

Abstract
Objective
To evaluate whether the systemic sclerosis (SSc)-associated IRAK1 non-synonymous single-nucleotide polymorphism rs1059702 is responsible for the Xq28 association with SSc or whether there are other independent signals in the nearby methyl-CpG-binding protein 2 gene (MECP2).
Methods
We analysed a total of 3065 women with SSc and 2630 unaffected controls from five independent Caucasian cohorts. Four tag single-nucleotide polymorphisms of MECP2 (rs3027935, rs17435, rs5987201 and rs5945175) and the IRAK1 variant rs1059702 were genotyped using TaqMan predesigned assays. A meta-analysis including all cohorts was performed to test the overall effect of these Xq28 polymorphisms on SSc.
Results
IRAK1 rs1059702 and MECP2 rs17435 were associated specifically with diffuse cutaneous SSc (PFDR=4.12×10−3, OR=1.27, 95% CI 1.09 to 1.47, and PFDR=5.26×10−4, OR=1.30, 95% CI 1.14 to 1.48, respectively), but conditional logistic regression analysis showed that the association of IRAK1 rs1059702 with this subtype was explained by that of MECP2 rs17435. On the other hand, IRAK1 rs1059702 was consistently associated with presence of pulmonary fibrosis (PF), because statistical significance was observed when comparing SSc patients PF+ versus controls (PFDR=0.039, OR=1.30, 95% CI 1.07 to 1.58) and SSc patients PF+ versus SSc patients PF− (p=0.025, OR=1.26, 95% CI 1.03 to 1.55).
Conclusions
Our data clearly suggest the existence of two independent signals within the Xq28 region, one located in IRAK1 related to PF and another in MECP2 related to diffuse cutaneous SSc, indicating that both genes may have an impact on the clinical outcome of the disease.
INTRODUCTION
Autoimmune diseases are complex polygenic conditions in which multiple susceptibility genes interact with epigenetic and environmental factors for their predisposition and progression. In some cases, part of the genetic component is shared among different immune disorders, suggesting that these pathologies may be influenced by disease-specific and common molecular pathways.1–4 For instance, most of the genetic associations described for systemic sclerosis (SSc), a fibrotic autoimmune disease of skin and internal organs, have also been reported to play a role in the susceptibility to systemic lupus erythematosus (SLE).5,6
Remarkably, a wide spectrum of autoimmune diseases shows a significant female preponderance. SSc represents a clear example of a sex biased immune disorder, with women reaching almost 90% of total affected individuals in some populations. Different factors have been proposed to explain this marked sexual dimorphism, including reproductive and sex hormones, fetal microchimerism, and gender differences in the immune system and lifestyle. Nevertheless, definitive evidences are still lacking and cumulative knowledge points to a major role of sex chromosomes in the immune system homeostasis. Due to the statistical complexity of testing for association between phenotype and genetic markers on the sex chromosomes, very few associations of sex-linked genes with the development of autoimmunity have been reported to date.7–9
One of the shared risk loci between SSc and SLE within the non-homologous region of the X chromosome is the interleukin-1 receptor-associated kinase 1 gene (IRAK1),10,11 that encodes a serine/threonine protein kinase with a special relevance in the signalling pathways of Toll-like receptors (TLRs)/IL-1R.12,13 However, IRAK1 is in the same haplotypic block as the methyl-CpG-binding protein 2 gene (MECP2) on Xq28, which is also associated with SLE,14,15 and functional genetic variants of the latter locus may explain the association signals with SLE observed in IRAK1.16
In this study we aimed to evaluate whether the SSc-associated IRAK1 polymorphism rs1059702 (Phe196Ser) described by Dieudé et al10 is the causal variant of the Xq28 association or whether it reflects another association signal from the nearby MECP2.
PATIENTS AND METHODS
Study population
Since the IRAK1/MECP2 genes are located in a sex-linked region, only women were included in the study. Informed written consent from all participants and approval from the local ethical committees were obtained in accordance with the tenets of the Declaration of Helsinki.
We analysed a total of 3065 female SSc patients and 2630 female unaffected controls of Caucasian ancestry, from an initial discovery cohort of Spain (1016 SSc and 1520 controls) and four additional replication cohorts from USA (965 SSc and 489 controls), Germany (490 SSc and 180 controls), The Netherlands (235 SSc and 278 controls) and UK (359 SSc and 163 controls). Since most samples have not been subjected to genome-wide association study platforms, population substructure analysis could not been performed and this may represent a potential limitation. In all cases, SSc patients were classified based on their skin involvement into limited cutaneous SSc (lcSSc) or diffuse cutaneous SSc (dcSSc) according to the criteria by Leroy et al17 A subset of the German cohort (43.73%) overlaps with that included in the study of IRAK1 by Dieudé et al10 To perform the subphenotype analyses, they were also phenotypically characterised accordingly with the presence or absence of anticentromere antibodies (ACA), antitopoisomerase antibodies (ATA) and pulmonary fibrosis (PF). ACA were determined by their characteristic distinctive pattern on HEP-2 cells, and ATAs were detected by passive immunodiffusion against calf thymus extract. PF was diagnosed by high resolution CT in all the European cohorts. However, the PF status of our US cohort was obtained by pulmonary function test (patients were considered to have PF if they showed a forced vital capacity (FVC) less than 70%). Since two different methodologies were used for the diagnosis, the meta-analyses on PF data were performed only in the European cohorts. The clinical characteristics of the analysed cohorts are summarised in table 1.
