Br ion and a U-5-yl radical, rather than a Br radical and U-5-yl anion.31 This reaction is also enhanced owing to the strong repulsive character of the dissociative radical anion state of 5BrU.31–33 The idea that DEA is favored in the reaction of LEE with TBrUT is supported by the high yield of TUT (53.5) compared to the yield of base release (25.8) and phosphodiester bond cleavage (17.7) in units of molecules per 1000 target molecules. According to theoretical studies, C-Br cleavage is the lowest energy process (about 1.88 kcal/mol)31 whereas the energy for C-O bond cleavage and C-N bond cleavage are higher by at least 3-fold and 10-fold, respectively.34,35 Interestingly, the yield of damage by these three pathways is inversely proportional to the distance between the nucleobase moiety and the bond undergoing cleavage, i.e., the farther away from the nucleobase, the lower the yield of damage. Similarly, the lack of damage at sites next to abasic sites in short oligonucleotides may be attributed to initial capture of LEEs by the nucleobase and subsequent transfer to other positions of the molecule.12
The chemistry of U-5-yl radicals has been examined in detail in aqueous solution.36–46 UV photolysis of 5BrU leads to C-Br cleavage and the formation of U-5yl radicals. The major reaction of U-5-yl radicals in single and double stranded DNA, as well as in various other forms, involves H-atom abstraction from C1′ or C2′ of the neighboring 5′-phosphate 2-deoxyribose moiety.36,37 Typically, H-atom abstraction does not immediately give a strand break but gives an abasic site that subsequently transforms into a break (e.g., 2′-deoxyribonolactone; half-life > 20 h at 37ºC)38–40. Nonmodified bases are released as a result of sugar damage induced by H-abstraction.41,42 The products from the reaction of LEE with T5BrUT in the condensed phase were similar to those from photolytic studies in the aqueous phase in that TUT as the major product (this work). However, the yield of products arising from H-atom abstraction at the neighboring sugar moiety appeared to be much less in the case of LEE reactions in the condensed phase. For example, the yield of T was only 50% of TUT and the yield of trinucleotides containing abasic sites, which likely elute between TT and TTT (see Figure 1), was only 25% of TUT. The lack of products arising from H-atom abstraction points to alternative pathways in the formation of TUT in experiments with LEE (Pathways a–c; Figure 4). The formation of TUT may arise from H–atom abstraction by an intramolecular or intermolecular reaction (pathway a). Alternatively, TUT may be formed by electron transfer and reduction of U-5-yl radicals involving either thermalized electrons or radical species that are trapped in the film during irradiation (Pathway b). These reactions would be thermodynamically favorable because of the high oxidation potential of U-5-yl radicals. In addition to pathways a and b, the formation of TUT may be explained by the reaction of hydride ions at C5 of 5BrU followed by elimination of Br− to reform the 5,6-double bond of U (Pathway c). It should be noted that LEE generates relatively large amounts of hydride ions in the condensed phase as demonstrated by electron-induced desorption studies of nucleic acid components.13
Figure 4.
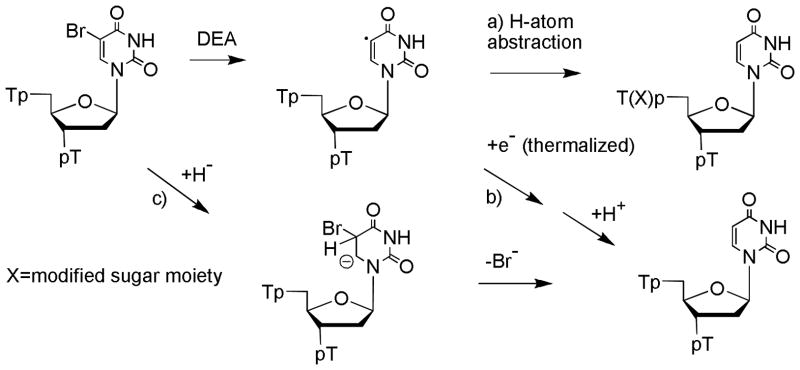
Proposed mechanism of formation of TUT by the reaction LEE with T5BrUT.
