

Abstract
Intracellular calcium concentration ([Ca2+]i) plays an important role in regulating most cellular processes, including apoptosis and survival, but its alterations are different and complicated under diverse conditions. In this study, we focused on the [Ca2+]i and its control mechanisms in process of hydrogen peroxide (H2O2)-induced apoptosis of primary cultured Sprague-Dawley (SD) rat retinal cells and 17β-estradiol (βE2) anti-apoptosis. Fluo-3AM was used as a Ca2+ indicator to detect [Ca2+]i through fluorescence-activated cell sorting (FACS), cell viability was assayed using MTT assay, and apoptosis was marked by Hoechst 33342 and annexin V/Propidium Iodide staining. Besides, PI3K activity was detected by Western blotting. Results showed: a) 100 μM H2O2-induced retinal cell apoptosis occurred at 4 h after H2O2 stress and increased in a time-dependent manner, but [Ca2+]i increased earlier at 2 h, sustained to 12 h, and then recovered at 24 h after H2O2 stress; b) 10 μM βE2 treatment for 0.5-24 hrs increased cell viability by transiently increasing [Ca2+]i, which appeared only at 0.5 h after βE2 application; c) increased [Ca2+]i under 100 µM H2O2 treatment for 2 hrs or 10 µM βE2 treatment for 0.5 hrs was, at least partly, due to extracellular Ca2+ stores; d) importantly, the transiently increased [Ca2+]i induced by 10 µM βE2 treatment for 0.5 hrs was mediated by the phosphatidylinositol-3-kinase (PI3K) and gated by the L-type voltage-gated Ca2+ channels (L-VGCC), but the increased [Ca2+]i induced by 100 µM H2O2 treatment for 2 hrs was not affected; and e) pretreatment with 10 µM βE2 for 0.5 hrs effectively protected retinal cells from apoptosis induced by 100 µM H2O2, which was also associated with its transient [Ca2+]i increase through L-VGCC and PI3K pathway. These findings will lead to better understanding of the mechanisms of βE2-mediated retinal protection and to exploration of the novel therapeutic strategies for retina degeneration.
Introduction
Intracellular Ca2+ concentration ([Ca2+]i) plays a vital role in regulating many fundamental cellular processes, such as gene regulation, cell proliferation, cell survival, and apoptosis [1]. Ca2+ homeostasis is tightly regulated and the disturbances in Ca2+ homeostasis have been implicated in degenerative diseases such as Parkinson's disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD) [2,3]. The increase of [Ca2+]i is mediated by two closely related mechanisms: excessive release of Ca2+ from endoplasmic reticulum (ER) stores and store-operated Ca2+ entry (SOCE), the Ca2+ influx process through plasma membrane (PM) channels following the release of Ca2+ from the ER stores [4]. Specifically, [Ca2+]i alterations are different under diverse conditions. Accumulating evidence suggests that both the excessive elevation of [Ca2+]i and the loss of [Ca2+]i are crucial for degenerative diseases [5]. Increased [Ca2+]i leads to the inappropriate activation of Ca2+-dependent processes, which are normally inactive or operate at low Ca2+ levels, thus causing metabolic derangements that ultimately lead to cell death [6]. In contrast, chronic depletion of ER Ca2+ influences ER-dependent processes and also inhibits Ca2+-dependent cellular functions. Furthermore, loss of Ca2+ homeostasis leads to the ER stress response and apoptosis [7]. Alternatively, increased Ca2+ entry has been implicated in both cell survival and cell death processes, and Ca2+ has been shown to exert a biphasic effect on cellular growth. Furthermore, a modest increase in [Ca2+]i promotes cell proliferation, whereas relatively high [Ca2+]i leads to increased mitochondrial Ca2+ and accounts for the release of pro-apoptotic factors resulting in cell death [8,9]. Therefore, diverse Ca2+ actions in different cells must be dependent on the cellular concentration as well as the locations [8].
