

Abstract
A Nicaraguan isolate of Spodoptera frugiperda multiple nucleopolyhedrovirus is being studied as a possible biological insecticide. This virus exists as a mixture of complete and deletion genotypes; the latter depend on the former for the production of an essential per os transmission factor (pif1) in coinfected cells. We hypothesized that the virus population was structured to account for the prevalence of pif1 defector genotypes, so that increasing the abundance of pif1 produced by a cooperator genotype in infected cells would favor an increased prevalence of the defector genotype. We tested this hypothesis using recombinant viruses with pif1 expression reprogrammed at its native locus using two exogenous promoters (egt, p10) in the pif2/pif1 intergenic region. Reprogrammed viruses killed their hosts markedly faster than the wild-type and rescue viruses, possibly due to an earlier onset of systemic infection. Group success (transmission) depended on expression of pif1, but overexpression was prejudicial to group-specific transmissibility, both in terms of reduced pathogenicity and reduced production of virus progeny from each infected insect. The presence of pif1-overproducing genotypes in the population was predicted to favor a shift in the prevalence of defector genotypes lacking pif1-expressing capabilities, to compensate for the modification in pif1 availability at the population level. As a result, defectors increased the overall pathogenicity of the virus population by diluting pif1 produced by overexpressing genotypes. These results offer a new and unexpected perspective on cooperative behavior between viral genomes in response to the abundance of an essential public good that is detrimental in excess.
Introduction
Alphabaculoviruses (lepidopteran infecting nucleopolyhedroviruses) are insect pathogens, some of which form the active ingredient in a number of bioinsecticidal products [1], [2]. These viruses produce two types of virions: budded virions (BVs) for cell-to-cell transmission in infected insects and occlusion derived virions (ODVs) that are occluded within occlusion bodies (OBs) for insect-to-insect transmission. A number of per os infection factors (PIFs) are necessary for primary virus infection that involves binding to microvilli receptors and fusion between the ODV membrane and the microvilli of midgut cells [3]. This is a multistep process [4] that seems to involve a highly stable multimolecular complex of PIF factors [5], [6].
PIF1 was first identified at a very low level in ODV membranes [7]. The reason for the very low expression of pif1 [8] is uncertain, but might be related to some unique property of the protein. The level of expression of pif1 compared to that of other pif genes is unknown, although the amount of pif1 mRNA transcript was estimated to be 300 times lower than that of polh mRNA. In a Spodoptera frugiperda multiple nucleopolyhedrovirus population defective genotypes, SfNIC-C and –D, are not infectious per os due to a 16.4 kb deletion that includes the pif1 and pif2 genes. These defective genotypes survive by complementation with pif1/pif2-containing genotypes in cells infected by multiple genotypes [9], [10].
Occlusion bodies (OBs) of SfNIC-B, the dominant genotype in the population and the genotype with the largest genome [9]–[11], are less pathogenic than OBs of the wild-type mixture, in terms of concentration-mortality metrics. However when ODVs of complete and defective genotypes were mixed in near natural proportions (75% SfNIC-B:25% SfNIC-C) and co-occluded into OBs, the pathogenicity of the mixed genotype OBs was restored to that of the wild-type population [10], [12]. Moreover, when subjected to serial passage in insects, mixed genotype OBs, comprising non-natural proportions of complete and defective genotypes, rapidly converged to a common stable proportion that reflected the natural proportion of each type of genotype, suggesting that the wild-type population is genetically structured to increase the likelihood of transmission [9], [10], [12]–[14].
Near identical results were observed in experiments with mixtures of SfNIC-B and recombinant viruses based on SfNIC-B in which pif1 and pif2 had been deleted, indicating that the absence of pif1 and pif2 in a fraction of the population is both necessary and sufficient to explain the observed pathogenicity phenotype of mixed genotype OBs [15]. It seems that pif1/pif2 expression is regulated not only at transcriptional level but also at population level, accounting for a higher prevalence of defective genotypes, to maximize the transmissibility of the OBs. Accordingly, Clavijo et al. [15] predicted that enhancing pif1 expression would have two different effects. First, a reduction in the potency of OBs of the reprogrammed genotype due to an increase in the amount of PIF1 in ODVs that could adversely influence ODV entry into midgut cells. Second, a shift in the frequencies of pif1 reprogrammed and deletion genotypes in mixed infections would be required to restore OB potency to that of the wild-type population.
In the present study we explored the consequences of manipulating the expression of pif1 that represents a public good in cells infected by multiple genotypes. We examined the hypothesis that expression of this gene alters the pathogenicity of OBs and thereby determines the frequencies of cooperator and defector genotypes in the virus population. To test this, the weakly transcribed pif1 gene [7], [8] was reprogrammed under the control of an early promoter (egt promoter) [16], [17], or a strong late promoter (p10 promoter) [18], [19] originating from a closely-related nucleopolyhedrovirus. The pathogenicity of OBs produced in insects infected by mixtures of reprogrammed and defector genotypes was then analyzed and shown to follow the predicted response.
Materials and Methods
Insects, cells and viruses
Larvae from a laboratory colony of S. frugiperda were maintained on a wheatgerm-based semisynthetic diet [20] at 25°C. Sf9 cells were cultured at 28°C in TC100 medium supplemented with 10% fetal calf serum (FCS), penicillin (1 U/ml) and streptomycin (1 µg/ml). Occlusion bodies (OBs) of a Nicaraguan isolate (SfNIC) of SfMNPV were amplified in S. frugiperda fourth instars. The complete genotype, SfNIC-B, was obtained from plaque purified material [9] and was used to construct the bacmid SfNIC-BΔpifs, a virus with a 2.8 kb deletion encompassing the consecutive pif1 and pif2 genes [15].
Construction of promoter-exchange donor vectors
A pUC19-based transfer vector was constructed to insert the selectable pif1 and pif2 genomic region (nt 31,228 to 36,075) in the SfNIC-B genome [11] (accession number HM595733), that would subsequently be used to insert alternative p10 or egt promoters from Spodoptera exigua NPV (SeMNPV), into the SfMNPV pif2-pif1 intergenic region by homologous recombination (Fig. 1A). The primers used for the constructions are listed in Table S1. First, a plasmid was constructed that contains the left and right flanking regions of pif2 and pif1 from the SfMNPV genome. The left genomic-flanking region (1,006 bp; 31,228–32,233) of the donor cassette, amplified from SfNIC-B DNA using the Sfarif1.1/Sfpif2.4 primer sets, contained the full arif1 ORF and partial downstream sf32 ORF of unknown function. This genomic flanking region is located just upstream from the ATG start codon of pif2. The Sfpif2.4 primer sequence included a 30 bp (32,204-32,233) homologous region to the sequence upstream from the ATG of pif2 and a BglII restriction site, the promoter region of pif1 (33,431–33,447), and a BamHI restriction site. The introduced BglII restriction site was used to clone the pif2 ORF, whereas the BamHI site was inserted to favor ligation to the right genomic-flanking region, and afterwards used to clone the pif1 ORF. First, the sf32/arif1 containing PCR fragment was cloned into the multiple cloning site of pUC19 using the PCR primer-introduced KpnI and BamHI restriction sites to create the plasmid pUC19.sf32/arif1. The right genomic-flanking fragment (1,038 bp; 35,038–36,075), amplified from SfNIC-B DNA using the Sfpif1.12/Sffgf.1 primer set, contained the complete sf36 ORF of unknown function and the 3′ end of the fgf ORF. The sf36/fgf-containing amplicon was then cloned into the remaining MCS of pUC19.sf32/arif1 using the PCR primer-introduced BamHI and HindIII restriction sites. This plasmid, containing the right and left genomic flanking regions of the pif2/pif1 genes, was designated as pUC19.sf32/arif1-sf36/fgf. The complete pif2 gene (32,234–33,430) was amplified from SfNIC-B genome using the Sfpif2.5/Sfpif2.6 primer set. The pif2-containing PCR fragment was cloned into the pUC19.sf32/arif1-sf36/fgf plasmid using the primer-introduced BglII restriction site. The pif1 gene (33,448–35,037) was also amplified from the SfNIC-B genome using the Sfpif1.13/Sfpif1.14 primer set, and cloned into pUC19.sf32/arif1-pif2-sf36/fgf utilizing the introduced BamHI restriction site. The plasmid containing the right and left flanking regions and both pif genes, pUC19.sf32/arif1-pif2-pif1-sf36/fgf (designated pUC19.(pif1)pif1 in Fig. 1A; the parentheses indicate the promoter, whereas the coding sequences are indicated in italics) was used to construct the donor plasmids for the cotransfection with the SfNIC-BΔpifs virus [15].