Table 1.
Main clinical features of the female systemic sclerosis patients included in the study. Data are referred to the total of analysed individuals
| Clinical feature | Spain | USA | Germany | The Netherlands | UK |
|---|---|---|---|---|---|
| lcSSc | 0.70 | 0.66 | 0.63 | 0.71 | 0.76 |
| dcSSc | 0.30 | 0.34 | 0.37 | 0.29 | 0.24 |
| ACA positive | 0.47 | 0.33 | 0.40 | 0.27 | 0.41 |
| ACA negative | 0.49 | 0.66 | 0.56 | 0.71 | 0.57 |
| ATA positive | 0.22 | 0.18 | 0.28 | 0.24 | 0.15 |
| ATA negative | 0.72 | 0.81 | 0.68 | 0.74 | 0.82 |
| PF positive* | 0.22 | 0.09 | 0.26 | 0.36 | 0.19 |
| PF negative* | 0.70 | 0.37 | 0.60 | 0.49 | 0.18 |
| SSc patients without PF data* | 0.08 | 0.54 | 0.14 | 0.15 | 0.63 |
ACA, anticentromere antibody; ATA, antitopoisomerase antibody; dcSSc, diffuse cutaneous SSc; lcSSc, limited cutaneous SSc; PF, pulmonary fibrosis; SSc, systemic sclerosis.
Pulmonary fibrosis data were obtained by high resolution CT (HRCT) in the European cohorts and by pulmonary function test (FVC<70%) in the US cohort.
Single-nucleotide polymorphism selection
We followed a single-nucleotide polymorphism (SNP) tagging strategy using Haploview V.4.2 (http://www.broad.mit.edu/mpg/haploview)18 to identify taggers that may cover all the common genetic variation within MECP2 (r2≥0.8) in the Utah residents with ancestry from northern and western Europe (CEU) population of the HapMap database (http://hapmap.ncbi.nlm.nih.gov/). Four MECP2 polymorphisms were selected with this method: rs3027935, rs17435, rs5987201 and rs5945175 (see online supplementary figure S1). Additionally, we also included in the study the non-synonymous IRAK1 genetic variant rs1059702 (Phe196Ser), which was described as the SNP that best explains the SSc susceptibility IRAK1 haplotype.10 The location of the five genetic variants analysed within the IRAK1/MECP2 region is shown in online supplementary figure S2.
Genotyping methods
DNA samples were obtained from peripheral white blood cells of participants following standard procedures. Predesigned TaqMan 5′ SNP genotyping assays were used to genotype the selected Xq28 genetic variants (assay IDs: rs3027935: C__15765567_10, rs17435: C___2597094_20, rs5987201: C__30089704_10, rs5945175: C__30485633_20, rs1059702: C___8966367_30) in a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, California, USA).
Statistical analyses
The overall statistical power of our study, according to Power Calculator for Genetic Studies 2006,19 is shown in online supplementary table S1.
The Linux software Plink V.1.7 (http://pngu.mgh.harvard.edu/purcell/plink/)20 was used to perform 2×2 contingency tables, χ2 and/or Fisher’s exact tests, when appropriate. p Values lower than 0.05 were considered as statistically significant. ORs, and 95% CI, were calculated according to Woolf’s method. The Breslow-Day (BD) test was used to estimate the OR heterogeneity among the different cohorts. Combined data were analysed by Mantel-Haenszel tests under fixed effect, or random effect (DerSimonian–Laird) when the BD test reached statistical significance. Benjamini & Hochberg (1995) step-up false discovery rate (FDR) control correction21 for multiple testing was applied to the p values in the independent analysis and the combined meta-analysis.
Plink V.1.7 was also used to carry out a conditional logistic regression analysis to test for dependence between the associated SNPs. Haplotype analyses of allelic combinations with a frequency higher than 5% in the control groups were performed with Plink V.1.7, Haploview V.4.2 and StatsDirect (V.2.6.6; StatsDirect, Altrincham, UK).
RESULTS
The genotyping success rate was higher than 95% in all analysed cohorts. No statistically significant deviation from Hardy-Weinberg equilibrium (p≤0.01) was observed for every SNP in the controls sets, except for rs3027935 in the Spanish population (see online supplementary table S2). The IRAK1 rs1059702 allele frequencies were similar to those described by Dieudé et al,10 whereas the frequencies of the four analysed MECP2 SNPs were in agreement with the data of the HapMap project.
