

Abstract
We investigated the effect of β-carotene (bC) supplementation during pregnancy in a mouse model of severe vitamin A deficiency, i.e. Lrat−/−Rbp−/− dams maintained on a vitamin A-deficient diet during gestation. bC, a provitamin A carotenoid, can be enzymatically cleaved to form vitamin A for use by the developing embryo. We found that an acute supplementation (13.5 days post coitum, dpc) of bC to Lrat−/−Rbp−/− dams on a vitamin A-deficient diet activated transcriptional mechanisms in the developing tissues to maximize the utilization of bC provided to the dams. Nevertheless, these regulatory mechanisms are inefficient under this regimen, as the embryonic phenotype was not improved. We further investigated the effect of a repeated supplementation of bC during a crucial developmental period (6.5–9.5 dpc) on the above-mentioned mouse model. This treatment improved the embryonic abnormalities, as 40% of the embryos showed a normal phenotype. In addition, analysis of retinoic acid-responsive genes, such as Cyp26a1 in these embryos suggests that bC cleavage results in the production of retinoic acid which then can be used by the embryo. Taken together, these in vivo studies show that bC can be used as a source of vitamin A for severely vitamin A-deficient mammalian embryos.
Keywords: placenta, embryo, β-carotene, vitamin A deficiency, Lrat−/−Rbp−/−, retinoids
INTRODUCTION
Adequate maternal nutrition during pregnancy is critical for fetal health (1,2). One such essential nutrient is vitamin A, of which maternal dietary intake must be carefully monitored, as deficient or excessive vitamin A intake can cause a range of birth defects (3). Furthermore, a poor pre-pregnancy maternal vitamin A status (i.e. low stores of the fat-soluble vitamin in body tissues) reduces the availability of vitamin A to be mobilized from the maternal liver to the fetus during times of inadequate dietary intake of this nutrient, and thus increases the risk of fetal vitamin A deficiency (VAD) (4).
VAD among women of child-bearing age is a serious global health problem. According to the World Health Organization, nearly 10 million pregnant women worldwide suffer from night blindness, and ~20 million have low serum retinol levels (5). Even in more developed countries (e.g. UK), 30% of women between 19 and 34 years of age have reported vitamin A intakes below the recommended lower limit (6). In many countries where VAD is prevalent, there is limited access to preformed vitamin A (retinol, retinyl esters, and retinoic acid) from meat and dairy products. On the other hand, plant products containing the vitamin A precursors (provitamin A carotenoids such as β-carotene [bC]) are more abundant (6). Various human studies have reported neutral or positive maternal and fetal health outcomes upon vitamin A supplementation, but fewer studies have tested the effects of bC supplementation exclusively (7). The Hohenheim consensus conference of 2009 recommended that some of the dietary vitamin A be obtained as bC (at least 6 mg/day in the case of low retinoid intake) (6), but it is unclear under what conditions bC alone can deliver adequate amounts of vitamin A. Thus, it is important to understand whether supplementation with carotenoids is sufficient to support normal embryogenesis, under conditions of both maternal dietary VAD and vitamin A-deficient status.
Ingested vitamin A (as retinyl esters) and bC (up to 45% in its intact form, with the remainder being converted to vitamin A (8)) are taken up by enterocytes, and enter the lymphatic system packaged in chylomicrons (9). In the vascular endothelium, chylomicron hydrolysis by lipoprotein lipase (LPL) generates chylomicron remnants, which still contain bC and retinyl esters (10). The majority (~75%) of retinyl esters in chylomicron remnants is taken up by the liver (11) and hydrolyzed to retinol (12), either to be stored in stellate cells after re-esterification by lecithin:retinol acyltransferase (LRAT) (13,14), or to be re-secreted bound to retinol-binding protein (RBP) (15). The remaining 25% of retinyl esters from chylomicron remnants are taken up by peripheral tissues, including the placenta (11). In addition, exchange of bC and vitamin A may also occur among lipoprotein particles (HDL, LDL, VLDL) in the bloodstream (16). Tissue vitamin A needs are met either by uptake of retinoids and their precursors from circulating lipoproteins, or by uptake of RBP-retinol via STRA6, the cell-surface receptor for holo-RBP (17). Most cells (including those of the placenta and embryo (4)), are capable of esterifying retinol via LRAT for storage (18). Alternatively, cellular retinol can be reversibly oxidized to retinaldehyde via retinol dehydrogenases (e.g. RDH10 (19,20)). Retinaldehyde also can be generated by bC cleavage. Central cleavage by β-carotene-15,15′-oxygenase (CMO1) generates two molecules of retinaldehyde (21), while eccentric cleavage by β-carotene-9′,10′-oxygenase (CMO2) (22) followed by chain shorting of β-apo-carotenoids (23) ultimately can generate one molecule of retinaldehyde. The latter is irreversibly oxidized by retinaldehyde dehydrogenases (e.g. RALDH2) to retinoic acid (24), the biologically active vitamin A metabolite that regulates the transcription of hundreds of genes (25). Retinoic acid can be converted to non-transcriptionally active metabolites by CYP26A1 (26).
Previous studies in mice have elucidated that maternal circulating bC can be taken up by embryos via the placenta, to support a large degree of normal embryogenesis in the absence of other vitamin A sources (27). However, prior work has not demonstrated to what extent bC can contribute to embryonic vitamin A needs when the pregnant mother is vitamin A-deficient (i.e. by status). Our lab has generated and described a model of marginal VAD, the Lrat−/−Rbp−/− strain (L−/−R−/−). While phenotypically normal on a vitamin A-sufficient diet, these mice rapidly become vitamin A-deficient and generate highly malformed embryos when deprived of dietary vitamin A, due to their inability to store retinoids via LRAT or mobilize retinol via RBP (4). Recently, we showed that on a vitamin A-sufficient diet, placental bC uptake was regulated by different mechanisms in L−/−R−/− and wild-type (WT) mice, due to the marginal vitamin A-deficient status of the L−/−R−/− dams (28). These results indicated that the maternal vitamin A status affects bC uptake and metabolism in the developing tissues.
In the present study, we use WT and L−/−R−/− mice to investigate the effects of dietary VAD or a vitamin A-deficient tissue status, respectively, on the uptake and processing of bC by maternal and embryonic tissues as well as the extent to which such vitamin A-deficient tissues can generate retinoids from bC to support normal embryogenesis. Even severely vitamin A-deficient developing tissues respond to bC supplementation by maximizing proper utilization of the provitamin A as a source of retinoid to support embryogenesis. However, improvement of the embryonic malformations can be achieved only when bC is provided to the dams at early stages of development. This regimen is also effective at improving the maternal vitamin A status.