Oxidative stress-induced cell apoptosis has been implicated in various diseases such as degeneration of nervous system [10]. Hydrogen peroxide (H2O2) has been implicated in triggering apoptosis in various cell types and has become a well-established in vitro model for studying the pathology of oxidative stress in central nervous system (CNS) disorders [11]. The retina is a part of CNS [12]. Apoptosis has been described in many retinal degenerative diseases such as retinitis pigmentosa (RP) and age-related macular degeneration (AMD) [13]. Many studies have focused on [Ca2+]i increases in degenerative disorders of CNS [14,15]; however, the effects of [Ca2+]i reduction and deficiency have also been studied and shown to play a role in degenerative disorders of CNS [16]. These different results may be caused by temporal and spatial specificity. For example, an early increase and subsequent decline in [Ca2+]i may occur or Ca2+ may be reduced in specific cellular compartments and increased in other compartments [17].
Estrogen is an antioxidant that exerts various role by itself or by regulating intracellular signaling pathways [18], and it has also been established that estrogen plays a role in Ca2+ homeostasis [19]. Nevertheless, the reports regarding the effects of estrogen on Ca2+ homeostasis in nervous system protection are inconsistent. Several studies showed that estrogen exerts neuroprotection by increasing [Ca2+]i [20–22], but other studies showed that the same result occurred via [Ca2+]i reduction [23,24]. These apparently conflicting results may be due to the differences in the study models, the intensity of injury or the timing of the [Ca2+]i assessment.
Several recent reports have shown that both estrogen receptor (ER) subtypes, ERα and ERβ, are present in the retina [25,26]. Evidence suggests that estrogen most likely plays a direct role in regulating the physiological processes of the retina [27]. Furthermore, 17β-estradiol (βE2), an extremely potent bioactive estrogen, attenuated the H2O2-induced apoptosis of retinal cells in vitro and inhibited light-induced photoreceptor apoptosis in vivo, suggesting that βE2 has retinal protective properties [28,29]. However, the roles of [Ca2+]i in apoptosis and anti-apoptosis in our study model remain unknown. In this study, we detected the [Ca2+]i of primary cultured Sprague-Dawley (SD) rat retinal cells treated with different concentrations of H2O2 or βE2 and at different time points after H2O2 or βE2 treatment. Next, we measured [Ca2+]i under βE2 and H2O2 co-treatment, and we explored the controlling mechanisms of [Ca2+]i. Consequently, we found that treatment with 100 μM H2O2 led to primary cultured SD rat retinal cell injury and apoptosis, while treatment with 10 μM βE2 played a protective role. Both completely different roles were mediated by increasing the [Ca2+]i, which occurred at the early stage of apoptosis and at 0.5 h after βE2 treatment. Furthermore, both of the increased [Ca2+]i under completely opposite conditions were partially due to extracellular [Ca2+]i. Importantly, the transient [Ca2+]i increase induced by βE2 was gated by the L-type voltage-gated Ca2+ channels (L-VGCC) and phosphatidylinositol-3-kinase (PI3K) was involved, but it was not involved in the H2O2-induced [Ca2+]i increase.
Materials and Methods
2.1: Animals and Chemicals
SD rats (obtained on postnatal days 0-3, body weights of 5-12 g) were housed in a controlled environment in a specific pathogen-free animal center. The temperature was maintained at 24±2°C, the humidity was 52±10% and fresh air was circulated continuously. All of the procedures used in the experiments were approved by the Institutional Animal Ethics Committee, Medical School of Xi’an Jiaotong University (permission No. 2009-12) and conformed to accepted ethical standards of the Animals in Research and the Association for Research in Vision and Ophthalmology statement for the use of animals in vision and ophthalmic research.
H2O2 was purchased from Xi’an Pure Chemical Industries (Xi’an, Shaanxi, China). Fetal Bovine Serum (FBS) and phenol red free 1:1 DMEM/F-12 were obtained from Hyclone (Logan, Utah, USA). Poly-lysine, βE2, Hoechst 333342 dye and nifedipine, an L-VGCC blocker, were purchased from Sigma (St. Louis, Missouri, USA). We used 95% ethanol as the solvent to make the βE2 stock solution at a concentration of 1x10-2 M. Fluo-3 AM, an indicator of intracellular Ca2+ levels, was purchased from Biotium (Hayward, Calif., USA). We used Dimethylsulfoxide (DMSO) as the solvent for making 5 mM Fluo-3 AM stock solution and 20% Pluronic F-127 (5900) (offered by Biotium) in DMSO to facilitate AM ester solubilization. Trypsin, DMSO, 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) and ethylene glycol tetraacetic acid (EGTA), an extracellular Ca2+ chelator, were purchased from Amresco (Solon, Ohio, USA). LY294002, a PI3K inhibitor, was purchased from Cayman (Ann Arbor, MI, USA). The Annexin V-FITC Apoptosis Assay Kit and bicinchoninic acid (BCA) Protein Assay Kit were purchased from Zhuhai Joincare Bioscience Ltd (Zhuhai, Guangdong, China), and radio immunoprecipitation assay (RIPA) buffer was purchased from Biotech (Biotechnology, Inc. of China). Anti-p-Akt and anti-Akt antibodies were purchased from Cell Signaling (Boston, Massachusetts, USA), and Anti-β-actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, Calif., USA).