Analysis of the discovery Spanish cohort
We first tested for association in a large Caucasian cohort of Spanish women (see online supplementary table S3). The statistical analysis of the allele frequencies indicated a significant association after FDR correction between IRAK1 rs1059702 and dcSSc (PFDR=0.028, OR=1.34, 95% CI 1.07 to 1.69). This genetic variant also showed trends towards association when the global SSc cohort and the PF+ patients were compared against the control set (PFDR=0.096, OR=1.16, 95% CI 0.99 to 1.35, and PFDR=0.058, OR=1.35, CI 95% 1.04 to 1.76, respectively). On the other hand, significant FDR-corrected p values were obtained for MECP2 rs17435 in the analyses of global disease (PFDR=0.014, OR=1.24, 95% CI 1.08 to 1.43), dcSSc (PFDR=0.014, OR=1.38, 95% CI 1.12 to 1.70), and PF (PFDR=0.047, OR=1.37, 95% CI 1.08 to 1.74). Other trends were suggested in the global SSc analysis for the MECP2 polymorphisms rs5987201 (PFDR=0.096, OR=1.32, 95% CI 0.97 to 1.79) and rs5945175 (PFDR=0.053, OR=1.50, 95% CI 1.06 to 2.11).
Mantel-Haenszel test
Considering the above described associations, we decided to include in the study three additional Caucasian cohorts from Europe (Germany, The Netherlands and UK) and one from the USA. The results of the independent analyses of these populations are summarised in the online supplementary tables S4–S7.
A meta-analysis including all five cohorts was then performed to increase the statistical power thus allowing a better estimation of disease susceptibility (table 2). The overall analysis yielded significant p values only for IRAK1 rs1059702 and MECP2 rs17435 (no heterogeneity of the ORs among the five populations was observed when applying the BD test in both cases). Regarding the IRAK1 non-synonymous SNP rs1059702, the allele frequencies of the dcSSc subgroup differed significantly from those of the control set in the combined analysis (PFDR=4.12×10−3, OR=1.27, 95% CI 1.09 to 1.47), and trends towards association were evident in the global SSc/control (PFDR= 0.070, OR=1.13, 95% CI 1.01 to 1.26) and ATA+/control (PFDR=0.087, OR=1.23, 95% CI 1.04 to 1.46) comparisons, consistent with previously published data.10 Similarly, the MECP2 rs17435 variant reached statistical significant after comparing the global disease group and dcSSc subgroup with the control population (PFDR=2.68×10−3, OR=1.19, 95% CI 1.08 to 1.31, and PFDR=5.26 ×10−4, OR=1.30, 95% CI 1.14 to 1.48, respectively). The ATA +/control comparison also suggested a trend for this SNP (PFDR=0.088, OR=1.18, 95% CI 1.01 to 1.38).
Table 2.
Combined analysis of the allele frequencies of Xq28 genetic variants in Caucasian systemic sclerosis patients and unaffected controls from Spain, USA, Germany, The Netherlands and UK
| Locus | rs# | Gene | 1/2 | Subgroup (N) | Genotype, N (%) | MAF (%) | M-H Allele test | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||
| 1/1 | 1/2 | 2/2 | p Value* | pFDR† | OR (95% CI)‡ | ||||||
| ChrX: 152937386 | rs1059702 | IRAK1 | A/G | Controls (n=2530) | 70 (2.77) | 599 (23.68) | 1861 (73.56) | 14.60 | |||
| SSc (n=2890) | 107 (3.70) | 729 (25.22) | 2054 (71.07) | 16.31 | 0.028 | 0.070 | 1.13 (1.01 to 1.26) | ||||
| lcSSc (n=1969) | 67 (3.40) | 476 (24.17) | 1426 (72.42) | 15.49 | 0.298 | 0.429 | 1.07 (0.95 to 1.20) | ||||
| dcSSc (n=921) | 40 (4.34) | 253 (27.47) | 628 (68.19) | 18.08 | 1.65E-03 | 4.12E-03 | 1.27 (1.09 to 1.47) | ||||
| ACA+ (n=1145) | 24 (2.10) | 279 (24.37) | 842 (73.54) | 14.28 | 0.734 | 0.734 | 0.98 (0.84 to 1.13) | ||||
| ATA+ (n=610) | 24 (3.93) | 169 (27.70) | 417 (68.36) | 17.79 | 0.017 | 0.087 | 1.23 (1.04 to 1.46) | ||||
| ChrX: 152957662 | rs3027935 | MECP2 | T/C | Controls (n=2539) | 13 (0.51) | 229 (9.02) | 2297 (90.47) | 5.02 | |||
| SSc (n=3003) | 12 (0.40) | 322 (10.72) | 2669 (88.88) | 5.76 | 0.311 | 0.389 | 1.09 (0.92 to 1.30) | ||||