Figure 1. Promoter-exchange donor constructs and intergenic viral sequences.

A) Donor plasmids used in this study. B) Viruses used in this study. The sequence of the complete genotype SfNIC-B is shown, where the right and left pif-2/pif-1 flanking regions are specified. First, a SfNIC-BΔpifs recombinant virus was constructed in which pif-2/pif-1 coding region was substituted by the LacZ operon. The primers used for the flanking regions amplifications are indicated below the figure. Recombinant viruses SfNIC-Bpif1 (rescue virus), SfNIC-Begt, in which the SeMNPV egt promoter would drive pif-1 transcription, and SfNIC-Bp10, in which the SeMNPV p10 promoter would drive pif-1 transcription, were constructed by homologous recombination between SfNIC-BΔpifs and donor plasmids pUC19.(pif1)pif1, pUC19.(egt)pif1 and pUC19.(p10)pif1 respectively. C) pif-2/pif-1 intergenic sequences from SfNIC-B, SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 viruses. The underlined portion in SfNIC-B containing the native pif-1 promoter was replaced by donor sequences.
The SeMNPV egt promoter (26,828–26,927) [20] was amplified by PCR from SeMNPV DNA using the SePregt.F-SePregt.R primer set (Table S1). The egt promoter PCR fragment containing primer-introduced BglII and BamHI sites was cloned to the pUC19.sf32/arif1-pif2-pif1-Sf36/fgf BamHI-digested plasmid, thus generating pUC19.Sf32/arif1-pif2-(egt)pif1-Sf36/fgf (designated pUC19.(egt)pif1 in Fig. 1A), so that the egt promoter would drive pif1 transcription. The sequence representing the SeMNPV p10 promoter (123,702–123,739) [21] was designed as two complementary oligomers (SePrp10.F-SePrp10.R) (Table S1) that, when annealed, had compatible overhangs for ligation of the pUC19.sf32/arif1-pif2-pif1-sf36/fgf BglII-BamHI digested plasmid, thus generating pUC19.sf32/arif1-pif2-(p10)pif1-sf36/fgf (designated pUC19.(p10)pif1 in Fig. 1A), so that the p10 promoter would drive pif1 transcription. The integrity of all cloned sequences was verified by sequencing.
Generation, isolation and screening of recombinant viruses
The DOTAP reagent and protocol (Roche, Basel, Switzerland) was used to cotransfect Sf9 cells with the LacZ+ SfNIC-BΔpifs viral genomic and plasmid transfer pUC19.(pif1)pif1, pUC19.(egt)pif1 and pUC19.(p10)pif1 DNAs (Fig. 1A). OBs of three recombinant viruses were generated: (i) SfNIC-Bpif1, representing both a rescue virus and a positive control for the recombinant construction methodology, (ii) SfNIC-Begt, SfNIC-B genotype in which pif1 was reprogrammed with the SeMNPV egt promoter and, (iii) SfNIC-Bp10, SfNIC-B genotype in which pif1 was reprogrammed with the SeMNPV p10 promoter (Fig. 1B). For this, cells were transfected with 1 µg of SfNIC-BΔpifs genomic DNA and 5 µg of the corresponding plasmid transfer vectors (Fig. 1A). Viral plaques were screened by adding 30 ng/µl X-gal reagent to the TC100 medium. A total of 20 white plaques were picked from each transfection and each plaque was amplified in Sf9 cells. DNA was extracted from amplified plaques and the authenticity of the recombinant viruses was confirmed by sequencing of PCR amplicons spanning the pif1 promoter region amplified using Sfpif1.7-Sfpif1.9 primers (Fig. 1C). OBs were produced by injecting 8 µl of each virus at 1×104 pfu/ml in S. frugiperda fourth instars. The authenticity of OBs produced in insects was also confirmed by sequencing of the PCR products obtained following amplification using Sfpif1.7-Sfpif1.9 primers (Table S1).
Temporal expression
Groups of 250 S. frugiperda second instars were inoculated with the 90% lethal concentration (LC90) of each of the following viruses SfNIC-B (1.65×106 OBs/ml), SfNIC-Bpif1 (8.98×105 OBs/ml), SfNIC-Begt (9.22×106 OBs/ml), SfNIC-Bp10 (1.77×107 OBs/ml) or mock-infected using the droplet feeding technique [22]. The experiment was performed three times. Total RNA was isolated from groups of 20 larvae at 0, 2, 4, 6, 8, 12, 24, 48 and 72 h post-infection (p.i.). The time zero h p.i. was defined as the moment that the larvae had ingested viral OBs. Total RNA was extracted from insect larvae using TRIzol isolation reagent (Invitrogen) according to manufacturer's protocol. The concentration and integrity of RNA samples were determined by measuring absorbance at 260 nm, and by agarose gel electrophoresis. RNA samples were stored at −80°C until required. The experiment was performed three times.
The temporal expression of pif1 under the control of homologous or heterologous promoters, was determined by qRT-PCR. For this, 1 µg RNA was treated with DNase I (Promega) following manufacturer's instructions. cDNA was synthesized by using Improm-II™ Reverse Transcriptase (Promega), according to the manufacturer's protocol. The absence of contaminant DNA was verified by performing PCR without a prior reverse transcription step. Three sets of specific primers that annealed in pif1 were designed based on the SfNIC-B genome sequence [11]. Non-template controls were analyzed for each set of primers designed in order to verify the absence of non-specific background signal. The qSfBpif1.F and qSfBpif1.R primer set (Table S1) was selected based on the presence of a single melting peak, an indicator of specific amplification. RNA isolated from mock-infected larvae, as well as the Milli-Q water used in all reactions, served as negative controls. All reactions were performed in triplicate.
A 1 µl volume of cDNA (1∶10 dilution) was used for qRT-PCR. All reactions were performed using SYBR Green fluorescence in an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). The reaction mixture (10 µl) contained 5 µl SYBR Premix Ex Taq (2×), 0.2 µl of ROX Reference Dye (50×), 0.1 µl of each SfMNPV primer (10 pmol/µl) (Table S1) and 1 µl of pooled cDNA. qPCR was performed under the following conditions: 95°C for 30 s, followed by 45 elongation cycles of 95°C for 5 s and 60°C for 30 s and finally a dissociation stage of 95°C for 15 s, 60°C for 15 s and 95°C for 15 s. Data acquisition and analysis were handled by Sequence Detector Version 2.2.2. software (Applied Biosystems).
SfNIC-B DNA was amplified in the pif1 region by conventional PCR using the qSfBpif1.F and qSfBpif1.R primer set. The resulting product was electrophoresed in 1% agarose, excised and purified using a DNA purification kit (Macherey-Nagel, Duren, Germany). Purified DNA was cloned into the pGEM-T Easy Vector (Promega, Madison, WI, USA), and its identity was checked by PCR and restriction endonuclease analysis with PstI. Volumes of 1 µl of plasmid DNA dilutions (10−1 to 10−8 ng/µl) were used as internal standards for each qPCR reaction. The number of target gene copies was calculated based on the DNA concentration and the molecular mass of the genome. Relative expression results at each time post-infection were subjected to analysis of variance (ANOVA) followed by Bonferroni means separation tests in SPSS ver. 17.0 (SPSS Inc.).
Virus growth kinetics
To examine budded virus (BV) production, 3×105 Sf9 cells were infected with 10 MOI of SfNIC-B, SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 BVs. Three supernatant samples were harvested from separate plates at 0, 2, 6, 12, 24, 48, 72, 96 and 120 h post-infection. Time zero was defined as the moment the virus inoculum was allowed to adsorb to the cells. The titers of supernatants were determined on Sf9 cells by end-point dilution [23]. Three independent infections were performed for each dilution. The experiment was performed three times. Results of BV production at different times post-infection were subjected to ANOVA followed by Bonferroni means separation tests; however for the samples taken at 2 h p.i. Kruskal-Wallis and Mann-Whitney tests were used as the data were not normally distributed. Critical α values were subjected to false discovery rate adjustment for multiple pairwise comparisons [24].