2.2: Primary Retinal Cells Cultures
We cultured primary retinal cells referencing other’s study [28] and making some revision. Neonatal SD rats were sacrificed (10–12 rats were needed for each 24-well or 6-well culture plate) and then the eyeballs were enucleated and immediately placed into a beaker containing D-Hanks solution. The retinas were removed from the pigment epithelium layer with the aid of a dissecting microscope under sterile conditions and were placed into a glass tube containing 1:1 Ham’s F-12-DMEM medium. The beaker containing the eyeballs and the tube containing the retinas were placed onto ice. The retina fragments were treated with 0.25% trypsin at 37°C for 8 mins and the digestion was terminated by adding three times the volume of 1:1 Ham’s F-12-DMEM containing 10% FBS. The suspension was filtered with a 200-mesh screen and centrifuged at 1000 rpm for 10 mins. After the supernatant was discarded, the cells were suspended, diluted with medium containing 10% FBS to 1x106 cells/ml and plated onto 24-well or 6-well plates (Corning Costar) with 1 ml or 3 ml of cell suspension per well. Before culturing, all the plates were coated with poly-lysine (0.1 mg/ml) and maintained in a humid incubator overnight. Next, we washed the plates three times with sterile double distilled water (ddH2O), once with D-Hanks balanced salt solution, and then with 200 μl of medium, which provided a pre-environment for cell growth. The cells were cultured at 37°C in a 5% CO2 atmosphere until they were used at 4-6 days in vitro, during which the medium was replaced according to the cell growth and metabolism conditions.
2.3: Drug Treatment
After the cultures were maintained for 4-6 days in vitro, H2O2 and/or βE2 were added by bath application. Overall, 1 M H2O2 was prepared from 30% H2O2 dissolved in sterile cool PBS and was diluted with the medium to 10 mM. Next, the 10 mM H2O2 was diluted with the essential medium gradually to 200-25 μM, and 0 μM was regarded as the control. The 0.5-100 μM βE2 was prepared from the 1x10-2 M βE2 stock solution with the medium and was added to the cultures. We considered 0 μM as the control. The βE2 stock solution was dissolved in 95% ethanol, and a small amount of ethanol was present in the medium (<1%), but it had no effect on the primary cultured SD rat retinal cells [28]. Except for analyzing the time and dose dependency of H2O2 or βE2, we used H2O2 at a final concentration of 100 μM for 2 hrs/24 hrs and βE2 at a final concentration of 10 μM for 0.5 hrs to perform the experiments. To discover the source of increased [Ca2+]i, different concentrations of EGTA were added directly to the medium 1 hr before the application of 100 μM H2O2 for 2 hrs or 10 μM βE2 for 0.5 hrs to chelate the extracellular Ca2+. Under the co-application, we pre-treated cells with 10 μM βE2 treatment for 0.5 hrs before the application of 100 μM H2O2 for 2 hrs. To conduct the channel experiments and the mechanism study, the cultures were pre-conditioned for 2 hrs by nifedipine or for 0.5 hrs by LY294002 before the other treatments.
2.4 Cell Viability Assay
To determine the cell viability of the primary cultured SD rat retinal cells, we performed an MTT assay. MTT was applied to the cultures at a final concentration of 0.5 mg/ml for 4 hrs at 37°C in 5% CO2, and the wells with no cells were used as blank controls. The medium was then removed, and DMSO was added to solubilize the colored formazan crystal product. The absorbance was determined at 490 nm on a Measurement Photometric multi-well plate reader (Electron Corporation Multiskan Spectrum, Thermo, Finland) with Skanlt RE for Mass 2.2 software after the plates were agitated at 37°C for 10 mins. All absorbance values were subtracted by the blank value, and the untreated cultures were considered as the control group. The mean cell viability for each condition was determined by averaging at least quadruplicate values, the fold change relative to the control was calculated, and the control values were normalized to 1. All experiments were performed using 3-5 separate experiments to confirm reproducibility.