| lcSSc (n=2056) | 7 (0.34) | 227 (11.04) | 1822 (88.62) | 5.86 | 0.343 | 0.429 | 1.09 (0.91 to 1.32) | ||||
| dcSSc (n=947) | 5 (0.53) | 95 (10.03) | 847 (89.44) | 5.54 | 0.778 | 0.778 | 1.04 (0.81 to 1.32) | ||||
| ACA+ (n=1178) | 1 (0.08) | 134 (11.38) | 1043 (88.54) | 5.77 | 0.469 | 0.596 | 1.08 (0.87 to 1.35) | ||||
| ATA+ (n=625) | 4 (0.64) | 52 (8.32) | 569 (91.04) | 4.80 | 0.348 | 0.435 | 0.87 (0.65 to 1.17) | ||||
| ChrX: 152965174 | rs17435 | MECP2 | T/A | Controls (n=2508) | 99 (3.95) | 744 (29.67) | 1665 (66.39) | 18.78 | |||
| SSc (n=2990) | 170 (5.69) | 952 (31.84) | 1868 (62.47) | 21.61 | 5.36E-04 | 2.68E-03 | 1.19 (1.08 to 1.31) | ||||
| lcSSc (n=2044) | 106 (5.19) | 638 (31.21) | 1300 (63.60) | 20.79 | 0.027 | 0.133 | 1.13 (1.01 to 1.26) | ||||
| dcSSc (n=946) | 64 (6.77) | 314 (33.19) | 568 (60.04) | 23.36 | 1.05E-04 | 5.26E-04 | 1.30 (1.14 to 1.48) | ||||
| ACA+ (n=1176) | 48 (4.08) | 368 (31.29) | 760 (64.63) | 19.73 | 0.459 | 0.596 | 1.05 (0.92 to 1.19) | ||||
| ATA+ (n=626) | 38 (6.07) | 199 (31.79) | 389 (62.14) | 21.96 | 0.035 | 0.088 | 1.18 (1.01 to 1.38) | ||||
| ChrX: 152983236 | rs5987201 | MECP2 | T/C | Controls (n=2449) | 6 (0.24) | 176 (7.19) | 2267 (92.57) | 3.84 | |||
| SSc (n=2985) | 8 (0.27) | 241 (8.07) | 2736 (91.66) | 4.31 | 0.468 | 0.468 | 1.07 (0.88 to 1.31) | ||||
| lcSSc (n=2036) | 6 (0.29) | 162 (7.96) | 1868 (91.75) | 4.27 | 0.728 | 0.728 | 1.04 (0.84 to 1.29) | ||||
| dcSSc (n=949) | 2 (0.21) | 79 (8.32) | 868 (91.46) | 4.37 | 0.641 | 0.778 | 1.06 (0.81 to 1.39) | ||||
| ACA+ (n=1181) | 1 (0.08) | 104 (8.81) | 1076 (91.11) | 4.49 | 0.477 | 0.596 | 1.09 (0.85 to 1.40) | ||||
| ATA+ (n=619) | 2 (0.32) | 40 (6.46) | 577 (93.21) | 3.55 | 0.297 | 0.435 | 0.84 (0.60 to 1.18) | ||||
| ChrX: 153011951 | rs5945175 | MECP2 | C/T | Controls (n=2550) | 3 (0.12) | 157 (6.16) | 2390 (93.73) | 3.20 | |||
| SSc (n=2991) | 7 (0.23) | 242 (8.09) | 2742 (91.68) | 4.28 | 0.063 | 0.104 | 1.22 (0.99 to 1.50) | ||||
| lcSSc (n=2038) | 5 (0.25) | 166 (8.15) | 1867 (91.61) | 4.32 | 0.065 | 0.162 | 1.23 (0.99 to 1.55) | ||||
| dcSSc (n=953) | 2 (0.21) | 76 (7.97) | 875 (91.82) | 4.20 | 0.270 | 0.450 | 1.17 (0.88 to 1.55) | ||||
| ACA+ (n=1167) | 3 (0.26) | 97 (8.31) | 1067 (91.43) | 4.41 | 0.036 | 0.178 | 1.32 (1.02 to 1.71) | ||||
| ATA+ (n=628) | 1 (0.16) | 39 (6.21) | 588 (93.63) | 3.26 | 0.590 | 0.590 | 0.91 (0.64 to 1.30) | ||||
All p values have been calculated for the allelic model.
ACA, anticentromere antibodies; ATA antitopoisomerase antibodies; dcSSc, diffuse cutaneous SSc; MAF, minor allele frequency; M-H, Mantel–Haenszel test under fixed effect; lcSSc, limited cutaneous SSc; SSc, systemic sclerosis.
Benjamini & Hochberg (1995) step-up FDR control correction.
OR for the minor allele.
To confirm that the genetic association between these two SNPs relies on dcSSc rather than on the global disease, we performed another meta-analysis but comparing this time dcSSc patients against those with lcSSc instead of unaffected controls, thus avoiding a possible effect of having SSc as a confounding variable (table 3). This new combined analysis showed statistically significant differences between dcSSc and lcSSc patients for the allele frequencies of IRAK1 rs1059702 (p=0.022, OR=1.19, 95% CI 1.02 to 1.37) and MECP2 rs17435 (p=0.026, OR=1.16, 95% CI 1.02 to 1.32).
Table 3.
Combined analysis of the allele frequencies of Xq28 genetic variants comparing limited cutaneous systemic sclerosis patients against diffuse cutaneous systemic sclerosis patients from Spain, USA, Germany, The Netherlands and UK
| rs# | Gene | Subgroup (N) | MAF (%) | M-H Allele test | |
|---|---|---|---|---|---|
|
| |||||
| p Value* | OR (95% CI)† | ||||
| rs1059702 | IRAK1 | lcSSc (n=1969) | 15.49 | ||
| dcSSc (n=921) | 18.08 | 0.022 | 1.19 (1.02 to 1.37) | ||
| rs17435 | MECP2 | lcSSc (n=2044) | 20.79 | ||
| dSSc (n=946) | 23.36 | 0.026 | 1.16 (1.02 to 1.32) | ||
All p values have been calculated for the allelic model.
