

Abstract
Objective
To define the relative frequencies of different mechanisms of viral escape.
Design
A population-based approach to examine the distribution of HIV polymorphism associated with diverse population human leucocyte antigens (HLAs) at sites within and flanking CD8 T-cell epitopes as a correlate of likely mechanisms of viral escape.
Methods
Sequence windows surrounding 874 HLA allele-specific polymorphisms across the full HIV-1 proteomic consensus sequence were scanned by an epitope-prediction programme. Either already known or probable CD8 T-cell epitopes with HLA restriction matching that of the proximal HLA association were identified and synthesized. These peptides were used as stimulating antigens in automated enzyme-linked immunospot (ELISpot) assays. Peptide arrays were customized to each individual based on their HLA genotype.
Results
Among HLA-associated HIV polymorphisms detected in the viral sequences of a cohort of 800 individuals with chronic subtype B HIV infection, those which were likely to affect HLA peptide binding were significantly more common than polymorphisms at nonanchor HLA binding sites. HIV epitopes with such polymorphisms were associated with reduced IFNγ responses in ELISpot assays. HIV escape at sites affecting T-cell receptor (TCR) engagement and epitope processing were also evident.
Conclusion
HIV escape from HLA-peptide binding predominates as an effective viral evasion strategy and therefore has implications for inclusion of HLA-adapted epitopes in vaccine immunogens.
Keywords: CD8, epitope, escape mutation, HIV-1, HLA-driven, polymorphism, T-cell immunity
Introduction
The selective outgrowth of HIV-1 genetic variants, which have mutations within or near CD8 T-cell epitopes, allows viral escape from CD8 T-cell immunity [1]. Viral escape from clonal antigen-specific responses plays a key role in establishing chronic replicative HIV infection [2] and subsequent progression of HIV-associated disease [3]. Mutations, which disrupt epitope binding to human leucocyte antigens (HLAs) occur at codons for anchor-binding sites within epitopes, at position 2 and the C-terminus residue for most epitopes. Those mutations occurring at other sites in the epitope disrupt contact between TCRs and the HLA-peptide complex. Mutations flanking or within epitopes may also allow viral escape by changing the proteosomal cleavage sites necessary for intracellular generation of the epitope from precursor proteins or TAP-dependent transport into the endoplasmic reticulum [4,5]. Primary escape mutations that reduce replicative capacity are often linked to secondary mutations that (partially) restore viral fitness under selection pressure and tend to be proximal to primary adaptation sites [6]. It is now well established that these phenomena occur throughout all HIV genes and in association with a wide array of population HLA genotypes. As such, CD8 T-cell escape is evident as mutational networks within the HIV genome, which are HLA allele-specific at the population level [7–10]. Vaccine concepts have been devised to incorporate HIV antigenic diversity into polyvalent immunogens in order to provide broad population-level immunogenicity [11]. Given the significant proportion of HIV diversity, which is driven by viral adaptation to HLA, viral adaptation which results specifically in loss of HLA-peptide binding or epitope processing in prevalent transmitting strains would remove these antigens as targets for vaccine-induced immune responses completely. In contrast, epitopes with TCR escape mutations may still bind cross reactive or new T-cell clonotypes. It is therefore of interest to know the relative frequency with which HIV escapes TCR recognition, versus HLA-peptide binding and epitope processing. Differences in the frequency of these different escape mechanisms between HIV proteins, which vary in cleavage site density, structural/functional constraint, plasticity and other biological characteristics, have not been described previously. In addition, differential susceptibilities of host-HLA loci may be revealed by examining preferential modes of viral evasion associated with them. To date, detailed mechanisms of viral escape have been described in the context of a small number of specified HLA-epitope combinations. Here, we used a population-based approach to examine the distribution of HIV polymorphism associated with diverse population HLA at sites within and flanking CD8 T-cell epitopes as a correlate of likely mechanisms of viral escape.