2.5: Assessment of Apoptosis
After exposure to 100 μM H2O2 for 0-24 hrs, apoptosis was assayed by Annexin V/Propidium Iodide (PI) staining and Hoechst 33342 staining. For Annexin V/PI staining, the cells were collected, centrifuged at 1000 rpm for 5 mins, suspended and diluted with 1×binding buffer (Annexin V-FITC Apoptosis Assay Kit) to 5×105 cells/ml. The 500 μl suspension was loaded with 5 μl Annexin V-FIFC and 10 μl PI for 15 mins. After incubated in the dark at room temperature, the cells were analyzed within one hour with a flow cytometer (San Jose, California, USA). For Hoechst 33342 staining, 40 μl of suspension was dropped onto the slide, fixed in 4% paraformaldehyde in PBS at room temperature for 20 mins and stained with 2 μg/ml Hoechst 33342 dye in the dark for 10 mins. The samples were then observed under a fluorescence microscope (Nikon, Eclipse Ti, Japan) with fluorescence excitation at 340 nm and emission at 510 nm. The cells with condensed DNA were counted as apoptotic cells, and the average apoptotic cells of each field were calculated. The sample fields with approximately 100 cells were randomly selected, and each sample was evaluated. The cells in 3-5 random fields/cultures were scored, and the counts were based on at least four separate cultures in each treatment condition.
2.6: Intracellular Ca2+ Measurement
[Ca2+]i detection was performed by FACS analysis [30]. After washing twice with PBS, the adherent cells were digested from plates with 300 μl 0.25% trypsin per well, and the digestion reaction was quenched by the addition of Ca2+-free medium containing 900 μl 10% FBS per well. The suspensions were collected and centrifuged at 1000 rpm for 10 mins. After discarding the supernatant, we suspended the cells with Ca2+-free PBS and incubated it in dark with 2 μM Fluo-3AM (Molecular Probes, Biotium) at 37°C for 30 mins and at room temperature for 15 mins. The sample without Fluo-3AM was considered as the blank control, whose fluorescence was represented as F0. Before detection, we washed the cells twice with PBS to minimize background fluorescence and nonspecific staining. The fluorescence was measured at FL-1 (526 nm) in a flow cytometer (Becton Dickinson, FACSCalibur-E4121, Becton Dickinson Immunocytometry systems driven by 2350 Qume, San Jose, California, USA) with an excitation laser at 488 nm, and at least 10,000 events per sample were acquired. The obtained image data were analyzed with Cell Quest Version 3.3 software and the Geo Mean of fluorescence (F) was used because its standard normal distribution was better compared to the mean fluorescence. All F values were subtracted by F0 to eliminate the background fluorescence and nonspecific staining. The relative F values of each treated group were expressed as the fold of control, with the F values of the control group normalized to 1. The changes of relative F values of Fluo-3AM represented the [Ca2+]i alteration. To confirm the reproducibility, all experiments were performed at least 3-5 times with separate cultures.
2.7: Western Blot Analysis
The primary cultured retinal cells lysates were made by mixing cold RIPA buffer at a pH of 7.0 (the RIPA buffer consists of 20 mM Tris/HCl, 2 mM ethyleneglycoltetraacetic acid, 25 mM 2-glycerophosphate, 1% Triton X-100, 2 mM dithiothreitol, 1 mM vanadate, 1 mM phenylmethylsulfonyl fluoride and 1% aprotinin) with a 1 mM solution of the serine protease inhibitor phenylmethanesulfonyl fluoride (PMSF) (Sigma-Aldrich, St. Louis, MO) and a 10% solution of phosphatase inhibitor mixture P1260 (Applygen Technologies Inc., Beijing, China). The mixture was then homogenized on ice for 5 mins and centrifuged at 12000 g at 4°C for 20 mins. The BCA protein assay reagents (Pierce, Rockford, USA) were used to assess the concentration of the cell lysates. The assays were performed in triplicate, and the cell lysates were subsequently loaded onto a 12% sodium dodecyl sulfate (SDS) polyacrylamide gel, underwent electrophoresis and were subsequently transferred to a nitrocellulose membrane (Millipore, Bedford, MA) that was blocked with 5% non-fat dry milk in Tris-buffered saline (TBS, pH7.4) and incubated with anti-p-Akt and anti-Akt (1:1000, Cell signaling, Boston, USA) at 4°C overnight. After washing the membrane with TBS/T (TBS with 0.1%Tween 20), we applied goat anti-rabbit IgG (1:5000) labeled with horseradish peroxidase (HRP) at room temperature for 4 hrs, and then washed the membrane with TBS. Anti-β-actin antibody (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to verify the protein concentration. The ECL system (Thermo, USA) was used to visualize the protein bands.