OR for the minor allele.
lcSSc, limited cutaneous systemic sclerosis; dcSSc, diffuse cutaneous systemic sclerosis; MAF, minor allele frequency; M-H, Mantel-Haenszel test under fixed effects; NA, not applicable.
Dependence analysis
Taking into account that both SNPs seemed to be specifically associated with risk to develop dcSSc, a conditional logistic regression analysis considering the populations as covariate was carried out to determine whether one of them could be dependent on the other one (table 4). The pairwise conditioning showed that the association of IRAK1 rs1059702 with dcSSc was explained by that of MECP2 rs17435, because only the latter SNP remained significant in the conditional analysis (rs1059702 conditioned p=0.786; rs17435 conditioned p=0.049). The fact that both polymorphisms were in moderate linkage disequilibrium (r2∼ 0.60) may explain the dependence.
Table 4.
Conditional logistic regression analysis for IRAK1/MECP2 polymorphisms accordingly with diffuse cutaneous systemic sclerosis considering the populations as covariate
| SNP | Gene | Uncorrected p value | Conditioned p value | Linkage disequilibrium (r2) | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Spain | USA | Germany | Netherlands | UK | ||||
| rs1059702 | IRAK1 | 1.65E-03 | 0.786 | 0.69 | 0.67 | 0.60 | 0.68 | 0.53 |
| rs17435 | MECP2 | 1.05E-04 | 0.049 | |||||
SNP, single-nucleotide polymorphism.
Analysis on PF data
Since the PF data of the SSc patients from the USA was estimated using a different methodology, we decided to include in the meta-analysis of this clinical feature only the European cohorts (Spain, Germany, The Netherlands and UK; table 5).
Table 5.
Combined analysis of the allele frequencies of Xq28 genetic variants accordingly with pulmonary fibrosis in the study cohorts from Spain, Germany, The Netherlands and UK
| rs# | Gene | Comparison | N | MAF (%) | M-H Allele test | |
|---|---|---|---|---|---|---|
|
| ||||||
| p Value* | OR (95% CI)†*** | |||||
| rs1059702 | IRAK1 | PF+ versus controls | 461/2043 | 18.33/14.46 | 8.46E-03‡ | 1.30 (1.07 to 1.58) |
| PF+ versus PF− | 461/1149 | 18.33/15.54 | 0.025 | 1.26 (1.03 to 1.55) | ||
| rs17435 | MECP2 | PF+ versus controls | 481/2035 | 22.66/18.72 | 0.015§ | 1.24 (1.04 to 1.48) |
| PF+ versus PF− | 481/1151 | 22.66/21.33 | 0.346 | 1.09 (0.91 to 1.31) | ||
MAF, minor allele frequency; M-H, Mantel–Haenszel test under fixed effect; PF+, patients with pulmonary fibrosis; PF−, patients without pulmonary fibrosis.
All p values have been calculated for the allelic model.
OR for the minor allele.
PFDR=0.039.
PFDR=0.039.
In a first step, we compared PF+ SSc patients with the unaffected control group, obtaining significant p values for IRAK1 rs1059702 (PFDR=0.039, OR=1.30, 95% CI 1.07 to 1.58) and MECP2 rs17435 (PFDR=0.039, OR=1.24, 95% CI 1.04 to 1.48). However, only IRAK1 rs1059702 showed a consistent association with PF susceptibility, because the allele frequencies were significantly different between SSc patients with PF and SSc patients without PF (p=0.025, OR=1.26, 95% CI 1.03 to 1.55). On the contrary, in the case to case comparison, MECP2 rs17435 was not associated with PF (p=0.346, OR=1.09, 95% CI 0.91 to 1.31).
Haplotype analysis
Finally, we further explore a possible haplotype effect within this genomic region by performing several allelic combinations. Although the combinations rs1059702*G-rs17435*A and rs1059702*A-rs17435*T showed clear association peaks (see online supplementary table S8), the strength of these associations did not differ from those of IRAK1 rs1059702 and MECP2 rs17435 independently.
DISCUSSION
This study represents an important step forward towards a better understanding of the complex genetic association between the Xq28 genomic region and the predisposition to autoimmunity. Because of the linkage disequilibrium structure of this locus, there is controversy about whether one of the two juxtaposed genes IRAK1 or MECP2 may harbour the causal variant of the described association with SLE and other autoimmune diseases including SSc.10,14–16,22 IRAK1 was considered as the most likely candidate, mainly due to its crucial involvement in the TLR pathway (in which it modulates the IL1-induced activation of NF-κB) and to the fact that this gene has been related with certain SLE manifestations in mouse models.11–13
On the other hand, MECP2 is also emerging as an interesting putative susceptibility gene in autoimmunity. It encodes a nuclear protein that binds specifically to methylated DNA, playing a central role in the transcriptional regulation of methylation-sensitive genes. Interestingly, there is increasing evidence pointing to epigenetic alterations, including those in DNA methylation, as relevant players in the pathogenesis of autoimmune diseases.23 For example, epigenetic dysregulation in T cells seems to be an important component of the SLE pathogenesis.24–26 Additionally, SLE patients containing the MECP2 SLE-risk haplotype show abnormal gene expression profiles of B cells, and this has been suggested to contribute to the interferon (IFN) signature observed in this pathology.15 Moreover, it has also been described in mouse models that MeCP2 protein may regulate the transcriptional activation of IRAK1 through microRNAs.27–29 Strikingly, the risk allele of the IRAK1 variant analysed here (rs1059702) has been recently associated with lower mRNA levels of MECP2 in SLE cases and unaffected controls of multiple ancestral groups, which has led to propose that both genes contribute independently to SLE susceptibility.30 Although no epistatic gene-gene interaction between MECP2 and IRAK1 was observed in our data, it could be possible that genetic variation within any of these two genes may be associated with the expression of the other one. In this regard, experimental or bioinformatic analysis on the association of MECP2 rs17435 and IRAK1 rs1059702 with expression levels of both genes would provide important clues that would help to completely unmask their inter-relation and its relevance in the genetic susceptibility to SSc and other related disorders.