Determination of phenotypic characteristics
The insecticidal properties of OBs produced after injection of larvae with BVs from SfNIC-B, SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 were determined by insect bioassay following the droplet feeding method [22]. Groups of S. frugiperda second instars were starved for 8-12 h at 25°C and were then allowed to drink from an aqueous suspension containing 10% (w/v) sucrose, 0.001% (w/v) Fluorella blue and one of the following five concentrations of OBs: 1.2×106, 2.4×105, 4.8×104, 9.6×103 and 1.9×103 OBs/ml. This range of concentrations was previously determined to kill between 95 and 5% of the experimental insects [9]–[14]. Larvae that ingested the suspension within 10 min were transferred to individual wells of a 25-well tissue-culture dish with semisynthetic diet. Bioassays were performed three times using groups of 25 larvae per virus concentration and 25 mock-infected control larvae. Larvae were reared at 26°C and mortality was recorded every 8 h until insects had either died or pupated.
Virus induced mortality results were subjected to probit analysis using the Polo-Plus program [25]. OB pathogenicity was expressed as the 50% lethal concentration (LC50). Time mortality results of the four viruses were subjected to Weibull survival analysis in GLIM 4 [26]. As OBs of the different viruses differed significantly in potency, the OB concentrations used for the time mortality analysis were 1.65×106 OBs/ml for SfNIC-B, 8.98×105 OBs/ml for SfNIC-Bpif1, 9.22×106 OBs/ml for SfNIC-Begt and 1.77×107 OBs/ml for SfNIC-Bp10, that resulted in comparable mortalities of 85, 84, 86 and 84%, respectively. The experiment was performed four times.
OB production by each virus was determined in vitro. Infected Sf9 cells were harvested from the BV production experiment at different intervals post-infection, and were pelleted by low-speed centrifugation and washed once with 500 µl PBS. Cell pellets were resuspended in 25 µl TE and mixed with 25 µl of cell lysis buffer (50 mM Tris-HCl pH 8.0, 5% 2-mercaptoethanol, 0.4% w/v SDS, 10 mM EDTA). The resulting OB suspensions were quantified by direct counting in a bacterial counting chamber. OB counts from each suspension were performed three times. OB production results at 120 h p.i. were subjected to ANOVA followed by Bonferroni tests with false discovery rate adjustment for multiple pairwise comparisons [24].
OB production was also determined in insects. For this, S. frugiperda second instars that died from polyhedrosis disease in the time to death experiment were randomly selected from groups of 19–23 insects for each virus treatment in each repetition, representing a total of ∼80 larvae per virus treatment. Virus killed insects were individually stored at −20°C until used for OB counting. Each larva was thawed at room temperature, homogenized using a plastic pestle in a volume of 100 µl distilled water and serially diluted in distilled water. OB counts from each insect were performed in triplicate using a Neubauer hemocytometer. The results were normalized by logarithmic transformation and subjected to ANOVA and Bonferroni means separation.
Physical characteristics of viral Obs
OBs of each virus were characterized for DNA content, nucleocapsid numbers per virion, and mean virion titer per OB. The DNA content of OBs was determined by qPCR. For this, OB suspensions containing 5×108 OBs were mixed with 100 µl of 0.5 M Na2CO3, 50 µl of 10% SDS in a final volume of 500 µl and incubated for 10 min at 60°C. Undissolved OBs and other debris were removed by low-speed centrifugation (3,800 x g, 5 min). The supernant fraction containing released virions was treated with 25 µl of proteinase K (20 mg/ml) for 30 min at 50°C. Viral DNA was extracted twice with TE buffer (pH 8.0) saturated phenol and once with chloroform. Viral DNA was isolated by alcohol precipitation. The pellet was resupended in 100 µl of TE buffer for 10 min at 60°C. DNA samples in volumes of 1 µl were diluted 1∶100 and quantified by qPCR as previously described using the qSfBpif1.F/qSfBpif1.R primer set and standard curve. DNA was extracted from a total of nine samples and all reactions were measured in triplicate. The results were subjected to ANOVA and Bonferroni means separation.
To compare the distribution of numbers of nucleocapsids in virions of each virus, ODVs were harvested by adding 5×108 OBs of each virus to an equal volume of 0.1 M Na2CO3. The resulting suspensions were layered onto a continuous 30–60% sucrose gradient and centrifuged at 76,800 x g for 1 h at 4°C in a Beckman Ti70 rotor. The banding patterns of each virus were visually inspected and photographed.
Mean numbers of ODV infectious units per OB were determined by end-point dilution as described previously [23]. Twenty four independent infections were performed for each dilution. The experiment was performed 12 times. Cells were examined daily for the presence of viral OBs in the nuclei for up to one week. TCID50 values were estimated by Spearman-Kärber method and were subsequently converted to infectious units per 5×108 OBs for presentation in the figures.
Production of OBs comprising co-occluded genotype mixtures
In order to determine the relationship between the proportions of recombinant and deletion viruses and OB potency, different co-occluded mixtures were created in which SfNIC-Begt:SfNIC-C and SfNIC-Bp10:SfNIC-C genotypes were co-enveloped into virions and subsequently co-occluded into OBs at the desired proportions following the methodology described previously [10], [12]–[14], in which ODVs released from OB mixtures were injected into S. frugiperda larvae. Previous studies demonstrated that co-envelopment of different genotypes in ODVs occurred following injection of mixtures of genotypes into larvae [27]. This technique was found to be effective for the production of mixed genotype OBs that contained each genotype in approximately the same to the proportions in which they had been injected. For this, OBs of each recombinant virus (SfNIC-Begt or SfNIC-Bp10) were diluted to a concentration of 5×108 OBs/ml and were mixed with an identical concentration of deletion genotype SfNIC-C OBs in the following proportions: 90% recombinant:10% SfNIC-C, 75% recombinant:25% SfNIC-C, 50% recombinant:50% SfNIC-C, 25% recombinant:75% SfNIC-C, and 10% recombinant:90% SfNIC-C. ODVs were then released from OB mixtures by alkali disruption with a dissociation buffer (1 vol. OB suspension: 1 vol. 0.5M Na2CO3: 5 vol. H2O). Undissolved OBs were pelleted by low speed centrifugation at 2,700× g for 5 min. The ODV-containing supernatant was injected into groups of 50 S. frugiperda fourth instars (8 µl/larva). These larvae were individually maintained on semisynthetic diet until death. Extraction of OBs containing mixtures of co-occluded genotypes, OB purification and DNA extraction were then performed as previously described. DNA was extracted from nine independent samples of OBs.
qPCR reactions were performed to quantify SfNIC-Begt:SfNIC-C and SfNIC-Bp10:SfNIC-C ratios. For specific detection of SfNIC-Begt and SfNIC-Bp10 the qSfBpif1.F and qSfBpif1.R primers were used (Table S1). For detection of SfNIC-C, primers qSfCcath.F and qSfCsf36.F (Table S1) were designed around the 16.37 kb deletion that is characteristic of SfNIC-C genotype, located between nt 18,752 and 35,122 in the SfNIC-B genome [10], [11]. This primer set was selected based on the presence of a single melting peak. The SfNIC-C PCR product from a standard PCR reaction was cloned into pGEM-T Easy Vector as described above. Volumes of 1 µl of plasmid DNA containing the SfNIC-B and SfNIC-C PCR products were diluted (10−1–10−8 ng/µl), and used to construct standard curves. Non-template controls were also analyzed for each set of primers designed in order to verify the absence of non-specific background signal.
Prior to analysis, DNA samples from mixtures of SfNIC-Begt:SfNIC-C and SfNIC-Bp10:SfNIC-C OBs were used to calibrate the qPCR assay. DNA samples from SfNIC-Begt:SfNIC-C and SfNIC-Bp10:SfNIC-C OBs, and a range of mixtures (1∶103-103∶1) in 10-fold intervals were quantified by qPCR. In order to standardize the OB quantification, the amounts of DNA in the OBs of SfNIC-Begt, SfNIC-Bp10 and SfNIC-C were determined in a previous qPCR assay using the primers sets for SfNIC-Begt or SfNIC-Bp10 and SfNIC-C, described above. No significant differences were observed in the amounts of genomic DNA in samples of 5×108 OBs between the different viruses (p>0.05). Triplicate samples of the calibration mixtures were also included with the SfNIC-Begt:SfNIC-C and SfNIC-Bp10:SfNIC-C co-occluded mixtures in the qPCR assay. All reactions were performed in triplicate.