The reaction of LEEs with T5BrUTT led to the release of nonmodified nucleobases (5BrU and U; 23%) as well as fragments containing a terminal phosphate (pT, Tp, pTT, TTp, p5BrUT, T5BrUp; 13%) (Table 1). The release of free nucleobase was greatly favored at the terminal compared to the central position of trinucleotides. For example, the release of T from T5BrUT was 26 molecules per 1000 target molecules whereas the combined release of 5BrU and U was 6.2. These results are very similar to those obtained previously with thymine derivatives and oligonucleotide trimers and tetramers.9–11 The release of non-modified bases can take place by DEA involving initial attachment of LEE to the base moiety followed by N-glycosidic bond cleavage. However, we cannot rule out H-atom abstraction reactions of U-5-yl radicals as another source of nonmodified T. In addition to base release, the reaction of LEEs with T5BrUT led to fragments containing a terminal phosphate. These products were identified by comparison of samples treated with and without alkaline phosphatase (Figure 1B). Blue arrows illustrate the conversion of products containing a terminal phosphate to those without a phosphate, i.e., those with a terminal hydroxyl group. For example, thymidine-5′-monophosphate (pT; peak 2) and thymidine-3′-monophosphate (Tp; peak 3) transform into thymidine (dThd; peak 1) and the dinucleotides, pUT (peak 6) and TUp (peak 7) transform into UT and TU (peaks 2 and 3), respectively. Following treatment with enzyme, p5BrUT (peak 8) and T5BrUp (peak 9) convert to 5BrUT (peak 13) and T5BrU (peak 14) (Figure 2). Thus, LEE induced phosphodiester-sugar cleavage gives fragments containing a terminal phosphate rather than a terminal hydroxyl group. These results are very similar to those obtained previously for short oligonucleotides.9–11 The formation of fragments containing 5BrU can take place by the initial capture of LEE through interaction with the base followed by transfer of the electron to the sugar-phosphate group and cleavage of the C-O bond.9,11,19 Likewise, the formation of fragments containing U may be explained by combination of LEE-induced events from a core-excited state (see below). Again, we cannot rule out H-atom abstraction reactions involving U-5-yl radicals as a source of mono- and dinucleotide fragments.
The formation of free U as well as fragments containing U, i.e., pUT and TUp, was observed from LEE irradiation of T5BrUT (Figure 2B). It is not likely that U arises from U-5-yl radicals because it implies that U-5-yl radicals efficiently abstract H-atoms from its own sugar moiety. To the contrary, several studies indicate that H-atom abstraction by U-5-yl radicals to abstract H-atoms involving its own sugar moiety is minor compared to that from neighboring nucleotides as indicated by the analysis of fragments as well as base release of U in oligonucleotides containing 5BrU.38,41–43 The extent of cleavage at 5BrU is also minor compared to that at neighboring sites within a single stranded mismatch region of DNA containing 5BrU and exposed to ionizing radiation.44 In contrast, the yield of U was 2-fold higher than that of T upon reaction of LEE with T5BrUT whereas one would expect more favourable release of T if H-atom abstraction was the main source of base release. Thus, we have considered other pathways for the formation of U in the case of LEE-induced damage. Furthermore, it should also be noted that U does not arise from secondary reactions, i.e., the reaction of one LEE with T5BrUT to give TUT followed by the reaction of another LEE with TUT. Even if one assumes that most of the damage occurs in the first monolayer, the amount of TUT molecules in the first monolayer (53.5) would be less than ¼ of initial T5BrUT molecules (200–53.5= 146.5). Thus, the yield of products arising from secondary reactions may be estimated to be less than 2.5/4 (0.6) molecules for base release (U) and 6.4/4 (1.6) molecules for phosphodiester cleavage (TUp and pUT). However, the yield of these products was much higher than predicted by secondary reactions: the yield of U was 4.4 molecules (7.3-fold higher than predicted) and the yield of pUT and TUp was 5.8 molecules (3.6-fold higher than predicted). To provide evidence that fragments containing U arise from the attack of a single electron, we examined the formation of these fragments as a function of the time of irradiation (Figure 3). Within the first 5 min of irradiation, the formation of fragments was linear as a function of dose. In this regime, the yield of products may be considered to result from a single electron hit because damage resulting from multiple successive collisions will necessarily be nonlinear with radiation exposure. Therefore, we conclude that fragments containing U do not likely arise from H-atom abstraction of U-5-yl radicals nor do they arise from secondary reactions involving two LEEs.