Consistent with the latter idea, our data clearly indicate the existence of independent association signals with different SSc clinical conditions in each gene. One of them, rs17435, is located in the intron 2 of MECP2 and tags the SLE-risk haplotype.15,31 The same genetic variant has also been associated with Sjögren’s syndrome (SS), one of the most frequent comorbidities in SSc patients.22,32,33 Specifically, we found a firm association of MECP2 rs17435 with the dcSSc subtype, which is the most severe form of the disease.9 It should be noted that SLE, SS and SSc are related autoimmune disorders that share some of their immunological and genetic component.1,2,34,35 For instance, a type I IFN signature similar to that described in SLE has also been identified in a subset of SSc patients.36 Taking into account the above, it is likely that MECP2 represents a real shared susceptibility factor for these systemic diseases probably affecting the epigenetic control of common pathogenic pathways (eg, the IFN signalling).
In addition, MECP2 could also be one of the genes contributing to the gender bias in autoimmune diseases. X chromosome inactivation in women involves processes that include DNA methylation.37 Considering that this chromosome contains a considerable number of important immune-related genes, such as inhibitor of κ light polypeptide gene enhancer in B cells kinase γ (IKBKG),38 interleukin 2 receptor gamma (IL2RG),39 CD40 ligand (CD40L),40 forkhead box P3 (FOXP3)41 or IRAK1,2,13 among others, some authors proposed that an abnormal gene dosage effect of X linked genes due to X chromosome defects may be involved in the female preponderance to develop autoimmunity.8 Supporting this idea, it has been recently reported that demethylation of regulatory elements of CD40L on the inactive X chromosome may be directly involved in the overexpression of this gene observed in CD4 T cells from SSc patients.42,43
On the other hand, we also found an independent association between the non-synonymous IRAK1 SNP rs1059702 and susceptibility to develop PF in SSc patients. This association is in agreement with that described by Dieudé et al10 However, these authors suggested that this locus is also associated with the dcSSc subtype and production of ATAs. In this regard, our data indicate that the association of IRAK1 rs1059702 with dcSSc is dependant of that for MECP2 rs17435, possibly due to the moderate linkage disequilibrium of both SNPs and the overlapping between this group of patients and those with pulmonary involvement (figure 1). The trend that we detected in the ATA analysis may also be a consequence of the latter cause. Since presence of ATAs and PF occur preferentially in dcSSc patients,9 it is not easy to determine specific associations with these three phenotypes if only cases versus controls are compared (having one of these features may represent a confounding variable). We observed statistically significant differences between SSc patients with PF and those without PF only for IRAK1 rs1059702 but not for MECP2 rs17435, thus suggesting that only IRAK1 rs1059702 is associated with PF predisposition.
Figure 1.
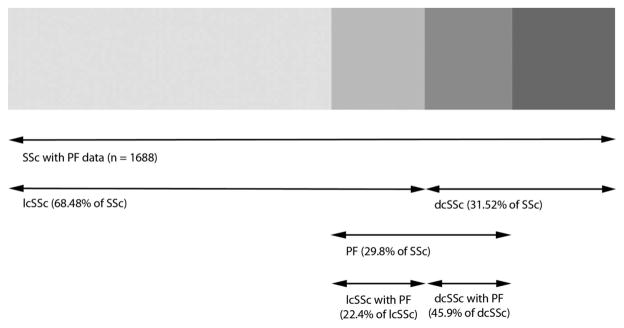
Degree of overlapping between different case subsets from Spain, Germany, The Netherlands and UK. SSc, systemic sclerosis; lcSSc, limited cutaneous systemic sclerosis; dcSSc, diffuse cutaneous systemic sclerosis; PF, pulmonary fibrosis. Note: ‘lcSSc with PF’ and ‘dcSSc with PF’ percentages are referred to the total number of lcSSc and dcSSc cohorts, respectively, with PF data.
Despite MECP2 and IRAK1 showing clear association signals with SSc, it should be noted that these associations are less robust than those described in SLE.11,14 It could be speculated that the risk variants in this genomic region may lead to a SLE-like phenotype, as it has been observed for other common susceptibility genes like ITGAM.44
In conclusion, the analysis of our data showed that MECP2 and IRAK1 are independent SSc susceptibility genes that influence distinct clinical phenotypes. MECP2 is associated with the most severe subtype of the disease, consistent with the major role that epigenetics seem to play in this kind of diseases, and IRAK1 may be a crucial gene underlying the pathogenic mechanisms leading to PF, probably by altering the TLR pathway.