Methods
The genetic analysis underpinning the functional data analyzed in this study was derived from a previously published genome-wide analysis of HLA-associated polymorphism in 800 antiretroviral-naive individuals from the US and Australia with chronic subtype B HIV-1 infection [10]. A subset of 290 individuals from the US cohort who had sufficient pretreatment cryopreserved peripheral blood mononuclear cells (PBMC) were then tested in a subsequent functional study in which the genetic selection imprints were used to characterize prevalent epitope-specific CD8 T-cell responses mediating selective pressure in the population [12]. Specific analysis of the epitope distribution of HLA-associated polymorphisms affecting a dataset of nonadapted/adapted epitope pairs identified in the original genetic study [10] and tested in the subsequent functional study [12] is presented here. All US study participants were co-enrolled in AIDS Clinical Trials Group (ACTG) protocol A5142 and A5128 [13,14] and provided written informed consent to HIV sequencing and cryopreservation of PBMC at a pretreatment time point as well as HLA-A, HLA-B and HLA-C genotyping.
Detailed methods for computation of HLA-HIV phylogeny-adjusted associations, epitope prediction and synthesis as well as the cellular testing protocol have been described previously [10,12]. Briefly, the epitope-prediction tool, Epipred, was used to scan sequence windows surrounding 874 HLA allele-specific polymorphisms across the full HIV-1 proteomic consensus sequence [15]. This prediction algorithm is trained on characteristics of known CD8 T-cell epitopes taking into account HLA-specific peptide-binding motifs, TCR contact residues, epitope length and flanking sequences to generate a probability score for any given peptide sequence being a CD8 T-cell epitope [15]. 8–11mer sequences that were either already known or probable CD8 T-cell epitopes with HLA restriction matching that of the proximal HLA association were identified [10]. Epitopes were then synthesized as peptides and used as stimulating antigens in automated enzyme-linked immunospot (ELISpot) assays at a final well concentration of 5 μg/ml. Peptide arrays were customized to each individual based on their HLA genotype and anti-CD3 antibody was used for positive controls. PBMCs cultured in media alone served as negative controls.
In order to rationalize testing of peptides in each individual when there was insufficient PBMCs to test all possible known and predicted epitopes, ranking criteria were used to prioritize testing of novel putative epitopes over already well characterised published epitopes or minor variants of known epitopes [12]. Overall we examined responses to 159 nonadapted epitopes together with their corresponding adapted (variant) sequences. As HLA-associated polymorphism could result in multiple alternative amino acid substitutions and occur at multiple positions within the epitope, there was more than one corresponding adapted sequence for a nonadapted epitope in many cases. Hence, there were 222 adapted peptide sequences in total. Of these, 194 cases reflected intra-epitopic variation, most likely impacting peptide binding to the HLA molecule or TCR recognition. There were also 28 instances in which HLA-associated polymorphism was predicted to create ‘neoepitopes’ which were overlapping or distant from the nonadapted epitope. We have previously defined neoepitopes as adapted peptide sequences which, though containing a HLA-associated polymorphism, were nevertheless predicted to elicit a T-cell response as identified by the epitope-prediction algorithm [10,12]. Of all paired peptides considered, 65% of the nonadapted epitopes investigated were known, published epitopes whereas, the remaining peptides were predicted to be epitopes by Epipred. There were 64 cases of putative antigen processing adaptations in which the amino acid under selection lay outside the nonadapted epitope and no alternative neoepitope was predicted, consistent with complete abrogation of epitope presentation in vivo.
IFNγ-producing cells were enumerated as spot forming units (SFUs) using an AID ELISPOT reader (Auto-immun-Diagnostika, Strassberg, Germany). All sample well SFU readings were determined after subtraction of negative control values. A response was considered positive if greater than twice the mean of the background and greater than or equal to 100 SFUs/106 PBMC [12].
The position of each HLA-associated HIV-viral polymorphism relative to the proximal known or predicted nonadapted epitope with matching HLA-restriction was recorded. The frequency distribution of polymorphism occurring within four amino acids flanking epitopes and from N-terminus to C-terminus positions within the epitope were determined and plotted. These distributions were determined for both the set of nonadapted CD8 T-cell epitopes tested in the study as well as the subset, which elicited positive IFNγ responses in our testing.
Anchor site polymorphism largely occurs at position 2 or C-terminus positions of CD8 T-cell epitopes (position 8 to position 11 depending on peptide length). In order to conduct formal statistical analyses of anchor site polymorphism enrichment, only epitopes of equivalent length (9mer) were included to allow equal probability of polymorphisms at all residues and 9mers represented the majority of epitopes in the study. We compared the observed proportions to the 2/9 expected if polymorphism was equivalent between anchor (position 2 and C-terminus) and nonanchor sites (Binomial test). Response rates were evaluated by linear regression within a generalized estimating equation framework, taking account of the multiple epitopes tested per individual. Included covariates considered site of polymorphism, HLA loci, relative reactivity of adapted versus nonadapted epitopes and impact of autologous viral sequence. Statistical analyses were undertaken using TIBCO Spotfire S+ 8.2 for Windows (TIBCO Software Inc., Palo Alto, California, USA).