Figure 3.

Time-course of double lesion formation by LEE impact. T5BrUT was exposed to 10 eV electrons during 0, 0.5, 1, 1.5, 3, 5 minutes at a constant electron beam flux of 10.6 μA. The yield of double lesions was determined by HPLC-UV. The data was fitted to a single exponential (dashed line) and to a straight line at initial times (solid line, R=0.999). Each point is the average of 3 independent measurements.
The release of U from T5BrUT upon irradiation with LEE suggests that single electrons are able to produce double events involving the dissociation of two bonds, e.g., C-Br and C-N bonds (Figure 5). The release of U may be explained by the decay of a localized core-excited resonance of T5BrUT into a dissociative electronically excited state of the C-N bond leaving a nearly thermalized electron in the molecule.11 Simultaneously, the remaining electron may transfer to the lowest potential (5BrU), where it forms a transient anion, which dissociates into a Br ion and a U-5-yl radical by DEA. The formation of fragments containing U (pUT and TUp) together with p5BrUT and T5BrUp may also be explained by a pathway involving double events in one T5BrUT molecule by a 10 eV electron, i.e., C-Br and C-O cleavage. In this case, the core-excited state may be localized on the phosphodiester bond, which leads to cleavage of the C-O bond, while the nearly thermalized electron is transferred to the 5BrU moiety. Several other scenarios may be proposed by different combinations of excited state bond dissociation together with DEA of lower energy involving C-N, C-O, and C-Br bonds. Interestingly, the release of pUT was 50% higher than that of p5BrUT. This suggests that double events are more efficient than single events leading to phosphodiester cleavage. Although double events were obvious in our analysis because of the change from 5BrU to U, it is reasonable to propose similar double events may occur in normal non-substituted DNA. For instance, the energy required to break the C-Br bond by LEE-induced DEA is close to that required to break the C-O. However, double events in previous studies with short oligonucleotides were not observed due to fact that a large percentage of the products with unknown structures have not been identified in the mixture of products.
Figure 5.

Proposed mechanism of formation fro double lesions from the reaction of LEE with T5BrUT. An initial core-excited state ([T5BrUT]*−) with energy (Eo) decays to an excited state ([T5BrUT]*) and a very low energy electron energy (e−t) with lower energy (E<Eo). The excited molecule [T5BrUT]* undergoes dissociative excitation (DE), which can involve C-N glycosidic or C-O phosphodiester bond cleavage, to give intermediate base fragments (e.g., 5BrU) or dinucleotide fragments (p5BrUT or T5BrUp), respectively. Very low energy electrons (e−t) interact with fragments (above) to give a transient anion that undergoes DEA, leading to C-Br bond cleavage, and giving rise to products not containing Br.
Conclusions
The reaction of LEE with a trinucleotide containing 5BrU (T5BrUT) increases total damage by 50% compared to TTT, and channels most of the additional damage into the formation of the dehalogenated derivative (TUT). The irradiation of T5BrUT also leads to the release of free nucleobases (T, 5BrU, U) together with mononucleotide and dinucleotide fragments containing either 5BrU or U. The majority of products may be explained by the initial formation of U-5-yl radicals, which arises from the reaction of LEE with DNA bases by a resonant process: DEA. The formation of U and fragments containing U, however, suggests that energetic LEEs (10 eV) induce the simultaneous cleavage of two bonds in the same molecule providing a novel mechanism to clustered damage.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada (J.R.W.), the Canadian Institutes of Health Research and the Marie Curie International Program of the European Commission.