Supplementary Material
Acknowledgments
The authors thank Sofía Vargas, Sonia García and Gema Robledo (from Instituto de Parasitología y Biomedicina ‘López-Neyra’, CSIC, Spain), Julio Charles and Marilyn Perry (University of Texas) for their excellent technical assistance, and all the patients and unaffected controls for kindly accepting their essential collaboration. Banco Nacional de ADN (University of Salamanca, Spain) is thanked for supplying the Spanish controls.
Funding This work was supported by the following grants: JM was funded by GEN-FER from the Spanish Society of Rheumatology, SAF2009–11110 from the Spanish Ministry of Science, CTS-4977, and CTS-180 from Junta de Andalucía, RETICS Program, RD08/0075 (RIER) from Instituto de Salud Carlos III (ISCIII), Spain, within the VI PN de I+D+i 2008–2011 (FEDER), and is sponsored by the Orphan Disease Program grant from the European League Against Rheumatism (EULAR). This study was also funded by PI-0590-2010, from Consejería de Salud y Bienestar Social, Junta de Andalucía, Spain. JLCR and JM are funded by Consejería de Salud, Junta de Andalucía, through PI-0590–2010. FDC was supported by Consejo Superior de Investigaciones Científicas (CSIC) through the program JAE-DOC. TRDJR was funded by the VIDI laureate from the Dutch Association of Research (NWO) and Dutch Arthritis Foundation (National Reumafonds). TW was granted by DFG WI 1031/6.1. Study on USA samples were supported by US National Institutes of Health and National Institute of Arthritis and Musculoskeletal Diseases (NIH-NIAMS) R01-AR-055258, Two-Stage Genome Wide Association Study in Systemic Sclerosis (MDM) and by the NIH-NIAMS Center of Research Translation (CORT) in SSc (P50AR054144) (MDM, FCA, FKT), the NIH-NIAMS SSc Family Registry and DNA Repository (N01-AR-0–2251) (MDM), NIH-KL2RR024149 (SA), K23AR061436 (SA), and the Department of Defence Congressionally Directed Medical Research Programs (W81XWH-07-01-0111) (MDM).
Footnotes
Collaborators See online supplementary note.
Contributors FDC, MCC, LMDG, TRDJR, MDM and JM were involved in the conception and design of the study and revised critically the submitted version of the manuscript. They also contributed in the analysis and interpretation of data. FDC drafted the manuscript. JCAB, CPS, PEC, JLCR, VFP, FJLL, MAGG, NH, GR, TW, AK, JHWD, RM, PS, JMvL, AJS, MCV, AEV, CF, CPD, AH, JW, FCA, FKT, SA, TRDJR and MDM, participated in the collection of samples and clinical information, in the analysis and interpretation of data, and revised critically the manuscript draft. All authors approved the final version of the manuscript.
Competing interests None.
Ethics approval Approval from the local ethical committees of all participating centres were obtained in accordance with the tenets of the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011;365:1612–23. doi: 10.1056/NEJMra1100030. [DOI] [PubMed] [Google Scholar]
- 2.Cotsapas C, Voight BF, Rossin E, et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 2011;7:e1002254. doi: 10.1371/journal.pgen.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zenewicz LA, Abraham C, Flavell RA, et al. Unraveling the genetics of autoimmunity. Cell. 2010;140:791–7. doi: 10.1016/j.cell.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhernakova A, van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet. 2009;10:43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- 5.Harley JB, Alarcon-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin JE, Bossini-Castillo L, Martin J. Unraveling the genetic component of systemic sclerosis. Hum Genet. 2012;131:1023–37. doi: 10.1007/s00439-011-1137-z. [DOI] [PubMed] [Google Scholar]
- 7.Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. 2010;10:594–604. doi: 10.1038/nri2815. [DOI] [PubMed] [Google Scholar]
- 8.Invernizzi P, Pasini S, Selmi C, et al. Female predominance and X chromosome defects in autoimmune diseases. J Autoimmun. 2009;33:12–16. doi: 10.1016/j.jaut.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 9.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 10.Dieude P, Bouaziz M, Guedj M, et al. Evidence of the contribution of the X chromosome to systemic sclerosis susceptibility: association with the functional IRAK1 196Phe/532Ser haplotype. Arthritis Rheum. 2011;63:3979–87. doi: 10.1002/art.30640. [DOI] [PubMed] [Google Scholar]
- 11.Jacob CO, Zhu J, Armstrong DL, et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci USA. 2009;106:6256–61. doi: 10.1073/pnas.0901181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Netea MG, Wijmenga C, O’Neill LA. Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol. 2012;13:535–42. doi: 10.1038/ni.2284. [DOI] [PubMed] [Google Scholar]