MATERIALS AND METHODS
Knockout Mice, Nutritional manipulation, and β-carotene supplementation
Wild-type (WT) and Lrat−/−Rbp−/− (L−/−R−/−) double knockout mice (4,28) were used in the current study. All mice had a mixed genetic background (C57BL/6 x sv129), with the L−/−R−/− being a model of VAD (4). Throughout the study, both water and diet were consumed ad libitum, and mice were maintained on a 12 hour light/dark cycle from 7 a.m. to 7 p.m. Experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (29) and were approved by the Rutgers University Institutional Committee on Animal Care.
Prior to nutritional manipulation, all mice were maintained on a non-purified diet copious in vitamin A (29 IU/g, Prolab Isopro RMH3000 5p75) but devoid of bC (trace to 2.6 μg/g). At 3 months of age, WT and L−/−R−/− females were mated with their respective males, and the presence of a vaginal plug was established as 0.5 days post coitum (dpc). Henceforward, dams were fed a purified vitamin A-deficient diet (Research Diets, <0.2 IU/g) until the time of sacrifice (14.5 dpc). Solutions of bC or its Vehicle (Veh) were prepared as previously described (28). Briefly, 50 mg bC (Type II, Sigma Aldrich) was mixed into 5 mL vehicle (ethanol : Cremophor : PBS, 1:11:18) by vortexing, and the concentration of the resulting solution was determined by spectrophotometry at 450 nm. Due to poor solubility of bC, the final concentration varied from 2–5 mg/mL.
For the acute bC supplementation study, WT and L−/−R−/− pregnant dams were randomly assigned to the Veh or bC treatment groups, and were injected with 250 μL of the assigned solution intraperitoneally (IP) at 13.5 dpc. For the repeated bC supplementation study, L−/−R−/− pregnant dams were randomly assigned to be given an IP injection of Veh or bC daily from 6.5–9.5 dpc. The resulting dose of one injection of bC given to the pregnant dams was ~40 μg/g body weight. Regardless of treatment, all dams were sacrificed at 14.5 dpc by CO2 inhalation between 9:30 and 11:30 a.m. Serum and tissues (livers, placentas, and embryos) were collected, frozen, and stored at −80°C until further processing.
HPLC
Retinoid (retinol and retinyl esters) and bC concentrations in maternal serum, liver, placenta and embryo were measured by reversed-phase HPLC analysis as described previously (4,30).
RNA Extraction, cDNA synthesis, and Quantitative Real-time RT-PCR
RNA Extraction, cDNA synthesis, and quantitative real-time RT-PCR (qRT-PCR) were performed on embryos and placentas as previously described (4). Primer sequences were as published for β-Actin, Stra6, Raldh2, Cyp26a1 (4), Cmo1 (28), Cmo2 (31), and Rdh10 (32). Changes in mRNA expression were analyzed by the ΔΔCT method.
Statistical analysis
Statistical analysis was performed using SPSS statistical software (IBM SPSS Statistics, version 16). Normal distribution of data was assessed by the Shapiro-Wilk test. Normally distributed data were analyzed by Student’s t test for comparisons of two groups, or by two-way ANOVA and post hoc analysis (least significant difference, LSD, for groups with equal variance; Tamhane’s for groups with unequal variance) for comparisons of genotype and treatment effects, followed by Student’s t test. Data that were not normally distributed were analyzed by the Mann-Whitney U test for comparisons of two groups, or by the Kruskal-Wallis test followed by Mann-Whitney U test for comparisons of three or more groups. P < 0.05 was considered significant. Data are presented as mean ± standard deviation (SD).
RESULTS
We previously showed that the placental uptake of bC is enhanced in an established model of marginal VAD such as the L−/−R−/− mice fed a vitamin A-sufficient diet (4,28). Here we employed the same strain to understand whether a severe maternal vitamin A-deficient status would affect the uptake and metabolism of bC in the developing tissues. Indeed, on a vitamin A-deficient diet, L−/−R−/− mice rapidly become severely vitamin A-deficient, whereas the retinoid stores of WT mice are not readily depleted (4).
Acute maternal supplementation with bC at 13.5 dpc
We injected ~40 μg/g bC or Veh at 13.5 dpc into WT and L−/−R−/− dams fed a VAD diet from the onset of gestation, and analyzed their embryos at 14.5 dpc. A single injection of Veh or bC at 13.5 dpc did not support normal embryogenesis in the L−/−R−/− group, which generated severely malformed embryos with a large percentage of resorptions (Table 1). Embryonic defects at 14.5 dpc included malformed eyes, peripheral edema, cleft face/palate, and/or shortened forelimbs (Fig. 1A–D). Under the same dietary regimen and treatment, WT embryos developed normally (data not shown).
Table 1.
Phenotype distribution of L−/−R−/− embryos from dams fed a vitamin A-deficient diet, supplemented with Veh or bC at 13.5 dpc
| Treatment | n [embryos, (dams)] | Percent Resorbed | Normal | Eye and/or Edema | Cleft |
|---|---|---|---|---|---|
| Veh 13.5 | 20 (7) | 56% (25/45) | 0% | 0% | 100% |
| bC 13.5 | 31 (10) | 54% (37/69) | 0% | 13% | 87% |
L−/−R−/− dams were fed a vitamin A-deficient diet from 0.5–14.5 dpc. Seven dams were injected with Veh at 13.5 dpc (Veh 13.5), and 10 dams were injected with bC at 13.5 dpc (bC 13.5). Percentage of resorbed embryos was calculated from the total number of implantations in the uterus.
Figure 1. Effects of β-carotene supplementation on gross morphology of L−/−R−/− embryos from dams fed a vitamin A-deficient diet from 0.5–14.5 dpc.
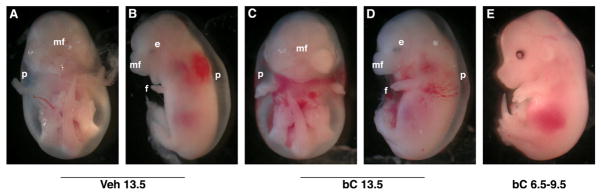
Embryos (14.5 dpc) from Lrat−/−Rbp−/− (L−/−R−/−) dams fed a vitamin A-deficient diet during pregnancy (0.5–14.5 dpc) were analyzed following a single dose at 13.5 dpc (C–D) or multiple doses daily at 6.5–9.5 dpc (E) of bC, compared with Veh (A–B). Embryonic defects include malformed eye (e), peripheral edema (p), shortened forelimbs (f), and abnormal midfacial region (mf).
HPLC analysis revealed that bC-supplemented WT and L−/−R−/− dams had similar concentrations of bC in maternal serum, liver, placenta and embryo (Table 2). L−/−R−/− liver bC displayed a non-significant tendency to increase compared to WT (p=0.065).
