

Abstract
Isoaspartate (isoAsp) formation is a major source of protein damage that is kept in check by the repair function of protein L-isoaspartyl methyltransferase (PIMT). Mice deficient in PIMT accumulate isoAsp-containing proteins, resulting in cognitive deficits, abnormal neuronal physiology and cytoarchitecture, and fatal epileptic seizures 30–60 days after birth. Synapsins I and II, dynamin-1, collapsin response mediator protein 2 (CRMP2), and α/β-tubulin are major targets of PIMT in brain. To investigate links between isoAsp accumulation and the neurological phenotype of the KO mice, we used Western blotting to compare patterns of in vivo phosphorylation or acetylation of the major PIMT targets listed above. Phosphorylations of synapsins I and II at Ser-9 were increased in female KO vs. WT mice, and acetylation of tubulin at Lys-40 was decreased in male KO vs. WT mice. Average levels of dynamin-1 phosphorylation at Ser-778 and Ser-795 were higher in male KO vs. WT mice, but the statistical significance (P>0.1) was low. No changes in phosphorylation were found in synapsins I and II at Ser-603, in CRMP2 at Ser-522 or Thr-514, in DARPP-32 at Thr-34, or in PDK1 at Ser-241. General levels of phosphorylation assessed with Pro-Q Diamond stain, or an anti-phosphotyrosine antibody, appeared similar in the WT and KO mice. We conclude that isoAsp accumulation is associated with altered functional status of several neuronal proteins that are highly susceptible to this type of damage. We also uncovered unexpected differences in how male and female mice respond to isoAsp accumulation in the brain.
Introduction
Isoaspartate (isoAsp) formation, through deamidation of asparaginyl residues or isomerization of aspartyl residues, constitutes a major source of spontaneous protein damage occurring in vivo and in vitro [1-7]. Generation of isoAsp sites is initiated by nucleophilic attack on the Asx side-chain carbonyl by the C-flanking amide bond nitrogen resulting in an intermediate succinimide, hydrolysis of which generates a mixture of α-linked L-aspartyl (~15-30%) and β-linked L-isoaspartyl (~70-85%) residues. Synthetic peptide studies have shown that altering the N+1 residue has a major influence on the propensity for isoAsp formation, with glycine, serine, and histidine most associated with “hot spots” of isoAsp formation. In structured proteins the same sequence effect is often found, but isoAsp formation is conformation dependent and usually restricted to flexible regions of the polypeptide.
Protein L-isoaspartate O-methyltransferase (PIMT; EC 2.1.1.77), selectively methylates the α-carboxyl group of L-isoaspartyl residues [8-10]. The isoAsp methyl esters produced are highly labile (T1/2 ≈ 7 min at pH 7.4, 37 °C for -isoAsp-Gly-) and spontaneously demethylate to reform a succinimide that can restore the normal α-linked Asp-Xaa bond [11,12]. Continuing cycles of PIMT action have been shown to efficiently repair L-isoAsp sites in a number of peptides and proteins [13-17]. A repair function for PIMT in vivo is supported by observations that reduction of PIMT activity in cultured cells or knockout (KO) mice dramatically increase the level of isoaspartyl proteins [18-21]. A critical need for PIMT action in the brain is indicated by its high specific activity in this tissue [22,23] as well as the overt neurological phenotype of PIMT KO mice: increased brain size with abnormal neuro-anatomical and electrophysiological properties, impaired learning, and fatal epileptic seizures beginning at 4 weeks of age [19,20,24-26].
Recent studies utilizing the PIMT KO mouse are shedding light on mechanisms by which isoAsp accumulation interferes with neuronal function. One mechanism may be disruption of gene expression. The flexible N-terminal region of histone H2B accumulates high levels of isoAsp in vivo, which could potentially alter chromatin function [27,28]. Bidinosti et al. [29,30] have shown that a brain-enriched regulator of mRNA translation, 4E-BP2, is highly susceptible to deamidation and isoAsp formation in vivo. Isoaspartyl 4E-BP2 exhibits decreased affinity for the mTORC1 complex, resulting in decreased protein synthesis that dramatically affects neuronal properties. Stimulated by the larger brain size of PIMT-KO mice, Farrar et al. [31] investigated the expression and phosphorylation state of proteins involved in insulin-dependent growth signaling. Using rapidly excised brain tissue from KO mice, they reported increased expression of the insulin receptor β-subunit and hyperphosphorylation of several protein kinases in the PI3K/Akt signaling pathway. Kosugi et al. [21] used a PIMT knockdown strategy to raise isoAsp levels in human embryonic kidney cells. In response to stimulation by epidermal growth factor, these cells exhibited hyperphosphorylation of Raf-1, ERK1/2, and MEK, all components of the mitogen-activated protein kinase cascade that regulates gene expression.
In 2006, our lab used a proteomic analysis of PIMT-KO mouse brain extracts to identify 22 proteins that are highly prone to isoAsp formation in vivo [32]. Prominent among this group were proteins involved in regulating synaptic transmission (synapsins I and II, and dynamin-1) and cytoskeletal structure (collapsin response mediator protein 2 (CRMP2/DPYSL2/ULIP2), and α/β-tubulin). All four of these proteins are regulated by reversible phosphorylation and one of them (tubulin) is also regulated by acetylation of a lysine residue. We wondered if isoAsp formation in these proteins alters their function in a manner that might contribute directly to the neurological deficits of the PIMT deficient mice. Toward this goal, we report here the use of modification-specific antibodies to monitor the effect of PIMT deficiency on site-specific phosphorylation of synapsins I and II, dynamin-1, and CRMP2, as well as acetylation of α-tubulin. In brain extracts of the PIMT KO mouse we found increased phosphorylation of synapsins I and II, decreased acetylation of α-tubulin, and a possible increase in the phosphorylation of dynamin-1. We propose that these changes in post-translational modification reflect functional deficits in the resident proteins, and that these deficits contribute to the abnormal synaptic physiology and cytoskeletal structures that are associated with the neurological phenotype of the PIMT KO mouse.
Materials and Methods
Reagents
Euthasol® and protease inhibitor cocktail were purchased from Virbac Corporation and Sigma-Aldrich, respectively. Sources and working dilutions of the primary antibodies used in Western blots are indicated in Table 1. HRP-conjugated donkey anti-rabbit IgG secondary antibody was purchased from GE Healthcare. HRP-conjugated rabbit anti-sheep secondary antibody and Pierce ECL Plus Substrate were purchased from Thermo Scientific. HRP-conjugated goat anti-mouse secondary antibody was from Santa Cruz Biotechnology. Pro-Q Diamond phosphoprotein stain and SYPRO Ruby protein stain were purchased from Invitrogen/Life Technologies. NaF was purchased from Fisher Scientific and Na3VO4 was purchased from Sigma-Aldrich.