Results
Overall data from 238 individuals were examined in this study. An average of 11.3 peptides were tested per individual with an average of 2.4 positive epitope-specific IFNγ responses detected per individual.
In total, 91 HLA-restricted epitopes (of the 159 included in this study) showed at least one positive IFNγ response to the nonadapted peptide. In order to exclude a bias associated with analysis of only tested nonadapted/adapted-paired epitopes, we compared average response rates in this dataset with that found in the overall dataset analysed in Almeida et al. [12] and found no significant difference (P =0.9).
The distribution of HLA-associated polymorphism across all analyzed epitopes is shown in dark grey bars in Fig. 1. Although polymorphisms occurred at all positions, position 2 and C-terminus positions (position 8 to position 11 depending on peptide length) were subject to HLA allele-specific polymorphism most frequently. Among peptides, which elicited a positive IFNγ response (light grey bars in Fig. 1), variation at position 2 was most common, followed by changes at the C-terminus position, and position 3.
Fig. 1.

Distribution of the positions of amino acid substitution against the nonadapted peptide across all proteins in all tested peptides (dark grey bars) and all peptides evoking an IFNγ positive response in at least one individual (light grey bars). N, N-terminus, P, intra-epitopic positions from N-terminus to C-terminus, C, C-terminus.
Formal analysis of enrichment of anchor site polymorphism was restricted to the dataset of 9mer epitopes with intra-epitopic polymorphism only (n =106). Overall, HLA-driven changes were significantly more frequent at the anchor-binding sites, position 2 and position 9, relative to the other sites (41 of 106 epitopes, 39%, P =0.0001), although the observed enrichment did not reach statistical significance when restricting the analysis to epitopes associated with positive IFNγ responses (19 of 61epitopes, 31%, P =0.12).
The polymorphism distributions were then visually compared across individual HIV proteins to observe if there were protein-specific patterns of adaptation. Plots of these distributions are shown for Gag, Pol and Nef as remaining HIV proteins had low numbers of epitopes tested (Fig. 2). Adaptation in Gag appeared to predominate at position 7 and position 9 in all tested epitopes and at position 9 for those epitopes associated with positive IFNγ responses (Fig. 2a). In Pol, position 2, position 3 and position 9 were most frequently subject to HLA-associated viral adaptation in all tested peptides although escape at position 2 remained predominant among IFNγ-inducing epitopes (Fig. 2b). Polymorphism in primary anchor binding sites therefore predominated across both Gag and Pol. Nef appeared to have a more evenly distributed profile of viral adaptation, across N-terminus and C-terminus flanking regions as well as at all positions within epitopes, with a predominance of polymorphism at position 2 and position 1. Among IFNγ responsive epitopes, polymorphism was most commonly observed at position 1 and position 9 (Fig. 2c). The 9mer epitopes restricted to HLA-A alleles showed significant predominance of anchor binding escape using the set of tested (17 of 35 epitopes, 49%, P =0.0007) and IFNγ-responsive epitopes (8 of 16 epitopes, 50%, P =0.01). However, this observed anchor site polymorphism enrichment was less evident amongst HLA-B restricted epitopes (22 of 61 epitopes, 36%, P =0.01, and 11 of 40 epitopes, 28%, P =0.4, for tested and responsive epitope sets, respectively), and lacking for HLA-C restricted epitopes (none of 10 epitopes, P >0.9, and none of eight epitopes, P =0.4, for tested and responsive epitope sets, respectively).
Fig. 2.

Distribution of the positions of amino acid substitution against the nonadapted peptide in all tested peptides (dark grey bars) and all peptides evoking an IFNγ positive response in at least one individual (light grey bars) in Gag (a), Pol (b) and Nef (c).