Table 2.
Serum and tissue β-carotene levels (14.5 dpc) of WT and L−/−R−/−dams fed a Vitamin A-deficient diet from 0.5–14.5 dpc, and injected with bC at 13.5 or 6.5–9.5 dpc
| Serum μg/dL | Liver μg/g | Placenta μg/g | Embryo μg/g | |
|---|---|---|---|---|
| WT, bC 13.5 | 541 ± 129 | 57 ± 27 | 1.53 ± 0.42 | 0.019 ± 0.01 |
| L−/−R−/−, bC 13.5 | 624 ± 502 | 117 ± 43 | 1.81 ± 0.60 | 0.018 ± 0.01 |
| L−/−R−/−, bC 6.5–9.5 | 1.37 ± 0.68* | 389 ± 131* | 2.59 ± 0.76 | 0.008 ± 0.007 |
WT and L−/−R−/− dams were fed a vitamin A-deficient diet from 0.5–14.5 dpc. Dams were injected with bC at 13.5 dpc (bC 13.5), or daily from 6.5–9.5 dpc (bC 6.5–9.5). Data presented as mean ± SD. Statistical comparisons were made between WT and L−/−R−/− bC 13.5, and between L−/−R−/− 13.5 and L−/−R−/− 6.5–9.5. Statistical analysis for WT vs. L−/−R−/− 13.5 data by Student’s t test [serum (equal variance not assumed), placenta, embryo], or Mann-Whitney U test (liver). Statistical analysis for L−/−R−/− 13.5 vs. 6.5–9.5 by Student’s t test. Sample size, n=3–11 dams/group; Placental and Embryonic levels include more than one embryo from each litter.
p<0.05 vs. L−/−R−/− bC 13.5.
Further HPLC analysis indicated that hepatic, placental, and embryonic retinol levels were significantly reduced in L−/−R−/− mice compared to WT, regardless of the treatment given at 13.5 dpc (Table 3). Hepatic retinol levels increased modestly but significantly following bC supplementation of L−/−R−/− dams, even though they did not reach the WT level. Placental retinol levels were significantly reduced upon bC injection of WT dams compared to Veh, whereas a trend towards an increase (p=0.057) was observed upon bC injection of L−/−R−/− dams compared to Veh. However, bC supplementation did not alter embryonic retinol levels either in WT or in L−/−R−/− mice.
Table 3.
Tissue Retinol levels (14.5 dpc) of WT and L−/−R−/− dams fed a Vitamin A-deficient diet from 0.5–14.5 dpc, and injected with Veh or bC at 13.5 dpc
| Liver μg/g | Placenta ng/g | Embryo ng/g | |
|---|---|---|---|
| WT, Veh 13.5 | 2.4 ± 0.6 | 166 ± 44 | 81.0 ± 13.0 |
| WT, bC 13.5 | 1.8 ± 0.4 | 112 ± 30# | 74.1 ± 9.3 |
| L−/−R−/−, Veh 13.5 | 0.6 ± 0.2* | 22 ± 2* | 2.7 ± 1.9* |
| L−/−R−/−, bC 13.5 | 0.9 ± 0.4*# | 79 ± 22* | 7.2 ± 7.2* |
WT and L−/−R−/− dams were fed a vitamin A-deficient diet from 0.5–14.5 dpc. Dams were injected with Vehicle (Veh 13.5) or β-carotene at 13.5 dpc (bC 13.5). Data presented as mean ± SD. Sample size: liver, n=5–8 dams/group; multiple placentas and embryos per dam (n=1–5 dams/group) were analyzed for n=3–12 placentas or embryos/group. Statistical analysis by two-way ANOVA + LSD/Tamhane’s post hoc, followed by Student’s t test (liver + embryo) or by Kruskal-Wallis + Mann-Whitney U test (placenta).
p<0.05 vs. WT;
p<0.05 vs. Veh.
Our lab has previously reported that acute bC supplementation of vitamin A-sufficient dams at 13.5 dpc alters the mRNA expression of genes involved in both uptake and metabolism of bC in placenta in order to maintain homeostatic retinoid levels in the placental-fetal unit (28). To determine whether such transcriptional events persist during maternal VAD, we performed qRT-PCR analysis on the placentas and embryos of WT and L−/−R−/− dams fed the vitamin A-deficient diet and injected with bC or Veh at 13.5 dpc. As shown in Figure 2A, Cmo1 mRNA expression in the placenta of Veh-treated L−/−R−/− dams was significantly greater than that of WT; however, upon bC injection, mRNA expression was reduced (p=0.05), though not to the WT level. The asymmetric bC cleavage enzyme, Cmo2, was similar in placentas of both genotypes, regardless of treatment (Fig. 2A). The mRNA expression of placental Rdh10 and Raldh2 were ~50% of WT Veh-treated in L−/−R−/− placentas, regardless of treatment (Fig. 2A). In contrast, bC treatment in WT dams reduced placental Rdh10 mRNA by ~50% compared to WT Veh-treated dams. Placental mRNA levels of Cyp26a1 in bC-injected L−/−R−/− dams showed a trend to differ from WT bC-treated (p=0.061), while Stra6 mRNA expression levels were similar in placentas of both genotypes, regardless of treatment (Fig. 2A).
Figure 2. Placental and embryonic mRNA expression levels of genes involved in β-carotene cleavage and retinoid homeostasis following β-carotene supplementation at 13.5 dpc.