Table 1. Antibodies used for Western blotting.
| Antigen | Host | P/Ma | Dilution | Supplier | Catalog no. |
|---|---|---|---|---|---|
| pPDK1 (Ser-241) | rabbit | P | 1: 1,000 | Cell Signaling | 3061 |
| Synapsin I | rabbit | P | 1:10,000 | Millipore | AB1543P |
| pSynapsin I (Ser-9) | rabbit | P | 1: 1,000 | Cell Signaling | 2311S |
| pSynapsin I (Ser-603) | rabbit | P | 1: 8,000 | Sigma-Aldrich | S8192 |
| pDARPP-32 (Thr-34) | rabbit | M | 1: 5,000 | Cell Signaling | 5393S |
| CRMP2 | rabbit | P | 1: 1,000 | Millipore | AB9218 |
| pCRMP2( Thr-514) | rabbit | P | 1: 1,000 | Cell Signaling | 9397 |
| pCRMP2 (Ser-522) | rabbit | P | 1: 1,000 | ECM Biosciences LLC | CP2191 |
| Dynamin-1 | mouse | M | 1:8,000 | Cell signaling | 4565 |
| pDynamin-1 (Ser-778) | sheep | P | 1: 2,000 | Thermo scientific | PA1-4621 |
| pDynamin-1 (Ser-795) | rabbit | P | 1: 2,000 | Santa Cruz Biotechnology | SC-12937 |
| Tubulin | rabbit | P | 1:16,000 | Cell Signaling | 2148 |
| Ac-Tubulin (Lys-40) | rabbit | P | 1: 1,000 | Cell Signaling | 3971S |
| Phosphotyrosine antibody (clone 4G10) | mouse | M | 1: 4,000 | Millipore | 05-321 |
| PIMT | rabbit | P | 1:2,500 | Custom | |
| β-actin | rabbit | M | 1:30,000 | Cell Signaling | 4970S |
a Polyclonal or Monoclonal
Mice
PIMT +/- (HZ) males used to start our mouse colony were kindly provided by Mark Mamula (Yale University, New Haven, CT) and were originally generated by inserting a neomycin resistance cassette into exon one of the PIMT gene [19]. PIMT -/- (KO) and PIMT +/+ (WT) C57BL/6 mice were obtained by intercrossing PIMT HZ C57BL/6 mice. Genotyping from tail clips was carried out by PCR at Transnetyx, Inc. (Cordova, TN) with probes for the neo cassette and the PIMT gene. Mice were monitored by on-site veterinarians, with all protocols undertaken in strict accordance with the recommendations for the Care and Use of Laboratory Animals, and approved by the University of California at Irvine Institutional Animal Care and Use Committee. Mice were anesthetized with Euthasol® and sacrificed by decapitation at an age of 4 weeks.
Brain Extract Preparation
Brains were homogenized using a Potter-Elvejhem tissue homogenizer in 9 vol of ice-cold lysis buffer containing 5 mM K-Hepes, 0.5 mM EDTA, pH 7.6, 10% sucrose, 50 mM NaF, 1 mM Na3VO4, 0.1 mM DTT (dithiothreitol) and 1% (v/v) protease inhibitor cocktail. After centrifugation at 800 × g for 30 min at 4°C, the supernatants (hereafter "brain extracts”) were collected and protein concentration was measured using the micro-BCA assay (Thermo Scientific) against a BSA standard.
Western Blots
Thirty microgram samples of brain extract protein were subjected to electrophoresis in 10% NuPAGE® Bis-Tris gels (Invitrogen/Life Technologies) after heating at 70°C for 10 min in SDS sample buffer. After semi-dry transfer to PVDF, the membrane was blocked with 5% non-fat milk in TBS-T (Tris-buffered saline, with 0.01% (v/v) Tween-20) and phosphatase inhibitors (50 mM NaF and 1 mM Na3VO4), and then incubated with antibodies diluted as indicated (Table 1) in the same buffer. Immunoreactive bands were developed with ECL Plus according to the manufacturer's directions. The chemiluminescent signals from immunoblots were acquired as 12-bit TIFF files using a digital SLR camera with exposure times ranging from 15 s to 4 min [33]. Band densities and background were quantified using ImageJ-64 (http://rsbweb.nih.gov/ij/index.html) for Mac OSX. For each primary antibody used, the ECL response was optimized for linearity over a 32X range with regard to protein load, antibody dilution, and camera exposure time.
Gel Staining
Phosphorylated and total proteins were stained in gel with the Pro-Q Diamond phosphoprotein stain and SYPRO Ruby stain, respectively, according to the manufacturers instructions. Brain extract proteins (2 µg per lane) were subjected to electrophoresis on NuPAGE 10% Bis-Tris gels. These were fixed and stained with Pro-Q Diamond and imaged on a Typhoon™ Variable Mode Imager (GE Healthcare). The same gels were then stained with SYPRO Ruby and imaged again on the Typhoon.
Statistical Analysis
Band densities were corrected for background and then normalized to the β-actin signal of the same lane. The mean value of the of all the WT bands on a given gel was calculated and used to normalize values of all the individual bands. After this double-normalization, paired t-tests were carried out using adjacent lanes (e.g., WT1 vs. KO1) for pairing. Pairwise analysis minimized errors due to subtle gradients of ECL background signal on the blots. Differences were considered significant at P ≤ 0.05.
Results
Effect of PIMT Deficiency on Phosphorylation of Synapsins I and II
The synapsins are important for regulation of neurotransmitter release [34] and are major in vivo substrates for PIMT [32]. Synapsin function is regulated by phosphorylation at several distinct sites, the most studied of which are Ser-9 (also known as site 1, modified by the cAMP-dependent protein kinase (PKA) and the type I calcium-dependent protein kinase (CaMKI)) and Ser–603 (also known as site 3, modified by the type II calcium-dependent protein kinase (CaMKII)). We employed Western blotting with phosphorylation-specific antibodies to see if modification of synapsin at these sites is altered in PIMT deficient mice, as this might help explain the neurological pathology of these mice. Pilot experiments, in which extracts of male and female mice were electrophoresed on the same gel, revealed increased phosphorylation in KO vs. WT brain, but unexpectedly suggested that the effect was stronger in females than males. We therefore continued the study using alternating lanes of WT vs. KO brain extracts from a single sex on each gel.