References
- 1.Pimblott SM, LaVerne JA. Radiat Phys Chem. 2007;76:1244–1247. [Google Scholar]
- 2.Sanche L. Eur Phys J D. 2005;35:367–390. [Google Scholar]
- 3.Abdoul-Carime H, Cloutier P, Sanche L. Radiat Res. 2001;155:625–633. doi: 10.1667/0033-7587(2001)155[0625:leeesd]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 4.Dugal PC, Huels MA, Sanche L. Radiat Res. 1999;151:325–333. [PubMed] [Google Scholar]
- 5.Pan X, Cloutier P, Hunting D, Sanche L. Phys Rev Lett. 2003;9020:8102–8102. doi: 10.1103/PhysRevLett.90.208102. [DOI] [PubMed] [Google Scholar]
- 6.Ptasinska S, Sanche L. J Chem Phys. 2006;125:44713–44713. doi: 10.1063/1.2338320. [DOI] [PubMed] [Google Scholar]
- 7.Abdoul-Carime H, Gohlke S, Illenberger E. Phys Rev Lett. 2004;9216:8103–8103. doi: 10.1103/PhysRevLett.92.168103. [DOI] [PubMed] [Google Scholar]
- 8.Denifl S, Ptasinska S, Cingel M, Matejcik S, Scheier P, Mark TD. Chem Phys Lett. 2003;377:74–80. [Google Scholar]
- 9.Zheng Y, Cloutier P, Hunting DJ, Sanche L, Wagner JR. J Am Chem Soc. 2005;127:16592–16598. doi: 10.1021/ja054129q. [DOI] [PubMed] [Google Scholar]
- 10.Li ZJ, Zheng Y, Cloutier P, Sanche L, Wagner JR. J Am Chem Soc. 2008;130:5612–5613. doi: 10.1021/ja077601b. [DOI] [PubMed] [Google Scholar]
- 11.Li ZJ, Cloutier P, Sanche L, Wagner JR. J Am Chem Soc. 2010;132:5422–5427. doi: 10.1021/ja9099505. [DOI] [PubMed] [Google Scholar]
- 12.Zheng Y, Wagner JR, Sanche L. Phys Rev Lett. 2006;9620:8101–8101. doi: 10.1103/PhysRevLett.96.208101. [DOI] [PubMed] [Google Scholar]
- 13.Park Y, Li ZJ, Cloutier P, Sanche L, Wagner JR. Radiat Res. 2011;175:240–246. doi: 10.1667/rr2381.1. [DOI] [PubMed] [Google Scholar]
- 14.Li XF, Sevilla MD. Adv Quantum Chem. 2007;52:59–87. [Google Scholar]
- 15.Gu JD, Xie YM, Schaefer HF. Chem Phys Lett. 2009;473:213–219. [Google Scholar]
- 16.Gu JD, Xie YM, Schaefer HF. J Am Chem Soc. 2005;127:1053–1057. doi: 10.1021/ja0400990. [DOI] [PubMed] [Google Scholar]
- 17.Bao XG, Wang J, Gu JD, Leszczynski J. Proc Natl Acad Sci USA. 2006;103:5658–5663. doi: 10.1073/pnas.0510406103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu JD, Wang J, Leszczynski J. J Am Chem Soc. 2006;128:9322–9323. doi: 10.1021/ja063309c. [DOI] [PubMed] [Google Scholar]
- 19.Simons J. Acc Chem Res. 2006;39:772–779. doi: 10.1021/ar0680769. [DOI] [PubMed] [Google Scholar]
- 20.Dewey WC, Humphrey RM. Radiat Res. 1965:538–553. [PubMed] [Google Scholar]
- 21.Ling L, Ward JF. 1990;121:76–83. [PubMed] [Google Scholar]
- 22.Limoli CL, Ward JF. Radiat Res. 1993;134:160–169. [PubMed] [Google Scholar]
- 23.Fasman GD. Handbook of Biochemistry and Molecular biology. CRC Press; Boca Raton, FL: 1995. [Google Scholar]