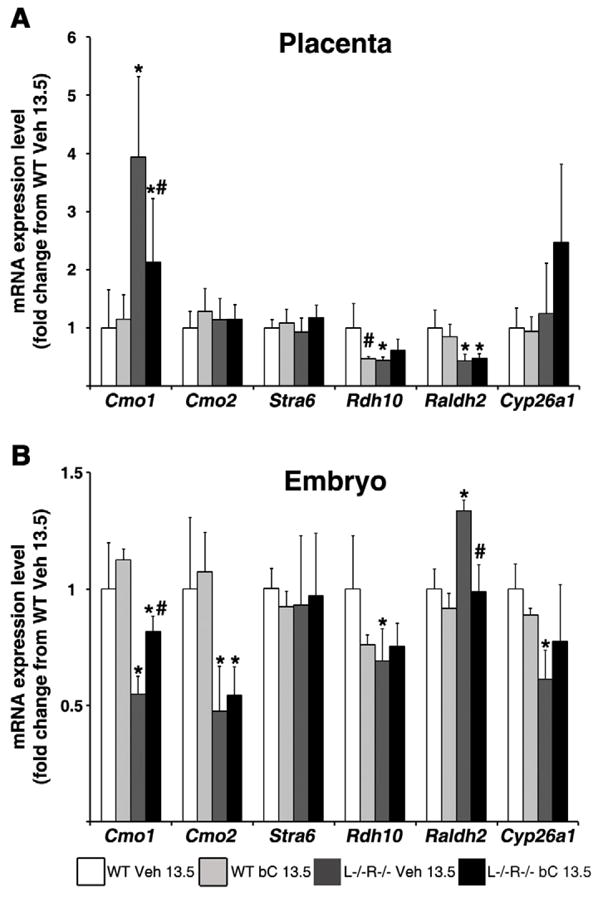
qRT-PCR analysis was performed using mRNA from 14.5 dpc placentas (A) and embryos (B) from wild-type (WT) and Lrat−/−Rbp−/− (L−/−R−/−) dams treated at 13.5 dpc with Vehicle (Veh 13.5) or β-carotene (bC 13.5). Tissues of WT Veh 13.5 were set as calibrator at 1. Data are presented as mean ± SD fold of WT Veh 13.5. Sample size, n=5–10 placentas or embryos/group (from 3–5 dams/group). Statistical analysis was performed by two-way ANOVA with genotype and treatment as factors, followed by LSD or Tamhane’s post hoc analysis. Individual comparisons then were made by Student’s t test. *, p<0.05 vs. WT; #, p<0.05 vs. Veh.
Analysis of mRNA expression in embryos (Fig. 2B) revealed that Cmo1 mRNA levels were ~50% lower in L−/−R−/− Veh compared to WT, and increased significantly upon bC injection without reaching the WT level. Cmo2 mRNA expression was half of the WT level in L−/−R−/− embryos, regardless of treatment (Fig. 2B). Rdh10 mRNA levels showed a non-significant tendency (p=0.076) to decrease in bC-treated WT embryos compared to WT Veh, and were reduced in L−/−R−/− Veh compared to WT but did not increase upon bC treatment (Fig. 2B). Raldh2 mRNA expression was significantly greater in L−/−R−/− Veh embryos compared to WT, but was reduced to the WT level upon bC treatment of the L−/−R−/− dams (Fig. 2B). Cyp26a1 mRNA was significantly reduced in L−/−R−/− Veh embryos compared to WT, whereas embryonic mRNA levels of Stra6 were unaffected by genotype or treatment (Fig. 2B).
All together these results suggest that even severely vitamin A-deficient developing tissues activate transcriptional mechanisms to maximize the utilization of bC provided to the dams. Nevertheless, these regulatory mechanisms appear to be rather inefficient under this regimen of maternal provitamin A supplementation as the embryonic phenotype was not improved.
Repeated maternal supplementation with bC from 6.5–9.5 dpc
As bC supplementation at mid-gestation (13.5 dpc) was subsequent to the majority of organogenesis (33), and thus unable to ameliorate the gross morphological defects of the vitamin A-deficient L−/−R−/− embryos, we next performed a more prolonged supplementation experiment during the critical window of mouse organ development (33). L−/−R−/− pregnant females were fed a vitamin A-deficient diet from 0.5–14.5 dpc, and supplemented with ~40 μg/g bC or Veh daily from 6.5–9.5 dpc by IP injection. bC supplementation dramatically improved the phenotype of L−/−R−/− embryos, as the cleft palate was no longer observed, 38% of embryos developed as grossly phenotypically normal (Fig. 1E), and the percentage of resorptions was reduced in comparison to Veh-treated animals (Table 4). At 14.5 dpc, L−/−R−/− dams injected from 6.5–9.5 dpc displayed higher concentrations of bC in liver, while serum bC levels were less than those described above for dams injected at 13.5 dpc (Table 2). Table 5 shows the increased hepatic retinol concentration of L−/−R−/− dams injected with bC from 6.5–9.5 dpc compared to Veh-injected controls. In contrast, embryonic and placental bC (Table 2) and retinol (Table 5) were not significantly altered by prolonged bC treatment.
Table 4.
Phenotype distribution of L−/−R−/− embryos from dams fed a vitamin A-deficient diet, supplemented with Veh or bC from 6.5–9.5 dpc
| Treatment | n [embryos, (dams)] | Percent Resorbed | Normal | Eye and/or Edema | Cleft | Exencephaly |
|---|---|---|---|---|---|---|
| Veh 6.5–9.5 | 22 (6) | 51% (23/45) | 0% | 18% | 82% | 0% |
| bC 6.5–9.5 | 39 (8) | 31% (19/58) | 38% | 59% | 0% | 3% |
L−/−R−/− dams were fed a vitamin A-deficient diet from 0.5–14.5 dpc. Six dams were injected with Veh daily from 6.5–9.5 dpc (Veh 6.5–9.5); 8 dams were injected with bC daily from 6.5–9.5 dpc (bC 6.5–9.5). Percentage of resorbed embryos was calculated from the total number of implantations in the uterus. Note that 3% exencephaly represents a single embryo, as n=39.
Table 5.
Tissue Retinol levels (14.5 dpc) of L−/−R−/−dams fed a Vitamin A-deficient diet from 0.5–14.5 dpc, and injected with Veh or bC 6.5–9.5 dpc
| Liver μg/g | Placenta ng/g | Embryo ng/g | |
|---|---|---|---|
| L−/−R−/−, Veh 6.5–9.5 | 0.4 ± 0.1 | 67 ± 32 | 5.6 ± 4.7 |
| L−/−R−/−, bC 6.5–9.5 | 0.7 ± 0.2# | 113 ± 42 | 26.2 ±12.1 |
L−/−R−/− dams were fed a vitamin A-deficient diet from 0.5–14.5 dpc, and injected with Vehicle (Veh) or β-carotene (bC) daily from 6.5–9.5 dpc. Data presented as mean ± SD. Statistical analysis by Student’s t test (liver, old placenta) or Mann-Whitney U test (old embryo). Sample size, n=3–7 dams; Placental and Embryonic levels include more than one embryo from each litter.
p<0.05 vs. L−/−R−/− Veh 6.5–9.5.
Since the external developmental defects were fully rescued in 38% of L−/−R−/− embryos from dams given bC from 6.5–9.5 dpc, we wondered whether key mediators of retinoid homeostasis had facilitated the improved phenotype. Thus, we performed qRT-PCR on malformed L−/−R−/− Veh embryos and normal L−/−R−/− bC embryos at 14.5 dpc, along with their respective placentas. Figure 3A shows similar placental mRNA expression levels of Cmo1, Stra6, Rdh10, Raldh2, and Cyp26a1 at 14.5 dpc between Veh and bC-treated dams; however, Cmo2 mRNA expression was significantly reduced in the placentas of bC-injected dams. Only Cyp26a1 embryonic mRNA expression increased significantly in bC-treated dams compared to Veh, whereas all other genes analyzed were similar in embryos between the groups (Fig. 3B).