Results
Genomic characterization of viruses
The identity of OBs produced in insects was confirmed by sequencing of the PCR products obtained following amplification using Sfpif1.7-Sfpif1.9 primers, which revealed that the genomic arrangement of the recombinant viruses SfNIC-B, SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 differed only at the pif1/pif2 intergenic locus (Fig. 1C).
Temporal transcription of pif1 in reprogrammed viruses
Temporal regulation of pif1 transcription was examined by quantitative RT-PCR (qRT-PCR) using total RNA isolated from infected S. frugiperda larvae at different times post-infection (Table 1). Control reactions, performed to ensure the absence of contaminant DNA, did not result in amplification. The efficiency of the qRT-PCR was 104% (r2 = 0.9916), which indicated that this technique generated accurate estimates of target nucleic acid copies [28]. Transcripts of pif1 were first detected at a very low level at 12 h p.i. in S. frugiperda larvae infected with SfNIC-B or SfNIC-Bpif1 rescue virus, with estimated mean (±SD) copy numbers of 104±8.56 and 100±5.67 cDNA copies/µg total RNA, respectively. The abundance of pif1 transcripts increased by approximately 25-fold at 72 h p.i., with copy numbers estimated at 2,838±183 or 2,559±175 cDNA copies/µg RNA in larvae infected with SfNIC-B or SfNIC-Bpif1 viruses, respectively. The transcription pattern of pif1 in larvae infected with SfNIC-B or SfNIC-Bpif1 did not differ significantly between these viruses over time (ANOVA, Bonferroni test, p>0.05). In contrast, in larvae infected with SfNIC-Begt, pif1 transcripts were detectable by 4 h p.i. (130±9.64 cDNA copies/µg RNA), increased by 6-fold (734±74 cDNA copies/µg RNA) and 2,700-fold (351,368±12,108 cDNA copies/µg RNA) between 6 and 72 h.p.i. Similarly, in larvae infected with SfNIC-Bp10, pif1 transcription was detected at 24 h p.i. (2,583±154 cDNA copies/µg RNA), increasing 97-fold at 48 h.p.i. (250,850±18,936 cDNA copies/µg RNA) and by approximately 480-fold at 72 h p.i. (1,252,025±109,814 cDNA copies/µg RNA).
Table 1. Relative expression of pif1 (cDNA copies/µg RNA) in larvae infected with SfNIC-B and SfNIC-Bpif1 rescue viruses and SfNIC-Begt and SfNIC-Bp10 recombinant viruses.
| Hours post infection | Viruses | |||
| SfNIC-B | SfNIC-Bpif1 | SfNIC-Begt | SfNIC-Bp10 | |
| 0 | - | - | - | - |
| 2 | - | - | - | - |
| 4 | - | - | 130±10 | - |
| 6 | - | - | 734±74 | - |
| 8 | - | - | 1153±80 | - |
| 10 | - | - | 1939±101 | - |
| 12 | 104±9 | 101±6 | 5734±272 | - |
| 24 | 429±33 | 426±13 | 40241±4913 | 2584±154 |
| 48 | 1655±124 | 1439±69 | 180465±10284 | 180465±10284 |
| 72 | 2838±183 | 2559±176 | 351368±109814 | 1252025±109814 |
Transcript amplifications were performed using qSfBpif.F and qSfBpif1.R primers. Target gene copy numbers were calculated based on SfMNPV genome molecular mass and the standard curve. Values indicate means ± SD of three different repetitions measured twice for each sample.
qRT-PCR analysis of pif1 was performed on total RNA extracted from larvae infected with the different viruses at indicated times post-infection.
Overall, pif1 transcription was 123 to 137-fold higher or 441 to 489-fold higher in larvae infected with SfNIC-Begt or SfNIC-Bp10 at 72 h p.i. than in larvae infected with SfNIC-B or the rescue virus, respectively. Reprogramming of pif1 transcription resulted in earlier and higher transcription for SfNIC-Begt and in later and extremely high transcription for the SfNIC-p10 virus.
BV production occurred earlier in reprogrammed viruses
Virus growth curves from three independent experiments were compared for SfNIC-B and reprogrammed viruses (Fig. 2A). No significant differences were observed in the final BV titers among the different viruses. However, the growth curve kinetics differed significantly between viruses; BV production occurred earlier in cells infected with SfNIC-Begt or SfNIC-Bp10 compared to the parental or rescue viruses. No significant differences were observed in the BV titer of viruses at 2 h p.i. (Kruskal-Wallis K = 4.96, df = 3, p = 0.175), 6 h p.i. (F3,32 = 0.105, p = 0.957), or 12 h p.i. (F3,32 = 0.324, p = 0.808). By 24 h p.i. SfNIC-Bp10 and SfNIC-Begt presented significantly increased BV production (F3,32 = 14.1, p<0.001), whereas by 48 h p.i SfNIC-Bp10 and SfNIC-Begt produced approximately 11 and 15-fold more BVs, respectively, than SfNIC-B (F3,32 = 270, p<0.001). By 72 h p.i. BV titers were slightly higher in SfNIC-Begt and SfNIC-Bp10 infected cells (F3,32 = 4.89, p = 0.007), but at 96 h p.i. (F3,32 = 0.268, p = 0.853) and 120 h p.i. (F3,32 = 1.279, p = 0.298), BV production was similar among all viruses.
Figure 2. Virus production dynamics.
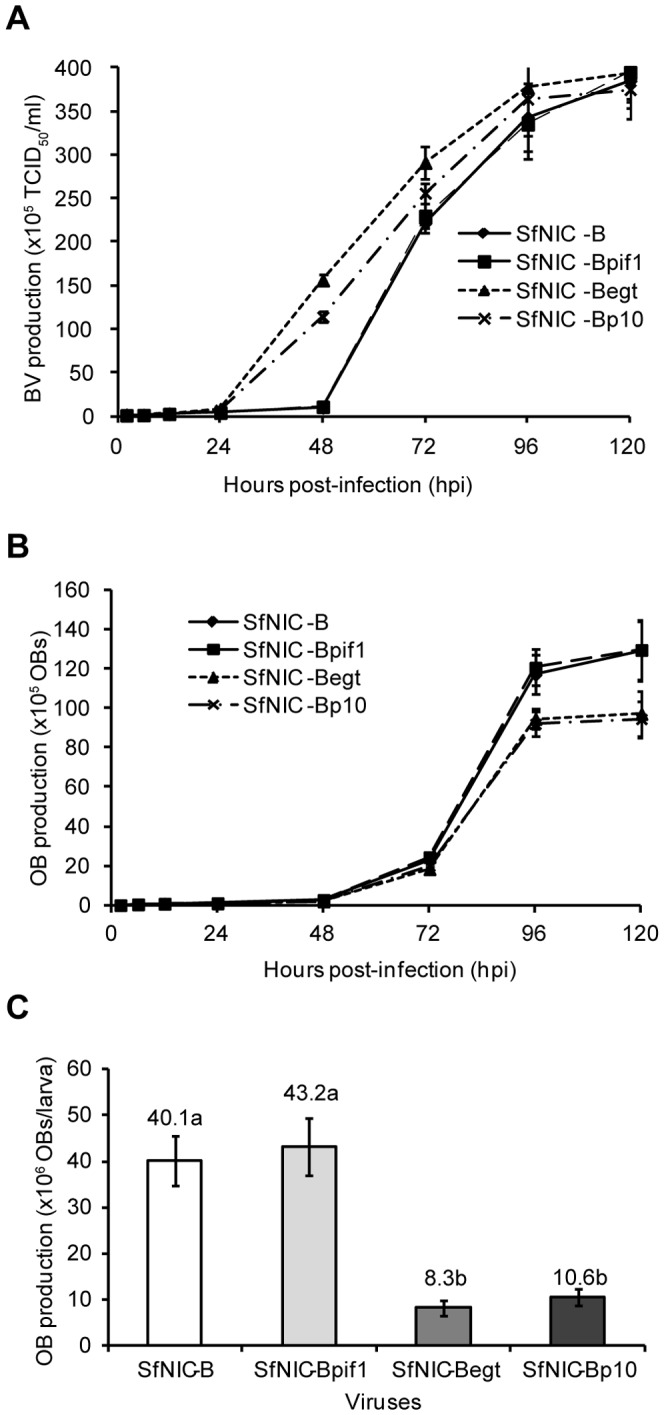
A) Budded virus (BV) production in Sf9 cells by one-step growth curve analysis. 3×105 cells were infected at 10 MOI using BV originating from SfNIC-B and SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 recombinant viruses. Supernatants harvested at indicated time points post infection were subjected to titer determination on Sf9 cells by end-point dilution. Each data point represents the average titer derived from three independent infections. The experiment was performed three times. Asterisks below hours-post infection values indicate significant differences in BV production between the viruses at indicated times. B) OB production values of SfNIC-B and SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 recombinant viruses at different times post infection in Sf9 cells (OBs/cell). Groups of 3×105 cells were infected at 10 MOI with BV originating from SfNIC-B and SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 recombinant viruses. Cells were collected at indicated time points post infection and subjected to OB production determination by direct counting in a Neubauer hemocytometer. Each data point represents the average titer derived from three independent infections. C) OB production values of SfNIC-B and SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 recombinant viruses in Spodoptera frugiperda second instars (OBs/larva). Figures above columns indicate the value of each column. Values followed by different letters were differed significantly (ANOVA, Bonferroni test, P<0.05).