2.8: Statistical Analysis
All results were based on 3-5 independent replications with 4-6 samples per condition per experiment. Values shown in this study were expressed as the mean ±SD. Data were analyzed using the T-test for independent samples, or One-way ANOVA and the LSD post hoc test were used for multiple comparisons. P<0.05 was considered statistically significant for all tests.
Results
3.1: H2O2 induced the apoptosis of primary cultured SD rat retinal cells, and the [Ca2+]i increased during the early apoptosis
Most cell culture models of oxidative stress employ H2O2 as the pro-oxidant to induce oxidative stress because it is capable of altering the intracellular redox state of a cell and causing oxidative damage by its conversion to the highly reactive hydroxyl radical OH [28,31,32]. Furthermore, 100 µM H2O2 treatment for 24 hrs induced retinal cell apoptosis [28]. To ascertain the role of [Ca2+]i in our study model and to dynamically observe the [Ca2+]i alteration during apoptosis under a modest treatment condition, we performed the following experiments. First, cell viability and the [Ca2+]i were assayed simultaneously at 2 h after treatment with different concentrations of H2O2. As shown in Figure 1, 25-200 µM H2O2 decreased cell viability (Figure 1A) but increased [Ca2+]i in a dose-dependent manner (Figure 1B, C), which was significant at 100-200 µM. This finding indicated that 2 hrs after the application, 100-200 µM H2O2 reduced cell viability and caused Ca2+ overload. Next and importantly, we used 100 µM as the H2O2 concentration to dynamically and continuously observe apoptosis by Hoechst 33342 staining and [Ca2+]i alteration during apoptosis, and cell viability was also assayed. The results showed that apoptosis was significant at 4 h, the significance increased over time (Figure 1G, H); however, the [Ca2+]i increased remarkably at 2 h and 4 h, and this increase remained until 12 h but then gradually recovered to the control level at 24 h (Figure 1E, F). Cell viability was reduced in a time-dependent manner from 0 to 24 hrs (Figure 1D). Compared with control group, the 100 µM H2O2 treatment for 2 hrs caused a dramatic increase in [Ca2+]i (P<0.001) and a slight decrease in cell viability; however, the 100 µM H2O2 treatment for 24 hrs caused a remarkable decrease in cell viability (P<0.001), but no significant alteration was discovered in [Ca2+]i (Figure 1D, E), suggesting that the [Ca2+]i increase occurs at the early stage of H2O2 induced apoptosis when cell injury is minimal.
Figure 1. 100 μM H2O2 induced primary cultured SD rat retinal cell apoptosis, which was associated with an increase in [Ca2+]i at the early stage of apoptosis.