Results obtained from four-week female mice are shown in Figure 1 and summarized in Table 2. Western blot images obtained with antibodies made against pan-synapsin I, the phosphorylated Ser-9 site, and the phosphorylated Ser-603 site are shown in Figure 1 (A-C). The bar graph in Figure 1D summarizes these results and indicates the WT vs. KO statistical significance based on a pairwise t-test. The signal data is normalized to β-actin expression within each lane. The pSer-9 antibody used in Western blot produced signals consistent with the molecular weights of synapsins Ia, Ib and II. Phosphorylation of all three synapsin isoforms was markedly higher in the KO extracts, with average increases of 149 %, 99%, and 70% for the Ia, Ib, and II isoforms, respectively. In contrast, we observed no significant difference in synapsin I expression or in phosphorylation of synapsin I at the Ser-603 site.
Figure 1. Phosphorylation of synapsins I and II in female mice.

Western blot of mouse brain extracts used a mixture of antibodies to synapsin I (Syn1) and β-actin (A), a mixture of antibodies to synapsins phosphorylated at Ser-9 and β-actin (B), and a mixture of antibodies to synapsins phosphorylated at Ser-603 and β-actin (C). Panel D shows quantitative measurements of band intensities after normalization to β-actin. Data are expressed as means ± SEM (n=5 for each genotype). A two-tailed t-test with pairwise matching of adjacent lanes (WT vs. KO) was employed. *P < 0.05, **P < 0.01, ***P < 0.001 for KO vs. WT.
Table 2. Summary and statistical analysis of Western blot data.
| Antigen | Sex | Na | WT signal | WT SE b | KO signal | KO SE | ttest c P |
|---|---|---|---|---|---|---|---|
| pPDK1 (Ser-241) | F | 5 | 1.00 | 0.03 | 0.94 | 0.04 | 0.307 |
| M | 5 | 1.00 | 0.04 | 0.95 | 0.04 | 0.570 | |
| Synapsin I | F | 5 | 1.00 | 0.07 | 0.94 | 0.08 | 0.493 |
| pSyn Ia (Ser-9)d | F | 5 | 1.00 | 0.21 | 2.45 | 0.34 | 0.018 |
| pSyn Ib (Ser-9) | F | 5 | 1.00 | 0.27 | 1.97 | 0.21 | <0.001 |
| pSyn IIb (Ser-9) | F | 5 | 1.00 | 0.16 | 1.69 | 0.08 | 0.028 |
| pSyn I (Ser-603) | F | 5 | 1.00 | 0.03 | 0.98 | 0.08 | 0.832 |
| Synapsin I | M | 5 | 1.00 | 0.06 | 1.03 | 0.02 | 0.683 |
| pSyn Ia (Ser-9) | M | 5 | 1.00 | 0.13 | 1.22 | 0.21 | 0.354 |
| pSyn Ib (Ser-9) | M | 5 | 1.00 | 0.16 | 1.13 | 0.25 | 0.402 |
| pSyn II (Ser-9) | M | 5 | 1.00 | 0.08 | 1.13 | 0.09 | 0.318 |
| pSyn I (Ser-603) | M | 5 | 1.00 | 0.08 | 1.03 | 0.06 | 0.774 |
| pDARPP-32 (Thr-34) | F | 6 | 1.00 | 0.05 | 0.83 | 0.11 | 0.292 |
| M | 6 | 1.00 | 0.12 | 1.00 | 0.10 | 0.998 | |
| CRMP2-A | F | 5 | 1.00 | 0.05 | 0.98 | 0.03 | 0.679 |
| CRMP2-B | F | 5 | 1.00 | 0.05 | 1.02 | 0.03 | 0.701 |
| pCRMP2 (Thr-514) | F | 5 | 1.00 | 0.05 | 1.01 | 0.03 | 0.842 |
| pCRMP2 (Ser-522) | F | 6 | 1.00 | 0.05 | 1.03 | 0.07 | 0.713 |
| CRMP2-A | M | 5 | 1.00 | 0.05 | 0.98 | 0.04 | 0.667 |
| CRMP2-B | M | 5 | 1.00 | 0.04 | 1.05 | 0.07 | 0.428 |
| pCRMP2 (Thr-514) | M | 5 | 1.00 | 0.04 | 1.00 | 0.03 | 0.855 |
| pCRMP2 (Ser-522) | M | 6 | 1.00 | 0.05 | 0.97 | 0.06 | 0.588 |
| Dynamin-1 | F | 6 | 1.00 | 0.08 | 1.05 | 0.16 | 0.633 |
| pDyn1 (Ser-778) | F | 6 | 1.00 | 0.19 | 0.96 | 0.33 | 0.902 |
| pDyn1 (Ser-795) | F | 6 | 1.00 | 0.31 | 0.90 | 0.44 | 0.852 |
| Dynamin-1 | M | 6 | 1.00 | 0.09 | 1.04 | 0.12 | 0.634 |
| pDyn1 (Ser-778) | M | 6 | 1.00 | 0.35 | 1.73 | 0.58 | 0.342 |
| pDyn1 (Ser-795) | M | 6 | 1.00 | 0.35 | 1.78 | 0.39 | 0.148 |
| Tubulin | F | 5 | 1.00 | 0.04 | 0.96 | 0.02 | 0.486 |
| Ac-αTub (Lys-40) | F | 5 | 1.00 | 0.16 | 0.81 | 0.11 | 0.360 |
| Tubulin | M | 5 | 1.00 | 0.05 | 1.01 | 0.05 | 0.688 |
| Ac-αTub (Lys-40) | M | 5 | 1.00 | 0.12 | 0.76 | 0.12 | 0.003 |
aNumber of animals per genotype.
bStandard Error
cTwo-tailed, with matched pairing of adjacent lanes.
dBold face indicates data where the KO signal is ≥20% above or below the WT signal.
Extracts from 4-week male mice showed qualitatively similar results, but the degree of enhanced phosphorylation at Ser-9 in the KO mice was much smaller; 22%, 14%, and 14%, for synapsins Ia, Ib, and II, respectively, with P > 0.05 in all three cases (Figure 2). This suggests an inherent sex difference in synapsin phosphorylation. To test this idea, we electrophoresed brain extracts of WT female and male mice in alternating lanes of the same gel prior to Western blotting for PIMT, pan-synapsin I, and pSer-9 synapsin (Figure 3A-C). As shown in Figure 3D, there was no significant sex difference in the expression of PIMT or synapsin I, yet the phosphorylation of synapsin at Ser-9 was 37- 40% lower in males than females for all three detected isoforms of synapsin. Overall these results indicate that female mice have a greater average level of phosphorylation of Ser-9 than males, and that they also show a greater increase in phosphorylation at this site in response to PIMT deficiency.
Figure 2. Phosphorylation of synapsins I and II in male mice.

Panels A-D are otherwise the same as in Figure 1.
Figure 3. Levels of PIMT, synapsin I, and phospho-synapsin (Ser-9) in WT female and male mice.