Reprogramming pif1 expression resulted in reduced OB potency and OB production but increased virulence
The biological activity of OBs was compared by lethal concentration metrics (LC50) and mean time to death analysis in S. frugiperda second instars. No significant differences were observed in the potency of SfNIC-B and SfNIC-Bpif1 rescue OBs (Table 2). Reprogramming the pif1 promoter resulted in significantly lower potencies of SfNIC-Begt and SfNIC-Bp10 OBs, that were reduced by approximately 5 and 10-fold, respectively, compared to SfNIC-B or SfNIC-Bpif1 OBs. The differences in the relative potencies of SfNIC-Begt and SfNIC-Bp10 OBs were not statistically significant (Table 2).
Table 2. Probit regression and time to death analysis for SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 recombinant viruses compared with the SfNIC-B parental virus in Spodoptera frugiperda second instars.
| Viruses | Intercept ± S.E. | LC50 (OBs/ml) | Relative potency | Fiducial limits (95%) | Mean time to death (h) | Fiducial limits (95%) | ||
| Low High | Low High | |||||||
| SfNIC-B | −4.592±0.427 | 7.25×104 | 1.00 | - | - | 124a | 121 | 127 |
| SfNIC-Bpif1 | −5.296±0.467 | 6.22×104 | 1.17 | 0.71 | 1.93 | 121a | 118 | 124 |
| SfNIC-Begt | −5.056±0.509 | 3.60×105 | 0.20 | 0.11 | 0.37 | 100b | 98 | 103 |
| SfNIC-Bp10 | −5.435±0.584 | 7.33×105 | 0.10 | 0.05 | 0.19 | 99b | 97 | 101 |
Probit regressions were fitted using the PoloPlus program [25]. A test for non-parallelism was not significant (χ2 = 2.61, d.f. = 3, P = 0.456). Lines were fitted with a common slope of 0.927±0.099 (S.E). Relative potencies were calculated as the ratio of effective concentrations relative to the SfNIC-B virus. Mean time to death (MTD) values were estimated by Weibull survival analysis [26]. MTD values labelled with different letters were significantly different (P<0.05). The hazard function (α) was 5.0577.
The mean time to death of insects infected with the rescue virus SfNIC-Bpif1 did not differ significantly from that of insects infected with SfNIC-B (Table 2), whereas insects infected by SfNIC-Begt and SfNIC-Bp10 died, on average, 21 - 25 h earlier than those infected by the wild-type or rescue viruses.
OB production was determined in Sf9 cells at intervals up to 120 h p.i. (Fig. 2B). No differences were observed in OB yields among the four viruses in the first 72 h p.i. However, at 120 h p.i. the cells infected by recombinant viruses with pif1 expression driven by heterologous promoters produced approximately 25% fewer OBs than SfNIC-B or the SfNIC-Bpif1 rescue virus (F3,32 = 8.41, p<0.001).
OB production was also determined in S. frugiperda larvae (Fig. 2C). Total OB production/larva was approximately 4-fold lower in insects infected by SfNIC-Begt (8.20×106 OBs/larva) and SfNIC-Bp10 (1.06×107 OBs/ml), compared to those infected by SfNIC-B (4.01×107 OBs/ml), or the rescue virus (4.32×107 OBs/ml) (F3,301 = 23.002; p<0.001).
Reprogramming pif1 expression did not alter the physical characteristics of Obs
Selected characteristics of OBs were determined in order to exclude them as a potential explanation for the observed differences in biological potencies of OBs among the pif1 reprogrammed and parental viruses. No significant differences were detected by qPCR in the mean amounts of DNA in samples of 5×108 OBs (F3,104 = 0.989, p = 0.401), suggesting similar numbers of genome copies in OBs of each of the different viruses (Fig. 3A). The efficiency of the qPCR technique was 99% (r2 = 0.9914). No gross differences were observed in the number of ODV bands or their intensity in samples originating from equal numbers of OBs of each of the viruses (Fig. 3B), indicating that these viruses did not differ appreciably in the distribution of numbers of nucleocapsids among ODVs. Finally, ODV titers from samples of 5×108 OBs were estimated by end-point dilution (Fig. 3C), and did not differ significantly between any of the viruses tested (F3,44 = 0.919, p = 0.440).
Figure 3. DNA and genome content of Occlusion Bodies.
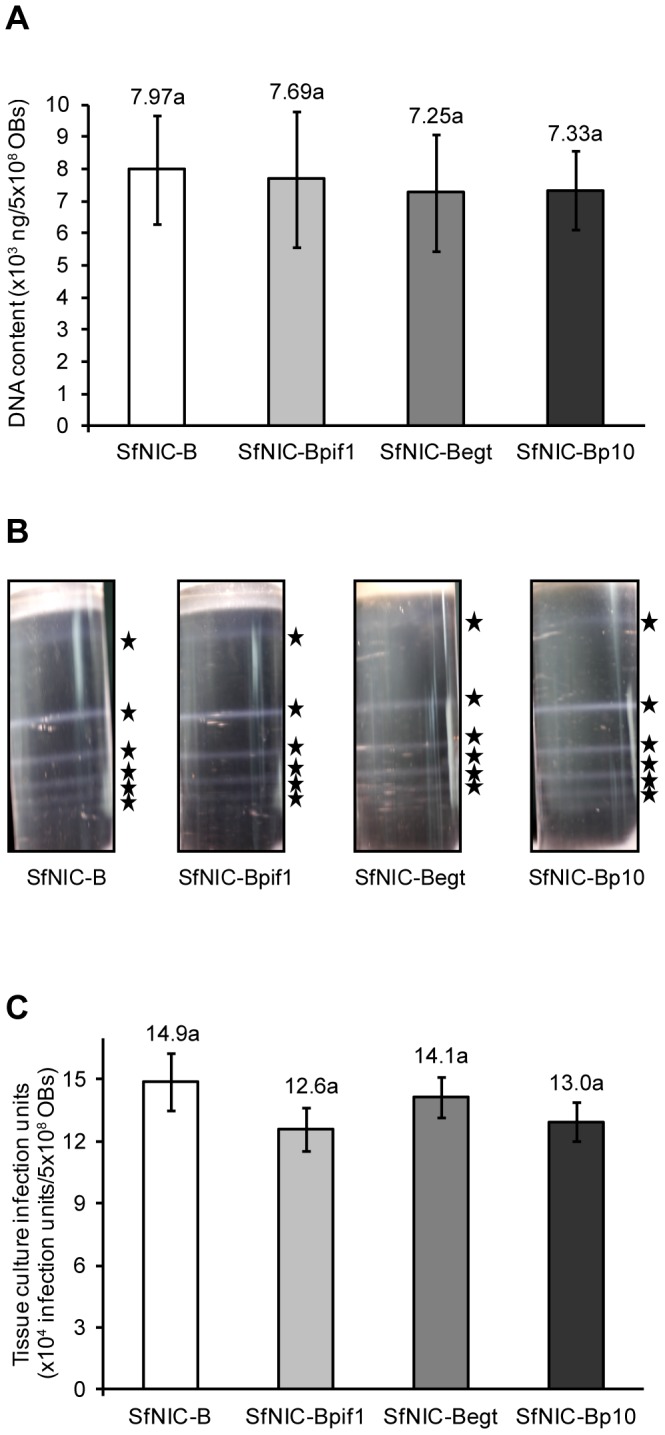
A) Mean amounts of DNA (ng/µl) extracted from samples of 5×108 OBs of SfNIC-B, SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 viruses and determined by qPCR. Mean amounts of DNA from OB samples did not differ significantly between viruses (ANOVA, P = 0.401). B) ODV banding patterns of SfNIC-B, SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 viruses after sucrose-gradient separation of ODVs from similar quantities of occlusion bodies (5×108 OBs). Stars indicate the positions of the observed ODV bands. C) ODV content in 5×108 OBs of SfNIC-B, SfNIC-Bpif1, SfNIC-Begt and SfNIC-Bp10 viruses. Sf9 cells were infected with serial dilutions (1∶10, 1∶50, 1∶250, 1∶1250, 1∶6250) of ODVs released from OBs. ODV titers (ODV/ml) were calculated by end-point dilution. No significant differences were observed in the ODV content of OBs of the different viruses (ANOVA, P = 0.440).