Figure 3. Placental and embryonic mRNA expression levels of genes involved in β-carotene cleavage and retinoid homeostasis following β-carotene supplementation from 6.5–9.5 dpc.

qRT-PCR analysis was performed using mRNA from 14.5 dpc placentas (A) and embryos (B) from L−/−R−/− dams treated from 6.5–9.5 dpc with Veh or bC. Tissues of L−/−R−/− Veh 6.5–9.5 were set as calibrator at 1. Data presented as mean ± SD fold of L−/−R−/− Veh 6.5–9.5. Sample size, n=5–6 embryos or placentas/group from different dams. Statistical analysis was performed by Student’s t test. #, p<0.05 vs. L−/−R−/− Veh 6.5–9.5.
Taken together, these results confirm the ability of severely vitamin A-deficient tissues to utilize bC provided to the dams at early stages of development. Such a developmental window (6.5–9.5 dpc) is optimal to achieve gross improvement in embryogenesis.
DISCUSSION
Both maternal dietary vitamin A intake and tissue status are critical determinants of embryonic development (4). Studies in both humans and mice have demonstrated that VAD is associated with cleft face/palate and defects in tissue development, including the heart and limbs (3,34). The world’s population relies on dietary intake of both preformed vitamin A and provitamin A carotenoids (including bC) to ensure adequate vitamin A levels (6). At term, correlations among maternal serum bC, placental and cord serum retinol have been observed only in mothers with subadequate serum vitamin A levels (35). Thus, scarce maternal tissue retinoid and carotenoid stores jeopardize the adequate supply of retinoids to the developing embryo, especially if dietary vitamin A is limiting. The L−/−R−/− mouse is a useful model to examine the uptake and function of supplemented bC during pregnancy when maternal vitamin A stores are limiting (28).
Our lab has previously shown that embryonic CMO1 can generate retinoids locally from maternal circulating bC, to support normal embryonic development (27). In the current study, we compared the ability of a single (13.5 dpc) or repeated (6.5–9.5 dpc) doses of bC to maintain embryonic retinoid homeostasis in a model of maternal dietary VAD (WT on vitamin A-deficient diet) and a model of severe maternal vitamin A-deficient status (L−/−R−/− on vitamin A-deficient diet). Given the dependence of embryonic patterning and organ development on retinoic acid (3), we were not surprised that the malformations in L−/−R−/− embryos persisted following a single maternal bC injection after the majority of organogenesis was complete (e.g. at 13.5 dpc (33); Table 1 and Fig. 1). Interestingly, despite the malformed appearance of the L−/−R−/− embryos following the single bC injection, supplemental bC improved the retinoid status of vitamin A-deficient dams to a limited degree, even at a time when it was too late to support normal embryogenesis (retinol levels increased in maternal liver in the supplemented group; Table 3). While a small percentage of embryos from dams supplemented with bC at 13.5 dpc displayed a milder phenotype than those treated with Veh (eye defect and edema, versus cleft face/palate, Table 1), we believe this difference is physiologically insignificant, as even embryos from dams treated with Veh from 6.5–9.5 dpc showed a low percentage of eye defects and edema (Table 4). In contrast, when bC was supplemented during the window of organogenesis (6.5–9.5 dpc) when the retinoid requirements are higher (36), L−/−R−/− embryos no longer showed cleft face/palate, but a shift towards a less severe phenotype (eye defect and edema) was observed, and nearly 40% of them appeared grossly normal (Table 4). In addition to this dramatic improvement of L−/−R−/− external morphology, the resorption percentage of these embryos became similar to our previously published results for vitamin A-sufficient L−/−R−/− dams (4), further reinforcing that a sustained bC supplementation during early development of otherwise vitamin A-deficient dams mimics a vitamin A-sufficient state.
We observed a lower recovery of normal embryos from bC-supplemented L−/−R−/− dams (38%) compared to our published results of Cmo1+/−Rbp−/− embryos from supplemented Cmo1−/−Rbp−/− dams (61%, (27)) – a result that can be understood in light of the maternal expression of CMO1. In the case of Cmo1−/−Rbp−/− dams, all of the injected bC can be delivered to their Cmo1+/−Rbp−/− embryos, to be cleaved locally via the enzyme produced by their single genomic copy of Cmo1. On the other hand, the L−/−R−/− dams express CMO1 in all of their tissues, and thus can cleave a significant portion of the supplemental bC (for example, in the liver) before it reaches the embryo. Indeed, the maternal serum bC concentration at 14.5 dpc in the L−/−R−/− dams (1.43 ± 0.65 μg/dL, Table 2) is approximately 5-fold lower than that of the reported Cmo1−/−Rbp−/− dams (7.0 ± 2.3 μg/dL, (27) injected under the same protocol. Furthermore, the bC-derived retinoids of L−/−R−/− mice cannot be transported to their embryos due to their lack of RBP.
We have previously shown that maternal dietary vitamin A intake and status alter embryonic mRNA expression of numerous retinoic acid-responsive genes responsible for maintaining retinoid homeostasis in the developing tissues, including Raldh2, Cyp26a1, and Stra6 (4). In addition, we recently demonstrated that maternally supplemented bC also regulates placental and embryonic mRNA expression of genes involved in bC cleavage and uptake to maintain retinoid homeostasis (28). Thus, we wondered to what extent such regulatory transcriptional events would take place (and be effective) upon bC supplementation in our model of severe maternal vitamin A-deficient intake and status.
We previously showed that the placenta of L−/−R−/− mice expresses high levels of Cmo1 under a vitamin A-sufficient diet compared to that of WT (28), and the results of the current study are in agreement with this finding, suggesting the likelihood that a constitutively elevated CMO1 activity may exist in the L−/−R−/− placenta. In this case, bC supplementation could lead to elevated retinoic acid levels (generated upon symmetric bC cleavage), which might then suppress Cmo1 via a negative feedback loop similar to that reported in intestine (37), leading to reduced levels of Cmo1 in the placenta of bC-supplemented L−/−R−/− (Fig. 2A). This down-regulation of placental Cmo1 may help preserve intact bC for delivery to the severely vitamin A-deficient L−/−R−/− embryos, which in turn could cleave bC locally and produce the much needed retinoic acid. Further studies investigating this mechanism in the placenta need to be conducted.