Mouse sex for each lane is indicated at the bottom on panel C. Western blots used a mixture of antibodies to PIMT and β-actin (A), a mixture of antibodies to synapsin 1 and β-actin (B), and a mixture of antibodies to synapsin 1 phosphorylated at Ser-9 and β-actin (C). Panel D shows quantitative measurements of band intensities in A-C after normalization to β-actin. Data are expressed as means ± SEM (n=5 for each sex) with statistics as in Figure 1.
Phosphorylation of Dynamin-1
Dynamin-1 is a neuronal GTPase implicated in endocytosis of synaptic vesicles [35] and is another major in vivo substrate for PIMT [32]. Dynamin-1 function is regulated by phosphorylation via cyclin-dependent protein kinase 1 (Cdk1) at Ser-778, and protein kinase C (PKC) at Ser-795. Figure 4 shows the status of these phosphorylation sites in brain extracts of WT, HZ, and KO mice. The mean of ECL signals for KO males at both phosphorylation sites was increased by 75% over WT males; however the high variance of these signals resulted in a low level of statistical significance (P > 0.05). In contrast, the ECL signal at these phosphorylation sites for female WT and KO mice were nearly identical. No difference in dynamin-1 expression was seen in either sex. Firm conclusions from these results are hampered by the high mouse-to-mouse variability of dynamin-1 phosphorylation, but strongly suggests PIMT deficiency may increase dynamin-1 phosphorylation in males, and not in females. This is an interesting contrast to the results found with the synapsins.
Figure 4. Phosphorylation of dynamin-1 at Ser-778 and Ser-795.

Panels A-C show Western blots of brain extracts from female mice for dynamin-1 (A), phosphorylation of dynamin-1 at Ser-778 (B), and phosphorylation of dynamin-1 at Ser-795 (C). Panels D-F: Same as with A-C for male mice. Panel G: Quantitative measurements of band intensities after normalization to β-actin. Data are expressed as means ± SEM (n=5 for each genotype) with statistics as in Figure 1. None of the tests showed a significant difference.
Phosphorylation of CRMP2 and DARPP-32
CRMP2 plays a key role in axonal guidance during brain development and plasticity of adult brain [36]. It also serves as a major in vivo substrate for PIMT [32]. CRMP2 function is regulated by phosphorylation via Cdk5 at Ser-522 and by PKC at Ser-795. Western blotting of brain extracts from KO vs. WT mice revealed no effect of PIMT deficiency at either phosphorylation site, or on overall CRMP2 expression, regardless of sex (Figure S1, panels E and F).
We also investigated the expression and phosphorylation of the 32 kDa dopamine and cAMP regulated phosphoprotein (DARPP-32) in KO vs. WT brain extracts. DARPP-32 was of interest because enhanced phosphorylation of synapsin I in KO mice was observed at the cAMP-regulated PKA site, but not at the CaMKII site. Although DARPP-32 is not known to be a substrate for PIMT in vivo, it serves as a major target for PKA and, in its phosphorylated state, enhances and prolongs the phosphorylation of other PKA targets by inhibiting their dephosphorylation [37]. If enhanced phosphorylation of synapsin at the Ser-9 site in the KO mice is related to a generalized increase in brain cAMP levels, then one might also see increased phosphorylation of DARPP-32 in the KO mice. As shown in Figure S1 (panels A and B) and Table 3, we saw no significant difference between WT and KO mice in expression or phosphorylation of DARPP-32 in either females or males.
Table 3. Summary and statistical analysis of Western blot for female and male wild-type mice.
| Antigen | Na | Female (WT) signal | Female(WT) SE | Male (WT) signal | Male (WT) SE | ttest b P |
|---|---|---|---|---|---|---|
| PIMT | 5 | 1.00 | 0.06 | 1.07 | 0.07 | 0.334 |
| Synapsin I | 5 | 1.00 | 0.11 | 0.90 | 0.16 | 0.352 |
| pSyn Ia(Ser-9) | 5 | 1.00 | 0.09 | 0.60 | 0.04 | 0.018 |
| pSyn Ib(Ser-9) | 5 | 1.00 | 0.17 | 0.61 | 0.09 | 0.041 |
| pSyn II(Ser-9) | 5 | 1.00 | 0.17 | 0.63 | 0.14 | 0.044 |
aNumber of animals per genotype
bTwo-tailed, with matched pairing of adjacent lanes.
Tubulin Acetylation
Tubulin is the single largest contributor to isoAsp formation in mouse brain [32,38], and abnormal bundling of microtubules is seen in cortical dendrites of the KO mouse brain [20]. Acetylation of tubulin at Lys-40 is associated with microtubule stability and may also play a role in regulating cell motility and differentiation, intracellular trafficking, and signaling [39]. To see if PIMT deficiency affects the acetylation of α-tubulin, brain extracts from WT and KO mice were subjected to Western blotting with a pan-tubulin antibody, and an antibody specific for tubulin acetylated at lysine-40 (Figure 5A and 5B). Tubulin expression was unaffected by genotype or sex, but acetylation of tubulin was decreased by ca. 20% in both male and female mice (Figure 5C).
Figure 5. Acetylation of α-tubulin.

Western blots for tubulin (pan-Tub) and acetyl-α-tubulin (Ac-Tub) in brain extracts of WT and KO mice, both female and male (A, B). Genotypes are indicated at the top of each lane and sex at the bottom. (C) Quantitative analysis of band intensities for tubulin and acetyl-α-tubulin respectively. Data are expressed as means ± SEM (n=10) with statistics as in Figure 1.
Phosphorylation of PDK1
PIMT KO mice have enlarged brains, a characteristic also found in mice that have defects in the PI3K/Akt growth signaling pathway. This prompted a study by Farrar et al. [31] who reported increased phosphorylation in PIMT KO vs. WT mouse brain of PDK1 and several other proteins in the insulin signaling pathway. Using extracts of dissected cerebral cortex and hippocampus, these workers reported that phosphorylation of PDK1 in the KO mouse was increased by 260% and 110% respectively, relative to the WT. We attempted to repeat these findings using our own protocol for brain extract preparation and Western blotting (Figure 6). As summarized in Table 2, we were unable to detect any significant difference between WT and KO in either male or female mice. Possible reasons for the discrepancy between our results and those of Farrar et al. are discussed later.
Figure 6. Phosphorylation of PDK1 at Ser-241 is unaffected by PIMT genotype or sex.
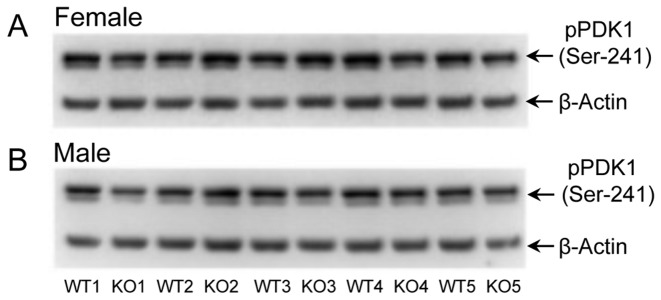
Western blots showing the phosphorylation of PDK1 at Ser-241 in brain extracts of WT and KO mice, both female and male mice. After normalization of the phospho-PDK1 signals to β-actin, statistical analysis revealed no significant difference between the KO and WT extracts.