Across all epitopes considered, average IFNγ response rates to adapted peptide sequences were significantly reduced for epitopes with adaptation at position 2 in particular compared with those having mutation at nonanchor positions (response rate 6.6% for position 2 adaptations, n =46, versus 19.7%, n =142, P <0.0001), whereas IFNγ responses to epitopes with the mutation at the C-terminus were comparable to those with the mutation at nonanchor positions (19.0%, n =34, P =0.8). In general, average IFNγ response rates to adapted epitopes were significantly lower than the nonadapted epitopes. This effect was observed in epitopes in which escape had occurred at a nonanchor position (response rate to adapted epitopes 21% versus nonadapted 29%, P <0.0001), as well as epitopes in which mutation had occurred at an anchor position (13 versus 16%, P =0.03), and irrespective of whether or not the peptide was present in the autologous sequence (Fig. 3). The disparity between the response rates was greatest when the nonadapted peptide matched the autologous viral sequence (response rates 26 versus 40%. P =0.0001 for nonanchor site polymorphism, 19 versus 30%, P =0.03 for anchor site polymorphism). Overall, the lower response rates to the adapted peptides were most evident amongst HLA-B restricted epitopes (n =130 adapted sequences, 17.9 versus 28.3%, P <0.0001) and HLA-A restricted epitopes (n =65, 14.6 versus 18.9%, P =0.01), with the differences remaining notable when taking account of the effects of the mutation position and similarity of the tested peptide to autologous viral sequence.
Fig. 3.
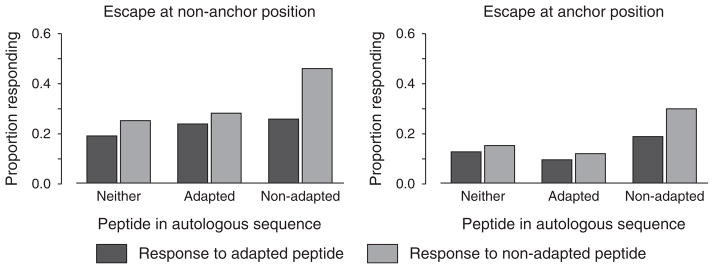
Response rates to nonadapted versus adapted peptides according to site of polymorphism and presence of peptide in autologous sequence.
Polymorphism was also observed commonly at TCR contact residues (Fig. 1). Contrasting with the general trend to reduced IFNγ responses elicited by adapted epitopes, there were a number of specific examples in which TCR variants elicited equivalent or even higher IFNγ responses than the corresponding nonadapted peptide. For example, carriage of HLA-B*08: 01 was associated with a change from valine (V) to methionine (M) at position 194 (position 5 within the epitope) in Nef AL9 (AFHHVAREL). IFNγ responses were investigated in eight individuals with HLA-B*08: 01 and there were no responders to the nonadapted peptide (mean magnitude =5 SFUs/106 PBMCs; range =0–40 SFUs/106 PBMCs) whereas responses to the adapted peptides (mean magnitude =92.5 SFUs/106 PBMCs; range =0–580 SFUs/106 PBMCs) included two above the cutoff of 100 SFUs/106 PBMCs.
Furthermore, more complex patterns of new epitope formation were observed. These included examples of intra-epitopic and extra-epitopic polymorphisms associated with formation of neoepitopes overlapping or distant from the nonadapted epitope, suggesting alteration of antigen-processing signals. There were 112 examples of epitopes associated with variation at TCR contact residues and 70 of these elicited a positive IFNγ response in at least one individual. There were 28 instances of antigen-processing adaptation associated with neoepitope formation, of which 12 elicited a positive IFNγ response to the neoepitope in at least one individual. Finally, 64 epitopes were associated with extra-epitopic mutation in either the N-terminus or C-terminus flanking regions with no discernable neo-epitope formation suggesting complete loss of epitope presentation in these cases. Twenty-nine of those 64 nonadapted epitopes elicited a positive IFNγ response in at least one individual.
Discussion
The overall epitope distribution of HLA allele-associated viral polymorphism over many epitopes and HIV sequences shown here suggests that multiple mechanisms of viral adaptation are evident at the population level. However, viral adaptation, which disrupts HLA-anchor site binding appears sufficiently frequent to lead to significant enrichment of polymorphism at primary HLA anchor residues compared with other residues. These results support the findings of a study involving independent datasets [16]. We could not show that this effect remained statistically significant when restricting to 9mer epitopes which elicited IFNγ responses in our own testing, however this may be because HIV adaptation by this mechanism occurs more commonly and earlier in infection, making ex-vivo detection of memory IFNγ responses in chronic infection more difficult for epitopes that are particularly subject to anchor-site polymorphism. Although we have used locations of mutations as a correlate of mechanism of escape in this analysis, the functional effects of position 2 mutation on binding is supported by the finding of significant reduction of IFNγ responses associated particularly with presence of the position 2 mutation (Fig. 1).