Rdh10 and Raldh2 control the two oxidative steps that result in the synthesis of retinoic acid from retinol (19,20,24). Specifically, Rdh10 catalyzes the conversion of retinol into retinaldehyde and Raldh2 the oxidation of retinaldehyde to retinoic acid. The placental mRNA expression of Rdh10 and Raldh2 in L−/−R−/− were lower than that of the WT Veh-treated placenta (Fig. 2A). The reduced level of Raldh2 is in agreement with our previous study (4). This transcriptional down-regulation may be a mechanism to help spare placental retinoids (including endogenous) for delivery to the severely vitamin A-deficient embryos, where they can ultimately be converted to retinoic acid. The lack of effect of bC supplementation on Rdh10 and Raldh2 in L−/−R−/− placentas may indicate that the 24-hour time point (14.5 dpc) is too early to observe feedback inhibition of retinoic acid (produced by bC cleavage) on these genes in such severely vitamin A-deficient tissues. Alternatively, this retinoic acid may be shunted to the developing embryo rather than used by the placenta. bC cleaved to retinaldehyde could be transported via CRBP1/RBP/albumin to the embryo or reduced to retinol, which too can be transported to the embryo via CRBP1/RBP/albumin for local production of retinoic acid in the vitamin A-deficient embryo (38–41).
The baseline reduction of Cmo1 in L−/−R−/− embryos (Fig. 2B) supports our results in vitamin A-sufficient dams lacking both LRAT and RBP (28), although the reduction is more dramatic in the vitamin A-deficient embryos. This result yet again indicates an effect of the lack of LRAT on Cmo1 that is tissue-specific (Cmo1 is reduced in the embryo [Fig. 2B] and increased in the placenta [Fig. 2A] in the absence of Lrat) – a phenomenon that is actively under investigation in our laboratory. The increase in L−/−R−/− embryonic Cmo1 mRNA levels upon bC supplementation is likely due to increased CMO1-mediated retinoic acid production (i.e. positive feedback). Indeed, at least in certain tissues, Cmo1 mRNA expression is positively regulated by PPARγ (42), and Pparγ itself is up-regulated by retinoic acid (43). However, this is the first report of a suppression of Cmo2 mRNA expression in the absence of embryonic LRAT (Fig. 2B). The bC-independent appearance of these mRNA reductions (note that bC is not present in the purified diet used in this experiments) suggests that a mechanism other than retinoic acid-mediated transcriptional regulation is responsible for the changes in mRNA levels. The lack of effect of bC supplementation on Cmo2 mRNA levels re-iterates the centrality of CMO1 as the main bC cleavage enzyme in vivo (44).
In a recent study by Sandell et al. (20), a potential retinoic acid response element (RARE) was identified in exon 5 of Rdh10, and reduced retinoic acid was shown to increase mRNA levels of embryonic Rdh10 in the vitamin A-deficient Raldh2−/− mouse at 9.5 dpc. Indeed, in our study, Rdh10 expression was reduced in WT embryos from bC-treated dams, suggesting that production of retinoic acid from bC cleavage may lower Rdh10 (Fig. 2B). Unlike our WT data or those reported by Sandell et al., Rdh10 embryonic mRNA levels did not differ between L−/−R−/− bC and Veh, although both were lower than WT Veh (Fig. 2B). We hypothesize that 24 hours post-injection is too early to detect negative feedback of Rdh10 in severely vitamin A-deficient tissues (L−/−R−/−).
The up-regulation of Raldh2 in vitamin A-deficient L−/−R−/− embryos (Fig. 2B) confirms our published results (4), while its reduction upon bC supplementation may indicate that increased retinoic acid production by CMO1 and the potential transport of retinol/retinaldehyde from the placenta lower Raldh2 to WT levels through negative feedback (24,45). Our earlier work also showed that embryonic Cyp26a1 mRNA levels were lower in vitamin A-deficient L−/−R−/− mice compared either to vitamin A-sufficient WT mice, or to L−/−R−/− mice on vitamin A-sufficient or vitamin A-excess diets (220 IU/g of diet) (4). The current study supports this finding, as L−/−R−/− Veh vitamin A-deficient embryos had lower Cyp26a1 mRNA levels than WT Veh (Fig. 2B), suggesting insufficient production of retinoic acid to support normal embryonic development.
When a sustained (6.5–9.5 dpc) maternal bC supplementation was performed we were only able to evaluate the transcriptional response of the developing tissues five days after the last injection of bC. This is likely the reason for the apparent difference in the transcriptional regulatory events observed in the developing tissues depending on the window of bC supplementation. In placenta, none of the tested genes showed changes in mRNA levels, except for Cmo2 (Fig. 3A). Given the persistence of circulating bC and the amount in the placenta at 14.5 dpc (Table 2), the down-regulation of Cmo2 is interesting, and may favor the delivery of intact bC to the embryo rather than scavenging it (44) in the placenta (Fig. 3A). Among all the genes tested in the embryo, only Cyp26a1, which encodes the enzyme that catabolizes retinoic acid into transcriptionally inactive forms (26), was significantly up-regulated when L−/−R−/− dams were injected four times with bC (Fig. 3B). This significant increase in embryonic Cyp26a1 at 14.5 dpc may signal that sufficient retinoic acid has been generated (supported by the normal phenotype of these embryos, Table 4), and that any further retinoic acid should now be catabolized. The more dramatic increase in embryonic Cyp26a1 mRNA levels when bC is supplemented for four days (Fig. 3B), compared to a single dose (Fig. 2B), is reinforced by the lack of phenotype improvement following the single dose (Table 1). Future studies using the same experimental design and analyzing genes involved in retinoid homeostasis at 10.5 dpc for example, would provide further information on the acute changes following four days of bC supplementation during the critical stages of development. It is noteworthy that the repeated supplementation with bC also improves the maternal vitamin A status as indicated by the elevated bC levels in the livers and placentas (mildly) of L−/−R−/− dams on the above-mentioned regimen.
Although our previous results indicated that Stra6 mRNA levels were reduced in vitamin A-deficient L−/−R−/− placentas and embryos compared to the same genotype on vitamin A-sufficient or vitamin A-excess diets (4), we did not observe any difference in Stra6 mRNA levels among any groups in the present study. It may be that concentrations of preformed vitamin A, rather than provitamin A carotenoids (e.g. bC), modulate the transcription of Stra6. While Stra6 is so named because it is “stimulated by retinoic acid”, our lab also has published evidence that Stra6 mRNA expression responds to the need to efflux excessive intracellular retinol (4). Thus, although bC is indeed generating retinoic acid in bC-supplemented embryos (supported by changes in Cyp26a1 mRNA levels and embryonic phenotype following the prolonged treatment of L−/−R−/− dams), cellular retinol is probably not accumulating in this system. However, in support of the previous study, WT and L−/−R−/− mRNA levels of Stra6 were similar in placentas and embryos on the vitamin A-deficient diet (Fig. 2).