Overall Protein Phosphorylation Levels are Similar in PIMT KO vs. WT mouse brain.
Attempts to determine if there are overall net changes in protein phosphorylation in the KO mouse brain were carried out in two ways. In the first approach we subjected brain extracts to SDS-PAGE, stained the gel with the phospho-specific protein stain Pro-Q® Diamond, and followed this by staining for total protein with SYPRO Ruby (Figure S2). The ratio of the Pro-Q® Diamond to SYPRO Ruby signals, obtained by image analysis, revealed that there was no significant difference between the WT and KO mice.
In the second approach, we carried out Western blotting of brain extracts from WT and KO mice using a primary antibody (4G10) that is specific for phospho-tyrosine in proteins. After obtaining an ECL image for phospho-tyrosine, the blot was then stained for total protein with Coomassie Blue R-250. (Figure S3). The ratio of ECL to Coomassie signals, obtained by image analysis, revealed that there was no significant difference in overall tyrosine phosphorylation between the WT and KO mice.
Discussion
This study was designed to test the hypothesis that specific isoaspartyl-rich proteins in the brains of PIMT-KO mice are functionally altered, and as such, might contribute to the neuropathology characteristic of these mice. Synapsins I and II, α/β-tubulin, dynamin-1, and CRMP2 have been identified previously as major targets for the PIMT repair enzyme in brain as they accumulate exceptional levels of isoAsp sites in the PIMT KO mouse. All four proteins play roles in synaptic function and all are subject to post-translational modifications (PTMs) that regulate those functions. We demonstrate here that in vivo phosphorylation of synapsins I and II and acetylation of α-tubulin, are significantly altered in PIMT KO mice, and that dynamin-1 phosphorylation may also be altered. These findings provide a potential basis for the mechanisms by which isoAsp accumulation leads to the neurological deficits characteristic of PIMT deficient mice.
Synapsin Phosphorylation
Synapsin modulates neurotransmitter release by reversibly anchoring synaptic vesicles to the cytoskeleton in a reserve zone of the pre-synaptic terminal [34]. Increased levels of cyclic-AMP and/or Ca2+ promote the release of synapsin from the cytoskeleton, allowing vesicles to migrate to the pre-synaptic active zone. Functional damage to synapsins could make an especially important contribution to the phenotype of the PIMT KO mice, as there are several similarities between the PIMT KO mice and synapsin KO mice. Both types of mutant mice are prone to epileptic seizures [19,40], have cognitive impairment [25,41], exhibit similar neurophysiological abnormalities and display an abnormal distribution of synaptic vesicles in the presynaptic terminal [25,42]
Purified synapsin aged in vitro accumulates isoaspartyl sites at 5-6 residues as indicated in Figure 7 [43]. All but one of these sites is clustered the C-domain, a multifunctional globular region that binds ATP, synaptic vesicles, and several components of the cytoskeleton. Three to four of the C-domain isoAsp sites coincide with sub-regions believed to mediate vesicle binding by insertion into the phospholipid bilayer [44-46]. This coincidence makes it tempting to speculate that isoaspartyl synapsins have a weakened affinity for vesicles. This could promote the redistribution of more synapsin to the cytosol, thereby leading to increased phosphorylation of synapsin at Ser-9, consistent with the phospho-switch model of Hosaka and Südhof [47]. Future experiments may establish the validity of this idea by comparing the affinity of isoaspartyl-rich and isoaspartyl-free forms of recombinant C domain with regard to their relative affinities for synaptic vesicles.
Figure 7. Domain structure of synapsin Ia.
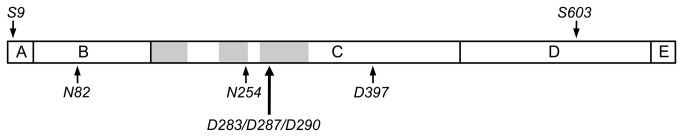
Positions of the Ser-9 and Ser-603 phosphorylation sites are shown along the top of the bar. Positions of the major isoAsp sites are shown along the bottom. Shaded areas in domain C indicate the three phospholipid membrane insertion regions.
Synapsin phosphorylation has been studied in various contexts for over 30 years, yet (with the exception of animals treated with sex hormones) we are unaware of any prior observations indicating differences between males and females in any animal species. Previous studies on the in vivo status of rodent synapsin phosphorylation used rats that were either all male [48,49] or all female [50]. One possible explanation for the dramatic differences we see between male and female brain extracts is that female mice undergo an overall higher level of synaptic activity in response to their handling just prior to, and during, the administration of anesthetic, and that this enhanced neural activity is exaggerated in females. Regardless of the cause, our findings add to a growing list of sex-specific changes in behavior and neurochemistry linked to gene knockouts in mice [51], and add support to the recommendation that possible sex differences in animal models of brain function should be given due consideration in designing experiments [52].
Tubulin Acetylation
Like the synapsins, α- and β-tubulin are known to be highly susceptible to isoAsp formation in vitro [53] and have been shown to be major targets for PIMT in mouse brain [32,38]. Tubulin is an essential element of the cytoskeleton in synaptic regions as well as other parts of neurons. Yamamoto et al. [20] found “deformed apical dendrites” in cortical pyramidal cells of the PIMT-KO mouse that correlated with the presence of “high density bundles” of microtubules not seen in the wild type mouse brain. This suggests that isoaspartyl-damaged tubulin may disrupt synaptic functions that require microtubule integrity.
The function and detailed mechanism of tubulin acetylation at lysine-40 is poorly understood, but seems to occur either during or after polymerization of α- and β-tubulin to form microtubules [39]. IsoAsp accumulation in tubulin could hinder microtubule assembly, thus explaining the decreased level of acetylation we observed here. It is noteworthy that decreased acetylation of tubulin seen in the KO mice was not significantly different between males and females. This demonstrates that the sex-dependent PTM alterations we observed in the synapsins, and possibly dynamin-1, does not apply to all substrates of PIMT.