Reprogramming pif1 expression shifted the composition of cooperator-defector mixtures in favor of defectors
To determine whether reprogramming of pif1 resulted in a shift in the proportions of genotype mixtures that resulted in a high potency OB phenotype, such as observed in the wild-type population, insects were injected with mixtures of ODVs from pif1 reprogrammed virus and a natural defector genotype (SfNIC-C) in different proportions. The resulting co-occluded mixed genotype OBs comprising SfNIC-Begt:SfNIC-C and SfNIC-Bp10:SfNIC-C were analyzed by qPCR to confirm that genotypes were present at the proportions in which they were inoculated (Fig. 4). For qSfBpif1.F and qSfBpif1.R primers, qPCR efficiency was 98.0% (r2 = 0.9818), whereas for qSfCcath.F and qSfCsf36.F the efficiency was 97.7% (r2 = 0.9841). As observed previously, SfNIC-WT OBs were 2.65-fold more pathogenic than SfNIC-B OBs (Table 3). The potencies of OBs produced in larvae co-infected with mixtures containing 90, 25 and 10% of SfNIC-Begt were not significantly different from that of SfNIC-B alone. However the potency of mixed genotype OBs comprising 50% SfNIC-Begt +50% SfNIC-C (potency of 3.38) was equivalent to that of SfNIC-WT OBs (Table 3). Similar results were obtained with the mixtures involving the SfNIC-Bp10 virus; the potencies of OBs involving 90, 75, 25 and 10% of SfNIC-Bp10 did not differ significantly from that of SfNIC-B, whereas the co-occluded OB mixture comprising 50% of SfNIC-Bp10 +50% SfNIC-C (potency 2.99), was as potent as SfNIC-WT OBs (Table 3).
Figure 4. Relative proportions of SfNIC-Begt or SfNIC-Bp10 and SfNIC-C viruses in OBs comprising A) SfNIC-Begt:SfNIC-C and B) SfNIC-Bp10:SfNIC-C co-occluded mixtures at proportions of 90∶10, 75∶25, 50∶50, 25∶75 and 10∶90.

The relative proportion of each virus was determined by qPCR using DNA extracted from co-occluded mixed genotype OBs obtained after injection of S. frugiperda fourth instars using ODVs released from OB mixtures at the desired proportions. Primers qSfBpif1.F and qSfBpif1.R were used for amplification of SfNIC-Begt and SfNIC-Bp10, that did not amplify in SfNIC-C. Primers qSfCcath1.F and qSfCsf361.R were used for amplification of SfNIC-C.
Table 3. Probit regression analysis of virus induced mortality in Spodoptera frugiperda second instars inoculated with (I.) Wild-type SfMNPV (SfNIC-WT) and purified variant SfNIC-B occlusion bodies (OBs). (II.) OBs comprising co-occluded mixtures of SfNIC-Begt and SfNIC-C defector genotype in the proportions indicated. (III.) OBs comprising co-occluded mixtures of SfNIC-Bp10 and SfNIC-C defector genotype in the proportions indicated.
| Viruses | Intercept ± S.E. | LC50 (OBs/ml) | Relative potency | Fiducial limits (95%) | |
| Low High | |||||
| (I.) SfNIC-B | −4.696±0.447 | 1.23×105 | 1 | - | - |
| SfNIC-WT | −4.719±0.431 | 4.65×104 | 2.66 | 1.56 | 4.51 |
| (II.) SfNIC-Begt + SfNIC-C | |||||
| 90∶10 | 5.074±0.463 | 9.22×104 | 1.34 | 0.79 | 2.29 |
| 75∶25∶00 | −5.381±0.474 | 7.15×104 | 1.73 | 1.03 | 2.89 |
| 50∶50 | −5.339±0.467 | 3.65×104 | 3.38 | 2.03 | 5.63 |
| 25∶75 | −5.058±0.457 | 8.71×104 | 1.42 | 0.83 | 2.41 |
| 10∶90 | −4.934±0.463 | 1.48×105 | 0.83 | 0.48 | 1.45 |
| (III.) SfNIC-Bp10 + SfNIC-C | |||||
| 90∶10 | 4.993±0.455 | 1.00×105 | 1.23 | 0.72 | 2.11 |
| 75∶25 | −5.104±0.457 | 8.28×104 | 1.49 | 0.88 | 2.53 |
| 50∶50 | −4.934±0.442 | 4.12×104 | 2.99 | 1.78 | 5.04 |
| 25∶75 | −5.192±0.467 | 1.05×105 | 1.71 | 0.69 | 1.99 |
| 10∶90 | −4.730±0.449 | 1.51×105 | 0.82 | 0.46 | 1.44 |
Probit regressions were fitted using the PoloPlus program [25]. A test for non-parallelism was not significant (χ2 = 6.66, d.f. = 11, P = 0.826). Lines were fitted with a common slope of 1.022±0.093 (S.E). Relative potencies were calculated as the ratio of effective concentrations relative to the SfNIC-B virus that was assigned a nominal potency of 1.0.
Discussion
In the present study, we hypothesized that the SfMNPV population was structured to optimize the prevalence of PIF1-producing genotypes (cooperators) in the infected cells and hence, in progeny OBs produced for virus transmission. Increasing the intracellular abundance of PIF1 due to higher expression by a cooperator genotype in infected cells would therefore favor an increased prevalence of the defector genotype. This was investigated by producing two recombinant viruses, each with pif1 expression reprogrammed at its native locus using exogenous promoters. This approach has also proved useful for gene function analysis of other NPVs [29]–[31]. The egt and p10 promoters were selected, as the egt gene is an early transcribed gene [16], [17], whereas p10 is a very late and strongly transcribed gene [18], [19]. The transcription of pif1 under its homologous promoter is extremely weak [8], as confirmed in the present study, which is likely to be responsible for the low quantity of PIF1 produced in infected cells [7]. The relative transcription level of pif1 in insects infected with SfNIC-B or SfNIC-Bpif1 rescue viruses was ∼3.0×103 cDNA copies/mg RNA at 72 h.p.i. When reprogrammed, pif1 transcription was temporally-advanced (20h) and ∼130-fold higher with the SeMNPV egt promoter, whereas transcription was increased by ∼450-fold and delayed by 12 h when under the control of the SeMNPV p10 promoter. These substantial modifications allowed us to examine the consequences on the potency of mixed genotype OBs at the population level.
The dynamics of BV production in pif1-reprogrammed viruses were temporally advanced compared to those of parental and rescue viruses, although final BV titers were similar among all viruses. Temporal shifts in the patterns of replication of pif1-modified virus were previously observed using a reporter gene based assay, which also indicated that final titers of PIF1 appeared to be similar in pif1-reprogrammed and parental viruses [32]. The reason for this is not clear. Modifying pif1 expression might affect the temporal expression of other genes that are directly or indirectly related to BV production, such as observed in another NPV gene [33], or may modify the course of the infection.