The current study highlights the importance of bC as a precursor of vitamin A during embryonic development. Even severely vitamin A-deficient developing tissues activate transcriptional mechanisms which aim to maximize the use of bC, the efficiency of which depends on the developmental window of bC supplementation. Our findings are important in view of the possibility that adequate amounts of bC given during pregnancy can attenuate the effects of VAD, a problem affecting millions of people worldwide.
Vitamin A-deficient (VAD) mouse embryos can use β-carotene (bc) for development.
Prolonged, rather than acute bC supplementation ameliorates cleft palate in VAD embryos.
bC treatments affect retinoic acid responsive genes in placenta and embryos.
bC treatment increases hepatic retinol in VAD dams.
Acknowledgments
This work was supported by grants R01HD057493 and R01HD057493-02S1 from the U.S. National Institute of Health (NIH).
Abbreviations
- dpc
days post coitum
- bC
β-carotene
- WT
Wild-type
- LRAT
lecithin:retinol acyltransferase
- RBP also known as RBP4
retinol-binding protein
- L−/−R−/−
Lrat−/−Rbp−/−
- VAD
vitamin A deficiency
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ramakrishnan U, Grant F, Goldenberg T, Zongrone A, Martorell R. Effect of women’s nutrition before and during early pregnancy on maternal and infant outcomes: a systematic review. Paediatr Perinat Epidemiol. 2012;26:285–301. doi: 10.1111/j.1365-3016.2012.01281.x. [DOI] [PubMed] [Google Scholar]
- 2.Monk C, Georgieff MK, Osterholm EA. Research Review: Maternal prenatal distress and poor nutrition-mutually influencing risk factors affecting infant neurocognitive development. Journal of Child Psychology and Psychiatry. 2013;54(2):115–130. doi: 10.1111/jcpp.12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clagett-Dame M, Knutson D. Vitamin A in Reproduction and Development. Nutrients. 2011;3(4):385–428. doi: 10.3390/nu3040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim YK, Wassef L, Hamberger L, Piantedosi R, Palczewski K, Blaner WS, et al. Retinyl Ester Formation by Lecithin: Retinol Acyltransferase Is a Key Regulator of Retinoid Homeostasis in Mouse Embryogenesis. J Biol Chem. 2008;283(9):5611–5621. doi: 10.1074/jbc.M708885200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. WHO global database on vitamin A deficiency. WHO; Geneva: 2009. Global Prevalence of Vitamin A Deficiency in Populations at Risk 1995–2005. [Google Scholar]
- 6.Grune T, Lietz G, Palou A, Ross AC, Stahl W, Tang G, et al. Beta-Carotene Is an Important Vitamin A Source for Humans. J Nutr. 2010;140(12):2268S–2285S. doi: 10.3945/jn.109.119024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorne-Lyman AL, Fawzi WW. Vitamin A and Carotenoids During Pregnancy and Maternal, Neonatal and Infant Health Outcomes: a Systematic Review and Meta-Analysis. Paediatr Perinat Epidemiol. 2012;26:36–54. doi: 10.1111/j.1365-3016.2012.01284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hickenbottom SJ, Lemke SL, Dueker SR, Lin Y, Follett JR, Carkeet C, et al. Dual isotope test for assessing beta-carotene cleavage to vitamin A in humans. Eur J Nutr. 2002;41(4):141–147. doi: 10.1007/s00394-002-0368-0. [DOI] [PubMed] [Google Scholar]
- 9.Goodman DS, Blomstrand R, Werner B, Huang H, Shiratori T. The intestinal absorption and metabolism of vitamin A and beta-carotene in man. J Clin Invest. 1966;45(10):1615–1623. doi: 10.1172/JCI105468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase-and CD36-mediated pathways. J Lipid Res. 2009;50:S86–S90. doi: 10.1194/jlr.R800085-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman DWS, Huang HS, Shiratori T. Tissue distribution and metabolism of newly absorbed vitamin A in the rat. The Journal of Lipid Research. 1965;6(3):390–396. [PubMed] [Google Scholar]
- 12.Harrison EH, Gad MZ, Ross AC. Hepatic uptake and metabolism of chylomicron retinyl esters: probable role of plasma membrane/endosomal retinyl ester hydrolases. J Lipid Res. 1995;36(7):1498–1506. [PubMed] [Google Scholar]
- 13.MacDonald PN, Ong DE. A lecithin: retinol acyltransferase activity in human and rat liver. Biochem Biophys Res Commun. 1988;156(1):157–163. doi: 10.1016/s0006-291x(88)80818-0. [DOI] [PubMed] [Google Scholar]
- 14.Matsuura T, Gad MZ, Harrison EH, Ross AC. Lecithin: retinol acyltransferase and retinyl ester hydrolase activities are differentially regulated by retinoids and have distinct distributions between hepatocyte and nonparenchymal cell fractions of rat liver. J Nutr. 1997;127(2):218–224. doi: 10.1093/jn/127.2.218. [DOI] [PubMed] [Google Scholar]
- 15.Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, et al. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999;18(17):4633–4644. doi: 10.1093/emboj/18.17.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zilversmit DB, Morton RE, Hughes LB, Thompson KH. Exchange of retinyl and cholesteryl esters between lipoproteins of rabbit plasma. Biochimica et Biophysica Acta (BBA)-Lipids and Lipid Metabolism. 1982;712(1):88–93. doi: 10.1016/0005-2760(82)90088-1. [DOI] [PubMed] [Google Scholar]
- 17.Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315(5813):820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 18.Zolfaghari R, Ross AC. Lecithin: retinol acyltransferase from mouse and rat liver: cDNA cloning and liver-specific regulation by dietary vitamin A and retinoic acid. J Lipid Res. 2000;41(12):2024–2034. [PubMed] [Google Scholar]
- 19.Sandell LL, Sanderson BW, Moiseyev G, Johnson T, Mushegian A, Young K, et al. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007;21(9):1113–1124. doi: 10.1101/gad.1533407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandell LL, Lynn ML, Inman KE, McDowell W, Trainor PA. RDH10 oxidation of Vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PloS one. 2012;7(2):e30698. doi: 10.1371/journal.pone.0030698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Lintig J, Vogt K. Filling the gap in vitamin A research. Molecular identification of an enzyme cleaving beta-carotene to retinal. J Biol Chem. 2000;275(16):11915–11920. doi: 10.1074/jbc.275.16.11915. [DOI] [PubMed] [Google Scholar]