- 13.Rao N, Nguyen S, Ngo K, et al. A novel splice variant of interleukin-1 receptor (IL-1R)-associated kinase 1 plays a negative regulatory role in Toll/IL-1R-induced inflammatory signaling. Mol Cell Biol. 2005;25:6521–32. doi: 10.1128/MCB.25.15.6521-6532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawalha AH, Webb R, Han S, et al. Common variants within MECP2 confer risk of systemic lupus erythematosus. PLoS One. 2008;3:e1727. doi: 10.1371/journal.pone.0001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Webb R, Wren JD, Jeffries M, et al. Variants within MECP2, a key transcription regulator, are associated with increased susceptibility to lupus and differential gene expression in patients with systemic lupus erythematosus. Arthritis Rheum. 2009;60:1076–84. doi: 10.1002/art.24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawalha AH. Xq28 and lupus: IRAK1 or MECP2? Proc Natl Acad Sci USA. 2009;106:E62. doi: 10.1073/pnas.0904068106. author reply E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- 18.Barrett JC, Fry B, Maller J, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 19.Skol AD, Scott LJ, Abecasis GR, et al. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–13. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 20.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 22.Cobb BL, Fei Y, Jonsson R, et al. Genetic association between methyl-CpG binding protein 2 (MECP2) and primary Sjogren’s syndrome. Ann Rheum Dis. 2010;69:1731–2. doi: 10.1136/ard.2009.122903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks WH, Le Dantec C, Pers JO, et al. Epigenetics and autoimmunity. J Autoimmun. 2010;34:J207–19. doi: 10.1016/j.jaut.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Sawalha AH, Jeffries M, Webb R, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008;9:368–78. doi: 10.1038/gene.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawalha AH, Jeffries M. Defective DNA methylation and CD70 overexpression in CD4+ T cells in MRL/lpr lupus-prone mice. Eur J Immunol. 2007;37:1407–13. doi: 10.1002/eji.200636872. [DOI] [PubMed] [Google Scholar]
- 26.Pan Y, Sawalha AH. Epigenetic regulation and the pathogenesis of systemic lupus erythematosus. Transl Res. 2009;153:4–10. doi: 10.1016/j.trsl.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Urdinguio RG, Lopez-Serra L, Lopez-Nieva P, et al. Mecp2-null mice provide new neuronal targets for Rett syndrome. PLoS One. 2008;3:e3669. doi: 10.1371/journal.pone.0003669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–9. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urdinguio RG, Fernandez AF, Lopez-Nieva P, et al. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics. 2010;5:656–63. doi: 10.4161/epi.5.7.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaufman KM, Zhao J, Kelly JA, et al. Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Ann Rheum Dis. 2013;72:437–44. doi: 10.1136/annrheumdis-2012-201851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suarez-Gestal M, Calaza M, Endreffy E, et al. Replication of recently identified systemic lupus erythematosus genetic associations: a case-control study. Arthritis Res Ther. 2009;11:R69. doi: 10.1186/ar2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hudson M, Rojas-Villarraga A, Coral-Alvarado P, et al. Polyautoimmunity and familial autoimmunity in systemic sclerosis. J Autoimmun. 2008;31:156–9. doi: 10.1016/j.jaut.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 33.Avouac J, Airo P, Dieude P, et al. Associated autoimmune diseases in systemic sclerosis define a subset of patients with milder disease: results from 2 large cohorts of European Caucasian patients. J Rheumatol. 2010;37:608–14. doi: 10.3899/jrheum.090815. [DOI] [PubMed] [Google Scholar]
- 34.Korman BD, Alba MI, Le JM, et al. Variant form of STAT4 is associated with primary Sjogren’s syndrome. Genes Immun. 2008;9:267–70. doi: 10.1038/gene.2008.1. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen CQ, Peck AB. Unraveling the pathophysiology of Sjogren syndrome-associated dry eye disease. Ocul Surf. 2009;7:11–27. doi: 10.1016/s1542-0124(12)70289-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Assassi S, Mayes MD, Arnett FC, et al. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis Rheum. 2010;62:589–98. doi: 10.1002/art.27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chow JC, Brown CJ. Forming facultative heterochromatin: silencing of an X chromosome in mammalian females. Cell Mol Life Sci. 2003;60:2586–03. doi: 10.1007/s00018-003-3121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–85. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 39.Noguchi M, Yi H, Rosenblatt HM, et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993;73:147–57. doi: 10.1016/0092-8674(93)90167-o. [DOI] [PubMed] [Google Scholar]
- 40.Cerutti A, Puga I, Cols M. Innate control of B cell responses. Trends Immunol. 2011;32:202–11. doi: 10.1016/j.it.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–62. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- 42.Valentini G, Romano MF, Naclerio C, et al. Increased expression of CD40 ligand in activated CD4+ T lymphocytes of systemic sclerosis patients. J Autoimmun. 2000;15:61–6. doi: 10.1006/jaut.2000.0387. [DOI] [PubMed] [Google Scholar]
- 43.Lian X, Xiao R, Hu X, et al. DNA demethylation of CD40L in CD4(+) T-cells from women with systemic sclerosis—A possible explanation for female susceptibility. Arthritis Rheum. 2012;64:2338–45. doi: 10.1002/art.34376. [DOI] [PubMed] [Google Scholar]
- 44.Carmona FD, Simeon CP, Beretta L, et al. Association of a non-synonymous functional variant of the ITGAM gene with systemic sclerosis. Ann Rheum Dis. 2011;70:2050–2. doi: 10.1136/ard.2010.148874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.