PDK1 Phosphorylation
Farrar et al. [31] compared brain extracts of PIMT KO vs. WT mice with regard to phosphorylation of proteins in the PI3K/Akt growth-signaling pathway because mice with certain defects in this pathway develop enlarged brains, a characteristic also found in the PIMT KO mouse [20]. Using Western blot analysis with phospho-specific antibodies, they found hyperphosphorylation in the KO brains for several proteins in this pathway. The greatest increase was with PDK1 in which the KO mice exhibited an average increase in phosphorylation of 260% and 110%, respectively, in extracts of cortex and hippocampus (P = 0.05 in both cases). In our attempt to repeat this result (Figure 6) we used the same commercial antibody (Table 1) as Farrar, but found no difference between KO and WT. There are three potentially significant differences between our experimental procedures and those of Farrar that, taken together, might contribute to the discrepant results (1). They compared 3 mice of each genotype, whereas we compared 5 mice of each genotype (2). We used mice that were all 28 days old, but they used mice that were 0-50 days old and did not indicate any age matching within a given Western blot (3). We used phosphatase inhibitors in the homogenization buffer and all Western blot solutions [54], whereas they did not indicate the use of phosphatase inhibitors in any of the solutions. The lack of inhibitors, especially in the homogenization buffer, may have allowed unintended dephosphorylation that varied significantly from sample to sample. Combined with the smaller number of mice use, and a possible lack of age matching, the significant difference observed by Farrar may have been a statistical anomaly.
None of the proteins in the current study have any known enzymatic activity, but some of the other PIMT targets identified in our isoAsp proteomic study [32], such as creatine kinase B and HSC70, do have measureable enzyme activity. It will be of interest to see if any of those activities are significantly altered in the KO mouse brain, and if so, are they sex-dependent.
Supporting Information
PIMT deficiency does not alter phosphorylation of DARPP-32 at Thr-34, or phosphorylation of CRMP2 at Ser-522 and Thr-514. A and B: Western analysis of phosphorylation of DARPP-32 at Thr-34 in brain extracts of female (A) and male (B) WT, HZ and KO mice. C and D: Western analysis of phosphorylation of CRMP2 at Ser-522 in brain extracts from female (C) and male (D) WT, HZ and KO mice. E and F: Western analysis of CRMP2 expression (CRMP2-A/B) and phosphorylation of CRMP2 at Ser-514 (pCRMP2) in brain extracts from female (E) and male (F) mice.
(TIF)
Global evaluation of protein phosphorylation in brain extracts of WT and KO PIMT mice by comparing Pro-Q Diamond gel stain for phosphoproteins with the Sypro Ruby general protein stain. A. Gel stained using Pro-Q Diamond dye to detect phosphoproteins.
B. The same gel post-stained with Sypro Ruby to visualize total protein. The mouse sex for each lane is indicated at the bottom on panel A and B.
(TIF)
Tyrosine phospho-proteome analysis of brain extracts from WT and KO PIMT mice. A, Western blot analysis to visualize the tyrosine-phosphorylated proteins with an anti-phosphotyrosine antibody. B, Coomassie Blue stain for total protein on the same membrane. The mouse sex for each lane is indicated at the bottom on panel A and B.
(TIF)
Acknowledgments
We thank Mark J. Mamula of Yale University School Medicine for providing mice of the PIMT +/- genotype to start our colony, and Larry Cahill of the University of California, Irvine, for alerting us to the growing number of studies with gene knockouts in mice that show distinct CNS phenotypes in males vs. females.
Funding Statement
This research was funded solely by National Institutes of Health grant NS-17269. http://grants.nih.gov/grants/oer.htm. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Desrosiers RR, Fanélus I (2011) Damaged proteins bearing L-isoaspartyl residues and aging: a dynamic equilibrium between generation of isomerized forms and repair by PIMT. Curr Aging Sci 4: 8-18. doi: 10.2174/1874612811104010008. PubMed: 21204776. [DOI] [PubMed] [Google Scholar]
- 2. Shimizu T, Matsuoka Y, Shirasawa T (2005) Biological significance of isoaspartate and its repair system. Biol Pharm Bull 28: 1590-1596. doi: 10.1248/bpb.28.1590. PubMed: 16141521. [DOI] [PubMed] [Google Scholar]
- 3. Robinson NE, Robinson ZW, Robinson BR, Robinson AL, Robinson JA et al. (2004) Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J Pept Res 63: 426-436. doi: 10.1111/j.1399-3011.2004.00151.x. PubMed: 15140160. [DOI] [PubMed] [Google Scholar]
- 4. Reissner KJ, Aswad DW (2003) Deamidation and isoaspartate formation in proteins: unwanted alterations or surreptitious signals? Cell Mol Life Sci 60: 1281-1295. doi: 10.1007/s00018-003-2287-5. PubMed: 12943218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clarke S (2003) Aging as war between chemical and biochemical processes: protein methylation and the recognition of age-damaged proteins for repair. Ageing Res Rev 2: 263-285. doi: 10.1016/S1568-1637(03)00011-4. PubMed: 12726775. [DOI] [PubMed] [Google Scholar]
- 6. Lindner H, Helliger W (2001) Age-dependent deamidation of asparagine residues in proteins. Exp Gerontol 36: 1551-1563. doi: 10.1016/S0531-5565(01)00140-1. PubMed: 11525877. [DOI] [PubMed] [Google Scholar]
- 7. Volkin DB, Mach H, Middaugh CR (1997) Degradative covalent reactions important to protein stability. Mol Biotechnol 8: 105-122. doi: 10.1007/BF02752255. PubMed: 9406181. [DOI] [PubMed] [Google Scholar]
- 8. Aswad DW (1984) Stoichiometric methylation of porcine adrenocorticotropin by protein carboxyl methyltransferase requires deamidation of asparagine 25. Evidence for methylation at the alpha-carboxyl group of atypical L-isoaspartyl residues. J Biol Chem 259: 10714-10721. PubMed: 6088513. [PubMed] [Google Scholar]
- 9. Murray ED Jr., Clarke S (1984) Synthetic peptide substrates for the erythrocyte protein carboxyl methyltransferase. Detection of a new site of methylation at isomerized L-aspartyl residues. J Biol Chem 259: 10722-10732. PubMed: 6469980. [PubMed] [Google Scholar]
- 10. Tyler-Cross R, Schirch V (1991) Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J Biol Chem 266: 22549-22556. PubMed: 1939272. [PubMed] [Google Scholar]