- 22.Kiefer C, Hessel S, Lampert JM, Vogt K, Lederer MO, Breithaupt DE, et al. Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J Biol Chem. 2001;276(17):14110–14116. doi: 10.1074/jbc.M011510200. [DOI] [PubMed] [Google Scholar]
- 23.Wang XD, Russell RM, Liu C, Stickel F, Smith DE, Krinsky NI. Beta-oxidation in rabbit liver in vitro and in the perfused ferret liver contributes to retinoic acid biosynthesis from beta-apocarotenoic acids. J Biol Chem. 1996;271(43):26490–26498. [PubMed] [Google Scholar]
- 24.Niederreither K, McCaffery P, Dräger UC, Chambon P, Dollé P. Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mech Dev. 1997;62(1):67–78. doi: 10.1016/s0925-4773(96)00653-3. [DOI] [PubMed] [Google Scholar]
- 25.Niederreither K, Dollé P. Retinoic acid in development: towards an integrated view. Nature Reviews Genetics. 2008;9(7):541–553. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 26.Niederreither K, Abu-Abed S, Schuhbaur B, Petkovich M, Chambon P, Dollé P. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat Genet. 2002;31(1):84–88. doi: 10.1038/ng876. [DOI] [PubMed] [Google Scholar]
- 27.Kim YK, Wassef L, Chung S, Jiang H, Wyss A, Blaner WS, et al. Beta-Carotene and its cleavage enzyme Beta-carotene-15, 15-oxygenase (CMOI) affect retinoid metabolism in developing tissues. The FASEB Journal. 2011;25:1641–1652. doi: 10.1096/fj.10-175448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wassef L, Shete V, Hong A, Spiegler E, Quadro L. Beta-Carotene Supplementation Decreases Placental Transcription of LDL Receptor-Related Protein 1 in Wild-Type Mice and Stimulates Placental beta-Carotene Uptake in Marginally Vitamin A-Deficient Mice. J Nutr. 2012;142(8):1456–1462. doi: 10.3945/jn.112.162677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garber JC, Barbee RW, Bielitzki JT, Clayton LA, Donovan JC, Hendriksen C, et al. Guide for the care and use of laboratory animals. Vol. 8. The National Academic Press; Washington DC: 2010. pp. 1–220. [Google Scholar]
- 30.Kim YK, Quadro L. Reverse-phase high-performance liquid chromatography (HPLC) analysis of retinol and retinyl esters in mouse serum and tissues. Methods in molecular biology (Clifton, NJ) 2010;652:263–275. doi: 10.1007/978-1-60327-325-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shmarakov I, Fleshman MK, D’Ambrosio D, Piantedosi R, Riedl KM, Schwartz SJ, et al. Hepatic stellate cells are an important cellular site for beta-carotene conversion to retinoid. Arch Biochem Biophys. 2010;504:3–10. doi: 10.1016/j.abb.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romand R, Kondo T, Cammas L, Hashino E, Dollé P. Dynamic expression of the retinoic acid-synthesizing enzyme retinol dehydrogenase 10 (Rdh10) in the developing mouse brain and sensory organs. J Comp Neurol. 2008;508(6):879–892. doi: 10.1002/cne.21707. [DOI] [PubMed] [Google Scholar]
- 33.Rugh R. Its reproduction and development. Minneapolis, Minn: Burgess Publishing Co; 1968. The mouse. [Google Scholar]
- 34.Wilson JG, Warkany J. Cardiac and aortic arch anomalies in the offspring of vitamin A deficient rats correlated with similar human anomalies. Pediatrics. 1950;5(4):708–725. [PubMed] [Google Scholar]
- 35.Dimenstein R, Trugo N, Donangelo CM, Trugo LC, Anastacio AS. Effect of subadequate maternal vitamin-A status on placental transfer of retinol and beta-carotene to the human fetus. Neonatology. 1996;69(4):230–234. doi: 10.1159/000244315. [DOI] [PubMed] [Google Scholar]
- 36.Satre MA, Ugen KE, Kochhar DM. Developmental changes in endogenous retinoids during pregnancy and embryogenesis in the mouse. Biol Reprod. 1992;46(5):802–810. doi: 10.1095/biolreprod46.5.802. [DOI] [PubMed] [Google Scholar]
- 37.Lobo GP, Hessel S, Eichinger A, Noy N, Moise AR, Wyss A, et al. ISX is a retinoic acid-sensitive gatekeeper that controls intestinal, beta-carotene absorption and vitamin A production. The FASEB Journal. 2010;24(6):1656–1666. doi: 10.1096/fj.09-150995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith JE, Milch PO, Muto Y, Goodman DWS. The plasma transport and metabolism of retinoic acid in the rat. Biochem J. 1973;132(4):821–827. doi: 10.1042/bj1320821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dancis J, Levitz M, Katz J, Wilson D, Blaner WS, Piantedosi R, et al. Transfer and metabolism of retinol by the perfused human placenta. Pediatr Res. 1992;32:195–199. doi: 10.1203/00006450-199208000-00014. [DOI] [PubMed] [Google Scholar]
- 40.Quadro L, Hamberger L, Gottesman ME, Colantuoni V, Ramakrishnan R, Blaner WS. Transplacental delivery of retinoid: the role of retinol-binding protein and lipoprotein retinyl ester. American Journal of Physiology-Endocrinology And Metabolism. 2004;286(5):E844–E851. doi: 10.1152/ajpendo.00556.2003. [DOI] [PubMed] [Google Scholar]
- 41.Johansson S, Dencker L, Dantzer V. Immunohistochemical localization of retinoid binding proteins at the materno-fetal interface of the porcine epitheliochorial placenta. Biol Reprod. 2001;64(1):60–68. doi: 10.1095/biolreprod64.1.60. [DOI] [PubMed] [Google Scholar]
- 42.Boulanger A, McLemore P, Copeland NG, Gilbert DJ, Jenkins NA, Yu SS, et al. Identification of beta-carotene 15, 15 -monooxygenase as a peroxisome proliferator-activated receptor target gene. The FASEB Journal. 2003;17(10):1304–1306. doi: 10.1096/fj.02-0690fje. [DOI] [PubMed] [Google Scholar]
- 43.Reichert B, Yasmeen R, Jeyakumar SM, Yang F, Thomou T, Alder H, et al. Concerted action of aldehyde dehydrogenases influences depot-specific fat formation. Molecular Endocrinology. 2011;25(5):799–809. doi: 10.1210/me.2010-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amengual J, Lobo GP, Golczak M, Li H, Klimova T, Hoppel CL, et al. A mitochondrial enzyme degrades carotenoids and protects against oxidative stress. The FASEB Journal. 2010;25(3):948–959. doi: 10.1096/fj.10-173906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobbs-McAuliffe B, Zhao Q, Linney E. Feedback mechanisms regulate retinoic acid production and degradation in the zebrafish embryo. Mech Dev. 2004;121(4):339–350. doi: 10.1016/j.mod.2004.02.008. [DOI] [PubMed] [Google Scholar]