The extreme diversity of HLA class I binding motifs has underpinned individual and population defences against a great diversity of intracellular pathogens over human evolution. In particular, HLA class I appears to preferentially bind conserved elements in viral pathogens with evidence of more efficient targeting of such elements in RNA viruses by HLA-B alleles compared with HLA-A alleles [17]. This is consistent with the dominant role of HLA-B mediated selection pressure in HIV evolution [18], and the recent confirmation from genome-wide association studies that the protective effects of particular HLA-B alleles including HLA-B*57: 01, HLA-B*27: 05 and HLA-B*14 derive from the presence of key pocket residues in their peptide binding grooves [19]. These data would predict that HLA-peptide binding escape would be a dominant or favoured counter-evolutionary strategy by HIV, as shown here. Furthermore, while adaptation at TCR contact sites is also common, such variants remain potentially visible to cross-reactive or variant-specific T cells. Indeed a greater precursor pool and cross-reactivity tolerance to point mutations in epitope targets associated with HLA-B*57: 01 and –B*27: 05 may also underpin their protective effects [20]. Host molecules associated with antigen-processing machinery are also relatively non-specific to accommodate epitope generation from many proteins and HLA types, and this may limit the frequency of antigen-processing escape, relative to other mechanisms.
Population-based analysis is limited by the fact that many of the adaptation imprints are putative here, based on statistical HLA associations and epitope prediction rather than proven by functional studies in every case. The large array of HLA types and viral polymorphisms limits experimental validation for all possible cases. Although all HLA types are not equally represented, the HLA distribution of datasets analyzed here is comparable to the HLA distribution of the full study cohort, and indeed comparable to that found in general US population studies [10], and therefore representative of the prevalent selection forces operating at the population level on HIV subtype B diversity in such populations. Limitations in numbers of cryopreserved PBMCs also imposed a restriction to the numbers of peptides which could be tested and it is possible our prioritization of putative over known epitopes for testing led to a lower overall rate of IFNγ responses than found in other epitope mapping studies. As previously mentioned, memory responses to adapted epitopes with anchor site polymorphisms may also be more likely to have waned in vivo. However, the fact that response rates between paired nonadapted/adapted epitopes used in this analysis did not differ from overall response rates for all (including unpaired) epitopes argues against a specific bias introduced by the datasets used here.
Notwithstanding these limitations, this ‘reverse genomics’ approach has provided biological evidence that many epitopes proximal to HLA associations are targets of CD8 T-cell selection pressure in vivo, including up to 50 novel epitopes [12]. The noise imposed by using putative signals of viral escape and predicted epitopes should affect all types of adaptation signatures and would not account for our central finding that there is significant enrichment of selection at HLA-binding sites. Aside from reinforcing the importance of the quality of HLA-antigen presentation in achieving strong CD8 T-cell immunogenicity against HIV, these data could inform strategies that include escape variants in polyvalent vaccine immunogens.
Acknowledgments
The authors would like to thank Dr Richard Haubrich, Dr Sharon Riddler, co-chairs of US Adult ACTG A5142 (NCT00050895) study team and Dr David Haas, chair of A5128 (NCT00031408) protocol as well as the protocol study teams, study sites and participants. We also thank colleagues within the Institute for Immunology and Infectious Diseases. The content of this study is the responsibility of the authors and does not necessarily represent the official views of NIAID or the National Institutes of Health (US)
This study was conceived by M.J. Experiments were designed by C.B., C.M.A, S.G.R., M.J. and performed by C.B., C.M.A., S.G.R. Data were analysed by C.B., C.M.A., E.M., M.J. N.M.K., A.C., J.M.C., D.H., S.M. contributed reagents/materials/analysis tools. The article was written by C.B., C.M.A., E.M., M.J. and the article was reviewed critically by N.M.K., A.C., J.M.C., D.H., S.M. The article’s revision was written by C.B., C.M.A., E.M., M.J.