- 11. Johnson BA, Aswad DW (1985) Enzymatic protein carboxyl methylation at physiological pH: cyclic imide formation explains rapid methyl turnover. Biochemistry 24: 2581-2586. doi: 10.1021/bi00331a028. PubMed: 4016073. [DOI] [PubMed] [Google Scholar]
- 12. Geiger T, Clarke S (1987) Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem 262: 785-794. PubMed: 3805008. [PubMed] [Google Scholar]
- 13. Johnson BA, Murray ED Jr., Clarke S, Glass DB, Aswad DW (1987) Protein carboxyl methyltransferase facilitates conversion of atypical L-isoaspartyl peptides to normal L-aspartyl peptides. J Biol Chem 262: 5622-5629. PubMed: 3571226. [PubMed] [Google Scholar]
- 14. Johnson BA, Langmack EL, Aswad DW (1987) Partial repair of deamidation-damaged calmodulin by protein carboxyl methyltransferase. J Biol Chem 262: 12283-12287. PubMed: 3624258. [PubMed] [Google Scholar]
- 15. McFadden PN, Clarke S (1987) Conversion of isoaspartyl peptides to normal peptides: implications for the cellular repair of damaged proteins. Proc Natl Acad Sci U S A 84: 2595-2599. doi: 10.1073/pnas.84.9.2595. PubMed: 3472227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Galletti P, Ciardiello A, Ingrosso D, Di Donato A, D'Alessio G (1988) Repair of isopeptide bonds by protein carboxyl O-methyltransferase: seminal ribonuclease as a model system. Biochemistry 27: 1752-1757. doi: 10.1021/bi00405a055. PubMed: 3365422. [DOI] [PubMed] [Google Scholar]
- 17. Brennan TV, Anderson JW, Jia Z, Waygood EB, Clarke S (1994) Repair of spontaneously deamidated HPr phosphocarrier protein catalyzed by the L-isoaspartate-(D-aspartate) O-methyltransferase. J Biol Chem 269: 24586-24595. PubMed: 7929130. [PubMed] [Google Scholar]
- 18. Johnson BA, Najbauer J, Aswad DW (1993) Accumulation of substrates for protein L-isoaspartyl methyltransferase in adenosine dialdehyde-treated PC12 cells. J Biol Chem 268: 6174-6181. PubMed: 8454593. [PubMed] [Google Scholar]
- 19. Kim E, Lowenson JD, MacLaren DC, Clarke S, Young SG (1997) Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proc Natl Acad Sci U S A 94: 6132-6137. doi: 10.1073/pnas.94.12.6132. PubMed: 9177182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamamoto A, Takagi H, Kitamura D, Tatsuoka H, Nakano H et al. (1998) Deficiency in protein L-isoaspartyl methyltransferase results in a fatal progressive epilepsy. J Neurosci 18: 2063-2074. PubMed: 9482793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kosugi S, Furuchi T, Katane M, Sekine M, Shirasawa T et al. (2008) Suppression of protein l-isoaspartyl (d-aspartyl) methyltransferase results in hyperactivation of EGF-stimulated MEK-ERK signaling in cultured mammalian cells. Biochem Biophys Res Commun 371: 22-27. doi: 10.1016/j.bbrc.2008.03.109. PubMed: 18381200. [DOI] [PubMed] [Google Scholar]
- 22. Diliberto EJ Jr., Axelrod J (1976) Regional and subcellular distribution of protein carboxymethylase in brain and other tissues. J Neurochem 26: 1159-1165. doi: 10.1111/j.1471-4159.1976.tb07001.x. PubMed: 6627. [DOI] [PubMed] [Google Scholar]
- 23. Boivin D, Bilodeau D, Béliveau R (1995) Immunochemical characterization of L-isoaspartyl-protein carboxyl methyltransferase from mammalian tissues. Biochem J 309 ( 3): 993-998. PubMed: 7639720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim E, Lowenson JD, Clarke S, Young SG (1999) Phenotypic analysis of seizure-prone mice lacking L-isoaspartate (D-aspartate) O-methyltransferase. J Biol Chem 274: 20671-20678. doi: 10.1074/jbc.274.29.20671. PubMed: 10400700. [DOI] [PubMed] [Google Scholar]
- 25. Ikegaya Y, Yamada M, Fukuda T, Kuroyanagi H, Shirasawa T et al. (2001) Aberrant synaptic transmission in the hippocampal CA3 region and cognitive deterioration in protein-repair enzyme-deficient mice. Hippocampus 11: 287-298. doi: 10.1002/hipo.1043. PubMed: 11769310. [DOI] [PubMed] [Google Scholar]
- 26. Vitali R, Clarke S (2004) Improved rotorod performance and hyperactivity in mice deficient in a protein repair methyltransferase. Behav Brain Res 153: 129-141. doi: 10.1016/j.bbr.2003.11.007. PubMed: 15219714. [DOI] [PubMed] [Google Scholar]
- 27. Carter WG, Aswad DW (2008) Formation, localization, and repair of L-isoaspartyl sites in histones H2A and H2B in nucleosomes from rat liver and chicken erythrocytes. Biochemistry 47: 10757-10764. doi: 10.1021/bi8013467. PubMed: 18795804. [DOI] [PubMed] [Google Scholar]
- 28. Young AL, Carter WG, Doyle HA, Mamula MJ, Aswad DW (2001) Structural integrity of histone H2B in vivo requires the activity of protein L-isoaspartyl O-methyltransferase, a putative repair enzyme. J Biol Chem 276: 37161-37165. doi: 10.1074/jbc.M106682200. PubMed: 11479322. [DOI] [PubMed] [Google Scholar]
- 29. Bidinosti M, Ran I, Sanchez-Carbente MR, Martineau Y, Gingras AC et al. (2010) Postnatal deamidation of 4E-BP2 in brain enhances its association with raptor and alters kinetics of excitatory synaptic transmission. Mol Cell 37: 797-808. doi: 10.1016/j.molcel.2010.02.022. PubMed: 20347422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bidinosti M, Martineau Y, Frank F, Sonenberg N (2010) Repair of isoaspartate formation modulates the interaction of deamidated 4E-BP2 with mTORC1 in brain. J Biol Chem 285: 19402-19408. doi: 10.1074/jbc.M110.120774. PubMed: 20424163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farrar C, Houser CR, Clarke S (2005) Activation of the PI3K/Akt signal transduction pathway and increased levels of insulin receptor in protein repair-deficient mice. Aging Cell 4: 1-12. doi: 10.1111/j.1474-9728.2004.00136.x. PubMed: 15659208. [DOI] [PubMed] [Google Scholar]
- 32. Zhu JX, Doyle HA, Mamula MJ, Aswad DW (2006) Protein repair in the brain, proteomic analysis of endogenous substrates for protein L-isoaspartyl methyltransferase in mouse brain. J Biol Chem 281: 33802-33813. doi: 10.1074/jbc.M606958200. PubMed: 16959769. [DOI] [PubMed] [Google Scholar]
- 33. Khoury MK, Parker I, Aswad DW (2010) Acquisition of chemiluminescent signals from immunoblots with a digital single-lens reflex camera. Anal Biochem 397: 129-131. doi: 10.1016/j.ab.2009.09.041. PubMed: 19788886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cesca F, Baldelli P, Valtorta F, Benfenati F (2010) The synapsins: key actors of synapse function and plasticity. Prog Neurobiol 91: 313-348. doi: 10.1016/j.pneurobio.2010.04.006. PubMed: 20438797. [DOI] [PubMed] [Google Scholar]