The pif1 reprogrammed viruses killed their hosts markedly faster than the SfNIC-B and rescue viruses, possibly due to the earlier onset of systemic infection in insects infected by the pif1 reprogrammed viruses. BV production following ingestion of high doses of OBs determines the rate of spread of infection that is positively correlated with speed of kill in other baculoviruses [33]–[35]. As a result of the rapid demise of pif1 reprogrammed virus-infected hosts, OB production in pif1 reprogrammed viruses was reduced by one quarter in vitro and by ∼4-fold in insects compared to the parental and rescue viruses, reflecting the well-established tradeoff between speed of kill and OB production in baculovirus infected insects [36]–[39]. BV production was advanced by 48 hours in these reprogrammed viruses.
Improvement of the speed of kill has been one of the major research objectives for the development of recombinant baculoviruses as the basis for bioinsecticidal products. Two main approaches have been employed: the expression of insecticidal toxins, enzymes or hormones [40]–[43], the deletion of life-stage manipulating virus genes [44], or a combination of both [45]. In the present study, we demonstrated that a different approach based on the modification of the expression of a virus core gene resulted in a significant improvement in speed of kill. Although in the case of pif1, reprogramming of expression resulted in reduced OB potency that is undesirable for the development of virus insecticides, the concept of reprogramming viral gene expression opens diverse possibilities in the improvement of baculoviruses for pest control.
The potency of OBs produced by pif1 reprogrammed viruses was approximately one logarithm lower than that of SfNIC-B and rescue viruses, in terms of concentration-mortality metrics. There could be two possible causes for this reduction in the insecticidal properties of OBs: first, that the physical composition of OBs was altered in reprogrammed viruses, although no significant differences were observed in the DNA content of OBs, distribution of numbers of nucleocapsids in ODVs, or the infectivity of the ODVs in cell culture. This leads us to favor the second hypothesis, that increased pif1 expression reduced the infectivity of ODVs compared to parental and rescue viruses.
A marked increase in the abundance of pif1 transcripts might result in an increase in the intracellular pool of PIF1. The resulting accumulation of PIF1 in ODVs may have influenced the functionality or integrity of the complex of PIF factors required for ODV infectivity during primary infection [5]. We suggest that this is likely to be the reason for the reduced potency of reprogrammed virus OBs in per os infected larvae.
Finally, in line with the concept that PIF1 concentration in ODVs is decisive in determining OB potency, co-occlusion of pif1 reprogrammed virus and a pif1/pif2 deficient genotype (SfNIC-C) resulted in an OB potency phenotype similar to that of the wild-type isolate, at a ratio of 50∶50 (cooperator: defector). As we predicted a priori, this ratio was shifted in favor of the defector genotype when compared to the 75∶25 mixture that previously restored wild-type potency to OBs comprising natural cooperator + defector genotypes (SfNIC-B + SfNIC-C, respectively) [12], [15]. In addition, as observed previously [13], [14], during five serial passages in larvae the proportions of each genotype in mixtures converged to an equilibrium ratio that maximized the likelihood of transmission. Moreover, once equilibrium frequencies of genotypes have been achieved, the proportions of genotypes in mixed genotype nucleopolyhedrovirus populations remains stable over successive passages [13], [14].
A significant proportion of ODVs contain a mixture of genotypes [24], and following ingestion of OBs multiple foci of primary infection are usually observed in the insect midgut [33]. These two factors favor transmission of a mixture of cooperator and defector genotypes. During the systemic phase of disease, each cell of a caterpillar is infected by multiple genomes (average 4.3 budded virions per cell) [46]. Such small group sizes tend to favor cooperative behavior among their members, as the costs of hosting defectors is proportionally higher than for large groups [47]. As PIF1 produced by cooperator genotypes was available to all genotypes in a particular cell, pif1 expression appears to modulate group-specific fitness and therefore represents a cooperative trait.
Game theory models often predict maximal group fitness when defectors are absent [48], [49]. Some exceptions to this general rule include excess production of goods leading to inefficient use and diminishing benefits to group members [50]. In the case of our study, PIF1 production represents an unusual case in which group success (transmission) depends on production of this component, but overproduction is highly prejudicial to group-specific transmissibility, both in terms of OB pathogenicity and total OB yield from each infected insect. Essential goods are usually a source of competition, as each individual tries to maximize the amount they can acquire [51]. In our model, however, this required resource, PIF1, appears to be deleterious if present in amounts higher than required. As a result, the presence of defectors that effectively dilute the intercelluar pool of PIF1 is necessary and beneficial to the entire virus population. When pif1 expression is manipulated, the level of defectors in the population shifts to compensate the variation in the amounts of PIF1 available. This is reflected in the genotypic composition of the OBs produced [13]. These results offer a new and unexpected perspective on cooperative behavior between viral genomes in response to the abundance of an essential public resource that is detrimental in excess.
Supporting Information
Primers used in this study.
(DOCX)
Acknowledgments
We thank N. Gorria and I. Ibáñez (Universidad Pública de Navarra, Pamplona, Spain) and Marcel Mariller (Baculovirus et Therapie, UPS 3044, CNRS, Saint Christol les Alés, France) for technical assistance.
Funding Statement
This study received financial support from the Spanish Ministry for Science and Technology projects AGL2005-07909-CO3-01 and AGL2008-05456-CO3-01/AGR. O.S. received a José Castillejo grant for postdoctoral mobility. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Moscardi F (1999) Assessment of the application of baculoviruses for the control of Lepidoptera. Annu Rev Entomol 44: 257–289. [DOI] [PubMed] [Google Scholar]
- 2. Caballero P, Murillo R, Muñoz D, Williams T (2009) The nucleopolyedrovirus of Spodoptera exigua (Lepidoptera; Noctuidae) as a biopesticide: analysis of recent advances in Spain. Rev Colomb Entomol 35: 105–115. [Google Scholar]
- 3. Ohkawa T, Washburn JO, Sitapara R, Sid E, Volkman LE (2005) Specific binding of Autographa californica M nucleopolyhedrovirus occlusion-derived virus to midgut cells of Heliothis virescens larvae is mediated by products of pif genes Ac119 and Ac022 but not by Ac115 . J Virol 79: 15258–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horton HM, Burand JP (1993) Saturable attachment sites for polyhedron-derived baculovirus on insect cells and evidence for entry via direct membrane fusion. J Virol 67: 1860–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peng K, van Lent JWM, Boeren S, Fang M, Theilmann DA, et al. (2012) Characterization of novel components of the baculovirus per os infectivity factor complex. J Gen Virol 86: 4981–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peng K, van Oers MM, Hu Z, van Lent JWM, Vlak JM (2010) Baculovirus per os infectivity factors form a complex on the surface of occlusion derived virus. J Virol 84: 9497–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kikhno I, Gutiérrez S, Croizier L, Croizier G, López-Ferber M (2002) Characterization of pif, a gene required for the per os infectivity of Spodoptera littoralis nucleopolyhedrovirus. J Gen Virol 83: 3013–3022. [DOI] [PubMed] [Google Scholar]
- 8. Gutiérrez S, Kikhno I, López-Ferber M (2004) Transcription and promoter analysis of pif, an essential but low-expressed baculovirus gene. J Gen Virol 85: 331–341. [DOI] [PubMed] [Google Scholar]
- 9. Simón O, Williams T, López-Ferber M, Caballero P (2004) Genetic structure of a Spodoptera frugiperda nucleopolyhedrovirus population: high prevalence of deletion genotypes. Appl Environ Microbiol 70: 5579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simón O, Williams T, López-Ferber M, Caballero P (2005) Functional importance of deletion mutant genotypes in an insect nucleopolyhedrovirus population. Appl Environ Microbiol 71: 4254–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simón O, Palma L, Beperet I, Muñoz D, López-Ferber M, et al. (2011) Sequence comparison between three geographically distinct Spodoptera frugiperda multiple nucleopolyhedrovirus isolates: detecting positively selected genes. J Invertebr Pathol 107: 33–42. [DOI] [PubMed] [Google Scholar]
- 12. López-Ferber M, Simón O, Williams T, Caballero P (2003) Defective or effective? Mutualistic interactions between virus genotypes. Proc R Soc B 270: 2249–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simón O, Williams T, Caballero P, López-Ferber M (2006) Dynamics of deletion genotypes in an experimental insect virus population. Proc R Soc B 273: 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simón O, Williams T, López-Ferber M, Caballero P (2008) Population genetic structure determines speed of kill and occlusion body production in Spodoptera frugiperda multiple nucleopolyhedrovirus. Biol Control 44: 321–330. [Google Scholar]
- 15. Clavijo G, Williams T, Simón O, Muñoz D, Lopez-Ferber M, et al. (2009) Mixtures of complete and pif1/pif2 deficient genotypes are required for increased potency of an insect nucleopolyhedrovirus. J Virol 83: 5127–5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evans OP, O'Reilly DR (1999) Expression and structural characterization of a baculovirus ecdysteroid UDP-glucosyltransferase. J Gen Virol 80: 485–492. [DOI] [PubMed] [Google Scholar]
- 17. O'Reilly DR, Miller LK (1990) Regulation of expression of a baculovirus ecdysteroid UDP-glucosyltransferase gene. J Virol 63: 1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Oers MM, Flipsen JTM, Reusken CBEM, Sliwinsky EL, Goldbach RW, et al. (1993) Functional domains of the p10 protein of Autographa californica nuclear polyhedrosis virus. J Gen Virol 74: 563–574. [DOI] [PubMed] [Google Scholar]
- 19. Weyer U, Possee RD (1989) Analysis of the promoter of the Autographa californica nuclear polyhedrosis virus p10 gene. J Gen Virol 70: 203–208. [DOI] [PubMed] [Google Scholar]
- 20. Greene GL, Leppla NC, Dickerson WA (1976) Velvetbean caterpillar: a rearing procedure and artificial medium. J Econ Entomol 69: 487–488. [Google Scholar]
- 21. IJkel WF, van Strien EA, Heldens JG, Broer R, Zuidema D, et al. (1999) Sequence and organization of the Spodoptera exigua multicapsid nucleopolyhedrovirus genome. J Gen Virol 80: 3289–3304. [DOI] [PubMed] [Google Scholar]
- 22.Hughes PR, Wood HA (1987) In vivo and in vitro bioassay methods for baculoviruses. In: Granados RR, Federici BA, editors. The biology of baculoviruses Boca Raton: CRC Press. pp. 1–30.