- 24.Meesungnoen J, Jay-Gerin JP, Filali-Mouhim A, Mankhetkorn S. Radiat Res. 2002;158:657–660. doi: 10.1667/0033-7587(2002)158[0657:leepri]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 25.Leclerc G, Goulet T, Cloutier P, Jay-Gerin JP, Sanche L. J Phys Chem. 1987;91:4999–5001. [Google Scholar]
- 26.Abdoul-Carime H, Huels MA, Bruning F, Illenberger E, Sanche L. J Chem Phys. 2000;113:2517–2521. [Google Scholar]
- 27.Abdoul-Carime H, Huels MA, Illenberger E, Sanche L. J Am Chem Soc. 2001;123:5354–5355. doi: 10.1021/ja003952d. [DOI] [PubMed] [Google Scholar]
- 28.Abdoul-Carime H, Limao-Vieira P, Gohlke S, Petrushko I, Mason NJ, Illenberger E. Chem Phys Lett. 2004;393:442–447. [Google Scholar]
- 29.Klyachko DV, Huels MA, Sanche L. Radiat Res. 1999;151:177–187. [PubMed] [Google Scholar]
- 30.du Penhoat MAH, Huels MA, Cloutier P, Jay-Gerin JP, Sanche L. J Phys Chem B. 2004;108:17251–17260. [Google Scholar]
- 31.Li XF, Sanche L, Sevilla MD. J Phys Chem A. 2002;106:11248–11253. doi: 10.1021/jp021669q. [DOI] [PubMed] [Google Scholar]
- 32.Wetmore SD, Boyd RJ, Eriksson LA. Chem Phys Lett. 2001;343:151–158. [Google Scholar]
- 33.Li XF, Sevilla MD, Sanche L. J Am Chem Soc. 2003;125:8916–8920. doi: 10.1021/ja034286u. [DOI] [PubMed] [Google Scholar]
- 34.Kumar A, Sevilla MD. J Phys Chem B. 2007;111:5464–5474. doi: 10.1021/jp070800x. [DOI] [PubMed] [Google Scholar]
- 35.Schyman P, Zhang RB, Eriksson LA, Laaksonen A. PCCP. 2007;9:5975–5979. doi: 10.1039/b711083a. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y, Sugiyama H. Angew Chem Int Ed Engl. 2006;45:1354–1362. doi: 10.1002/anie.200501962. [DOI] [PubMed] [Google Scholar]
- 37.Cook GP, Chen TQ, Koppisch AT, Greenberg MM. Chem Biol. 1999;6:451–459. doi: 10.1016/s1074-5521(99)80063-5. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe T, Bando T, Xu Y, Tashiro R, Sugiyama H. J Am Chem Soc. 2005;127:44–45. doi: 10.1021/ja0454743. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe T, Tashiro R, Sugiyama H. J Am Chem Soc. 2007;129:8163–8168. doi: 10.1021/ja0692736. [DOI] [PubMed] [Google Scholar]
- 40.Zheng Y, Sheppard TL. Chem Res Toxicol. 2004;17:197–207. doi: 10.1021/tx034197v. [DOI] [PubMed] [Google Scholar]
- 41.Sugiyama H, Tsutsumi Y, Saito I. J Am Chem Soc. 1990;112:6720–6721. [Google Scholar]
- 42.Sugiyama H, Tsutsumi T, Fujimoto K, Saito I. J Am Chem Soc. 1993;115:4443–4448. [Google Scholar]
- 43.Rokita SE, Ito T. J Am Chem Soc. 2003;125:11480–11481. doi: 10.1021/ja035952u. [DOI] [PubMed] [Google Scholar]
- 44.Dextraze ME, Wagner JR, Hunting DJ. Biochemistry. 2007;46:9089–9097. doi: 10.1021/bi062114e. [DOI] [PubMed] [Google Scholar]