- 35. Ferguson SM, De Camilli P (2012) Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 13: 75-88. PubMed: 22233676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hensley K, Venkova K, Christov A, Gunning W, Park J (2011) Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol 43: 180-191. doi: 10.1007/s12035-011-8166-4. PubMed: 21271304. [DOI] [PubMed] [Google Scholar]
- 37. Le Novère N, Li L, Girault JA (2008) DARPP-32: molecular integration of phosphorylation potential. Cell Mol Life Sci 65: 2125-2127. doi: 10.1007/s00018-008-8150-y. PubMed: 18488140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ohta K, Seo N, Yoshida T, Hiraga K, Tuboi S (1987) Tubulin and high molecular weight microtubule-associated proteins as endogenous substrates for protein carboxymethyltransferase in brain. Biochimie 69: 1227-1234. doi: 10.1016/0300-9084(87)90150-7. PubMed: 3129027. [DOI] [PubMed] [Google Scholar]
- 39. Perdiz D, Mackeh R, Poüs C, Baillet A (2011) The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell Signal 23: 763-771. doi: 10.1016/j.cellsig.2010.10.014. PubMed: 20940043. [DOI] [PubMed] [Google Scholar]
- 40. Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS et al. (1995) Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A 92: 9235-9239. doi: 10.1073/pnas.92.20.9235. PubMed: 7568108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Corradi A, Zanardi A, Giacomini C, Onofri F, Valtorta F et al. (2008) Synapsin-I- and synapsin-II-null mice display an increased age-dependent cognitive impairment. J Cell Sci 121: 3042-3051. doi: 10.1242/jcs.035063. PubMed: 18713831. [DOI] [PubMed] [Google Scholar]
- 42. Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM et al. (2004) Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci 24: 11368-11380. doi: 10.1523/JNEUROSCI.3795-04.2004. PubMed: 15601943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reissner KJ, Paranandi MV, Luc TM, Doyle HA, Mamula MJ et al. (2006) Synapsin I is a major endogenous substrate for protein L-isoaspartyl methyltransferase in mammalian brain. J Biol Chem 281: 8389-8398. doi: 10.1074/jbc.M510716200. PubMed: 16443604. [DOI] [PubMed] [Google Scholar]
- 44. Benfenati F, Greengard P, Brunner J, Bähler M (1989) Electrostatic and Hydrophobic Interactions of Synapsin I and Synapsin I Fragments with Phospholipid Bilayers. J Cell Biol 108: 1851-1862. doi: 10.1083/jcb.108.5.1851. PubMed: 2497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stefani G, Onofri F, Valtorta F, Vaccaro P, Greengard P et al. (1997) Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic vesicles. J Physiol 504: 501-515. doi: 10.1111/j.1469-7793.1997.501bd.x. PubMed: 9401959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheetham JJ, Hilfiker S, Benfenati F, Weber T, Greengard P et al. (2001) Identification of synapsin I peptides that insert into lipid membranes. Biochem J 354: 57-66. doi: 10.1042/0264-6021:3540057. PubMed: 11171079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hosaka M, Hammer RE, Südhof TC (1999) A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 24: 377-387. doi: 10.1016/S0896-6273(00)80851-X. PubMed: 10571231. [DOI] [PubMed] [Google Scholar]
- 48. Strömbom U, Forn J, Dolphin AC, Greengard P (1979) Regulation of the state of phosphorylation of specific neuronal proteins in mouse brain by in vivo administration of anesthetic and convulsant agents. Proc Natl Acad Sci U S A 76: 4687-4690. doi: 10.1073/pnas.76.9.4687. PubMed: 291996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamagata Y, Obata K, Greengard P, Czernik AJ (1995) Increase in synapsin I phosphorylation implicates a presynaptic component in septal kindling. Neuroscience 64: 1-4. doi: 10.1016/0306-4522(94)00492-N. PubMed: 7708197. [DOI] [PubMed] [Google Scholar]
- 50. Iwata S, Hewlett GH, Ferrell ST, Czernik AJ, Meiri KF et al. (1996) Increased in vivo phosphorylation state of neuromodulin and synapsin I in striatum from rats treated with repeated amphetamine. J Pharmacol Exp Ther 278: 1428-1434. PubMed: 8819530. [PubMed] [Google Scholar]
- 51. Jazin E, Cahill L (2010) Sex differences in molecular neuroscience: from fruit flies to humans. Nat Rev Neurosci 11: 9-17. doi: 10.1038/nrn2754. PubMed: 20019686. [DOI] [PubMed] [Google Scholar]
- 52. Cahill L (2012) A half-truth is a whole lie: on the necessity of investigating sex influences on the brain. Endocrinology 153: 2541-2543. doi: 10.1210/en.2011-2167. PubMed: 22492307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Najbauer J, Orpiszewski J, Aswad DW (1996) Molecular aging of tubulin: accumulation of isoaspartyl sites in vitro and in vivo. Biochemistry 35: 5183-5190. doi: 10.1021/bi953063g. PubMed: 8611502. [DOI] [PubMed] [Google Scholar]
- 54. Sharma SK, Carew TJ (2002) Inclusion of phosphatase inhibitors during Western blotting enhances signal detection with phospho-specific antibodies. Anal Biochem 307: 187-189. doi: 10.1016/S0003-2697(02)00008-8. PubMed: 12137799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PIMT deficiency does not alter phosphorylation of DARPP-32 at Thr-34, or phosphorylation of CRMP2 at Ser-522 and Thr-514. A and B: Western analysis of phosphorylation of DARPP-32 at Thr-34 in brain extracts of female (A) and male (B) WT, HZ and KO mice. C and D: Western analysis of phosphorylation of CRMP2 at Ser-522 in brain extracts from female (C) and male (D) WT, HZ and KO mice. E and F: Western analysis of CRMP2 expression (CRMP2-A/B) and phosphorylation of CRMP2 at Ser-514 (pCRMP2) in brain extracts from female (E) and male (F) mice.
(TIF)
Global evaluation of protein phosphorylation in brain extracts of WT and KO PIMT mice by comparing Pro-Q Diamond gel stain for phosphoproteins with the Sypro Ruby general protein stain. A. Gel stained using Pro-Q Diamond dye to detect phosphoproteins.
B. The same gel post-stained with Sypro Ruby to visualize total protein. The mouse sex for each lane is indicated at the bottom on panel A and B.
(TIF)
Tyrosine phospho-proteome analysis of brain extracts from WT and KO PIMT mice. A, Western blot analysis to visualize the tyrosine-phosphorylated proteins with an anti-phosphotyrosine antibody. B, Coomassie Blue stain for total protein on the same membrane. The mouse sex for each lane is indicated at the bottom on panel A and B.
(TIF)