This project was supported by grant number RO1 AI060460 from the National Institute of Allergy and Infectious Diseases (NIAID). The ACTG is supported by grant number AI-68636 and the Vanderbilt DNA Resources Core by grant number RR024975. The ACTG Clinical Trials Sites that collected DNA were supported by NIH grants AI64086, AI68636, AI68634, AI069471, AI27661, AI069439, AI25859, AI069477, AI069513, AI069452, AI27673, AI069419, AI069474, AI69411, AI69423, AI69494, AI069484, AI069472, AI069501, AI69467, AI069450, AI32782, AI69465, AI069424, AI38858, AI069447, AI069495, AI069502, AI069556, AI069432, AI46370, AI069532, AI046376, AI34853, and AI069434. The project was additionally supported by the Australian National Health and Medical Research Council (program grant #384702) and the Bill and Melinda Gates Foundation (grant #31844).
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8:619–630. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med. 1997;3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 4.Draenert R, Le Gall S, Pfafferott KJ, Leslie AJ, Chetty P, Brander C, et al. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J Exp Med. 2004;199:905–915. doi: 10.1084/jem.20031982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen TM, Altfeld M, Yu XG, O’Sullivan KM, Lichterfeld M, Le Gall S, et al. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J Virol. 2004;78:7069–7078. doi: 10.1128/JVI.78.13.7069-7078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med. 2001;193:375–386. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 2002;296:1439–1443. doi: 10.1126/science.1069660. [DOI] [PubMed] [Google Scholar]
- 8.Carlson JM, Brumme ZL, Rousseau CM, Brumme CJ, Matthews P, Kadie C, et al. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput Biol. 2008;4:e1000225. doi: 10.1371/journal.pcbi.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, et al. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One. 2009;4:e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.John M, Heckerman D, James I, Park LP, Carlson JM, Chopra A, et al. Adaptive interactions between HLA and HIV-1: highly divergent selection imposed by HLA class I molecules with common supertype motifs. J Immunol. 2010;184:4368–4377. doi: 10.4049/jimmunol.0903745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer W, Perkins S, Theiler J, Bhattacharya T, Yusim K, Funkhouser R, et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat Med. 2007;13:100–106. doi: 10.1038/nm1461. [DOI] [PubMed] [Google Scholar]
- 12.Almeida CA, Bronke C, Roberts SG, McKinnon E, Keane NM, Chopra A, et al. Translation of HLA-HIV associations to the cellular level: HIV adapts to inflate CD8 T cell responses against Nef and HLA-adapted variant epitopes. J Immunol. 2011;187:2502–2513. doi: 10.4049/jimmunol.1100691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riddler SA, Haubrich R, DiRienzo AG, Peeples L, Powderly WG, Klingman KL, et al. Class-sparing regimens for initial treatment of HIV-1 infection. N Engl J Med. 2008;358:2095–2106. doi: 10.1056/NEJMoa074609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas DW, Wilkinson GR, Kuritzkes DR, Richman DD, Nicotera J, Mahon LF, et al. A multiinvestigator/institutional DNA bank for AIDS-related human genetic studies: AACTG Protocol A5128. HIV Clin Trials. 2003;4:287–300. doi: 10.1310/MUQC-QXBC-8118-BPM5. [DOI] [PubMed] [Google Scholar]
- 15.Heckerman D, Kadie C, Listgarten J. Leveraging information across HLA alleles/supertypes improves epitope prediction. J Comput Biol. 2007;14:736–746. doi: 10.1089/cmb.2007.R013. [DOI] [PubMed] [Google Scholar]
- 16.Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, et al. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J Virol. 2012;86:13202–13216. doi: 10.1128/JVI.01998-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hertz T, Nolan D, James I, John M, Gaudieri S, Phillips E, et al. Mapping the landscape of host-pathogen coevolution: HLA class I binding and its relationship with evolutionary conservation in human and viral proteins. J Virol. 2011;85:1310–1321. doi: 10.1128/JVI.01966-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–775. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 19.Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosmrlj A, Read EL, Qi Y, Allen TM, Altfeld M, Deeks SG, et al. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature. 2010;465:350–354. doi: 10.1038/nature08997. [DOI] [PMC free article] [PubMed] [Google Scholar]