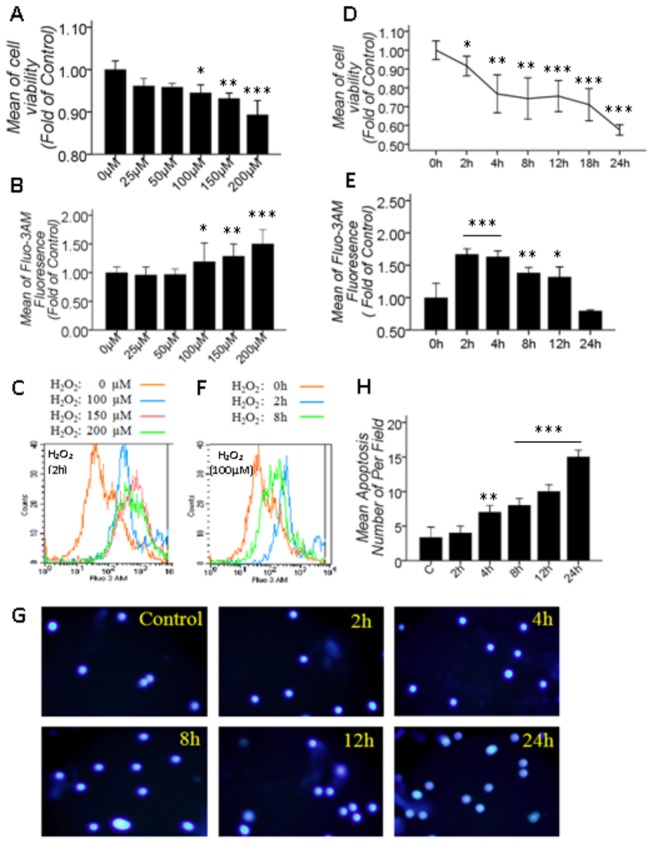
A, B: Quantitative data of cell viability and [Ca2+]i under different concentrations of H2O2 treatments for 2 hrs; D, E: Cell viability and [Ca2+]i quantitative data at different time points after 100 μM H2O2-induced stress; C, F: The overlay figure of the representative statistical significance for B and E; G: Apoptosis assay using Hoechst 33342 staining at different time points after 100 μM H2O2-induced stress; H: Quantitative data of G. Values shown are the Mean ±SD. *represents P<0.05, **represents P<0.01 and ***represents P<0.001 compared with the control group by one-way ANOVA statistical analysis. (A, D, H: n indicates 3 independent replicates with 4 samples per condition per experiment; B, E: n indicates 3 independent replicates with 5 samples per condition per experiment.).
3.2: βE2 increased cell viability and protected primary cultured SD rat retinal cells from H2O2 injury, and the transient [Ca2+]i increase was found to be involved in protection
Pretreatment with 10 µM βE2 for 0.5 hrs effectively protected retinal cells from 100 µM H2O2-induced apoptosis [28]. To confirm whether or not [Ca2+]i was involved in βE2-mediated protection in our model, we first observed the effects of different concentrations of βE2 treatment for 0.5 hrs and 10 µM βE2 treatment for different periods on cell viability and [Ca2+]i, respectively. The results showed that a range of 0.5-100 µM βE2 treatment for 0.5 hrs significantly increased [Ca2+]i in a dose-dependent manner (Figure 2B, C), and 5-50 µM βE2 significantly increased cell viability (Figure 2A). However, at lower (0.5 and 1 µM) or higher (100 µM) concentrations of βE2, the treatment only increased [Ca2+]i but had no effect on cell viability, which may be due to the concentration selectivity or because lower concentrations (0.5 and 1 µM) of βE2 are insufficient to increase cell viability and higher concentrations (100 µM) of βE2 are toxic for retinal cells. Interestingly, cell viability was significantly increased at 0.5-24 h after the application of 10 μM βE2 (Figure 2D), but the [Ca2+]i increased significantly and rapidly only at 0.5 h after 10 μM βE2 treatment, fluctuated near the control level at 1-18 h, and then restored to the control level at 24 h (Figure 2E, F). Furthermore, under 10 μM βE2 pretreatment for 0.5 hrs and then 100 μM H2O2 treatment for 2 hrs, 10 μM βE2 pretreatment for 0.5 hrs significantly restored the decreased cell viability but significantly sharpened the increased [Ca2+]i induced by 100 μM H2O2 for 2 hrs (Figure 2G,H), suggesting that βE2 increased cell viability and protected primary cultured SD rat retinal cells from H2O2 injury that is associated with immediate and transient [Ca2+]i increases.
Figure 2. 10 μM βE2 pretreatment for 0.5 hrs played a protective role in primary cultured SD rat retinal cells, which was associated with a transient and rapid increase in [Ca2+]i.