- 23. Lynn DE (1992) Improved efficiency in determining the titer of the Autographa californica baculovirus nonoccluded virus. Biotechniques 13: 282–5. [PubMed] [Google Scholar]
- 24. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc 57: 289–300. [Google Scholar]
- 25.LeOra Software (1987) Polo-PC a user's guide to probit or logit analysis. California: Berkeley.
- 26.Crawley MJ (1993) GLIM for ecologists. Oxford: Blackwell.
- 27. Clavijo G, Williams T, Muñoz D, Caballero P, López-Ferber M (2010) Mixed genotype transmission bodies and virions contribute to the maintenance of diversity in an insect virus. Proc R Soc B 277: 943–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bustin SA, Benes V, Garson JA, Hellmans J, Huggett J, et al. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chim 55: 611–622. [DOI] [PubMed] [Google Scholar]
- 29. Bonning BC, Roelvink PW, Vlak JM, Possee RD, Hammock BD (1994) Superior expression of juvenile hormone esterase and β-galactosidase from the basic protein promoter of Autographa californica nuclear polyhedrosis virus compared to the p10 protein and polyhedrin promoters. J Gen Virol 75: 1551–1556. [DOI] [PubMed] [Google Scholar]
- 30. Hodgson JJ, Arif BM, Krell PJ (2007) Reprogramming the chiA expression profile of Autographa californica multiple nucleopolyhedrovirus. J Gen Virol 88: 2479–2787. [DOI] [PubMed] [Google Scholar]
- 31. Knebel-Mörsdorf D, Flipsen JTM, Roncarati R, Jahnel F, Kleefsman AWF, et al. (1996) Baculovirus infection of Spodoptera exigua larvae: lacZ expression driven by promoters of early genes pe38 and me53 in larval tissue. J Gen Virol 77: 815–824. [DOI] [PubMed] [Google Scholar]
- 32. Gutiérrez S, Mutuel D, Grard N, Cerutti M, López-Ferber M (2005) The deletion of the pif gene improves the biosafety of the baculovirus. J Biotechnol 116: 135–143. [DOI] [PubMed] [Google Scholar]
- 33. Washburn JO, Chen EY, Volkman LE, Aumiller JJ, Jarvis DL (2003) Early synthesis of BV envelope fusion protein GP64 enhances AcMNPV virulence in orally-infected Heliothis virescens . J Virol 77: 280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Washburn JO, Lyons EH, Haas-Stapleton EJ, Volkman LE (1999) Multiple nucleocapsid packaging of Autographa californica nucleopolyhedrovirus accelerates the onset of systemic infection in Trichoplusia ni . J Virol 73: 411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McNeil J, Cox-Foster D, Gardner M, Slavicek J, Thiem S, et al. (2010) Pathogenesis of Lymantria dispar multiple nucleopolyhedrovirus in L. dispar and mechanisms of developmental resistance. J Gen Virol 91: 1590–1600. [DOI] [PubMed] [Google Scholar]
- 36. Cory JS, Green BM, Paul RK, Hunter-Fujita F (2005) Genotypic and phenotypic diversity of a baculovirus population within an individual insect host. J Invertebr Pathol 89: 101–111. [DOI] [PubMed] [Google Scholar]
- 37. Hernández-Crespo P, Sait SM, Hails RS, Cory JS (2001) Behavior of a recombinant baculovirus in lepidopteran hosts of different susceptibilities. Appl Environ Microbiol 67: 1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hodgson DJ, Hitchman RB, Vanbergen AJ, Hails RS, Possee RD, et al. (2004) Host ecology determines the relative fitness of virus genotypes in mixed-genotype nucleopolyhedrovirus infections. J Evol Biol 17: 1018–1025. [DOI] [PubMed] [Google Scholar]
- 39. Hodgson DJ, Vanbergen AJ, Watt AD, Hails RS, Cory JS (2001) Phenotypic variation between naturally coexisting genotypes of a lepidopteran baculovirus. Evol Ecol Res 3: 687–701. [Google Scholar]
- 40. Hammock BD, Bonning BC, Possee RD, Hanzlik TN, Maeda S (1990) Expression and effect of the juvenile hormone esterase in a baculovirus vector. Nature 344: 458–461. [Google Scholar]
- 41. Maeda S (1989) Expression of foreign genes in insects using baculovirus vectors. Annu Rev Entomol 34: 351–372. [DOI] [PubMed] [Google Scholar]
- 42. Stewart LMD, Hirst M, López-Ferber M, Merryweather AT, Cayley PJ, et al. (1991) Construction of an improved baculovirus insecticide containing an insect-specific toxin gene. Nature 352: 85–88. [DOI] [PubMed] [Google Scholar]
- 43. Tomalski MD, Miller LK (1991) Insect paralysis by baculovirus-mediated expression of a mite neurotoxin gene. Nature 352: 82–85. [DOI] [PubMed] [Google Scholar]
- 44. O'Reilly DR, Miller LK (1991) Improvement of a baculovirus pesticide by deletion of the egt gene. BioTechnol 9: 1086–1089. [Google Scholar]
- 45. Bonning BC, Hoover K, Booth TF, Duffey S, Hammock BD (1995) Development of a recombinant baculovirus expressing a modified juvenile hormone esterase with potential for insect control. Arch Insect Biochem Physiol 30: 177–194. [Google Scholar]
- 46. Bull JC, Godfray HCJ, O'Reilly DR (2001) Persistence of an occlusion-negative recombinant nucleopolyhedrovirus in Trichoplusia ni indicates high multiplicity of cellular infection. Appl Environ Microbiol 67: 5204–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hauert C, Holmes M, Doebeli M (2006) Evolutionary games and population dynamics: maintenance of cooperation in public goods games. Proc R Soc B 273: 2565–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gore J, Youk H, van Oudenaarden A (2008) Snowdrift game dynamics and facultative cheating in yeast. Nature 459: 253–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Travisano M, Velicer GJ (2004) Strategies of microbial cheater control. Trends Microbiol 12: 72–78. [DOI] [PubMed] [Google Scholar]
- 50. MacLean RC, Fuentes-Hernández A, Greig D, Hurst LD, Gudelj I (2010) A mixture of “cheats” and “co-operators” can enable maximal group benefit. PLoS Biol 8: e1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Frank SA (2010) A general model of the public goods dilemma. J Evol Biol 23: 1245–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study.
(DOCX)