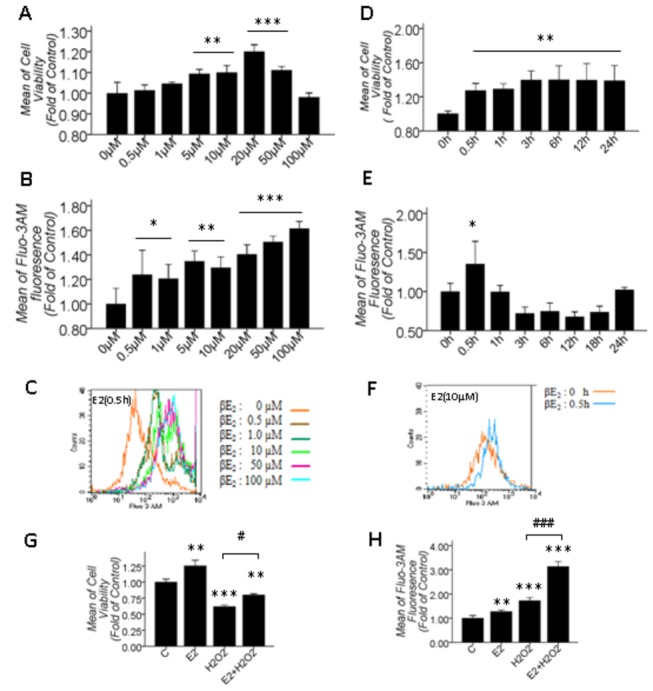
A, B: Cell viability and [Ca2+]i quantitative data under different βE2 concentrations for 0.5 hrs; D, E: Cell viability and [Ca2+]i quantitative data at different time points after 10 μΜ βE2 treatment; C, F: The overlay figure of representative statistical significance for B and E; G, H: Cell viability and [Ca2+]i quantitative data after 10 μM βE2 pretreatment for 0.5 hrs and 100 μM H2O2 treatment for 2 hrs. Values shown are the Mean ±SD. *represents P<0.05, **represents P<0.01 and ***represents P<0.001 compared with the control group; # represents P<0.05 and ### represents P<0.001 compared with the H2O2 application group by one-way ANOVA statistical analysis. (A, D: n indicates 3 independent replicates with 4 samples per condition per experiment; B, E: n indicates 3 independent replicates with 5 samples per condition per experiment; G, H: n indicates 3 independent replicates with 6 samples per condition per experiment.).
3.3: Both increased [Ca2+]i induced by 100 μM H2O2 treatment for 2 hrs and 10 μM βE2 treatment for 0.5 hrs were caused by extracellular Ca2+ influx
Ca2+ homeostasis is strictly controlled by channels, pumps and exchangers functioning as gates for Ca2+ entry and release. A cell becomes activated because of an external signal, which results in up to an 100-fold increase in the [Ca2+]i caused by the uptake of extracellular Ca2+ and/or the release of intracellular Ca2+ stores. To confirm whether the increased [Ca2+]i in our model treated with 100 μM H2O2 for 2 hrs or 10 μM βE2 for 0.5 hrs is due to the extracellular Ca2+ influx, we preliminarily detected the [Ca2+]i before and after adding EGTA, a chelator of extracellular Ca2+, in the presence and absence of H2O2 or βE2, respectively. Simultaneously, cell viability was assayed. As shown in Figure 3, 0.2-5 mM EGTA treatment for 24 hrs decreased cell viability (Figure 3A), treatment with 1-5 mM EGTA for 1 hr had no effect on the [Ca2+]i (Figure 3B, C). However, the effect of EGTA on the [Ca2+]i was different in the presence of H2O2 or βE2. Based on previous experiments, we selected to pretreat the cells with 0.1-5 mM EGTA for 1 hr to chelate the extracellular Ca2+ before H2O2 or βE2 treatment. The results showed that 1-5 mM EGTA significantly aggravated the decrease in cell viability (Figure 3D), but 0.5-5 mM EGTA significantly attenuated the increase in [Ca2+]i caused by the 100 μM H2O2-induced injury for 2 hrs (Figure 3E, F). This aggravating or attenuating effect was dose-dependent. Furthermore, 1-5 mM EGTA dose-dependently attenuated the increased cell viability and the increased [Ca2+]i caused by 10 μM βE2 treatment for 0.5 hrs (Figure 3G, H, I). The attenuating impact of EGTA on the increased [Ca2+]i induced by H2O2 or βE2 implicated that [Ca2+]i increases under the two conditions were, at least, caused by extracellular sources. In this experiment, we monitored the pH before and after EGTA application and found that the low dose of EGTA did not alter the pH value of the medium, eliminating the effect of a change in pH as the cause of the increase in [Ca2+]i.
Figure 3. Sources of increased [Ca2+]i induced by 100 μM H2O2 treatment for 2 hrs and 10 μM βE2 treatment for 0.5 hrs.
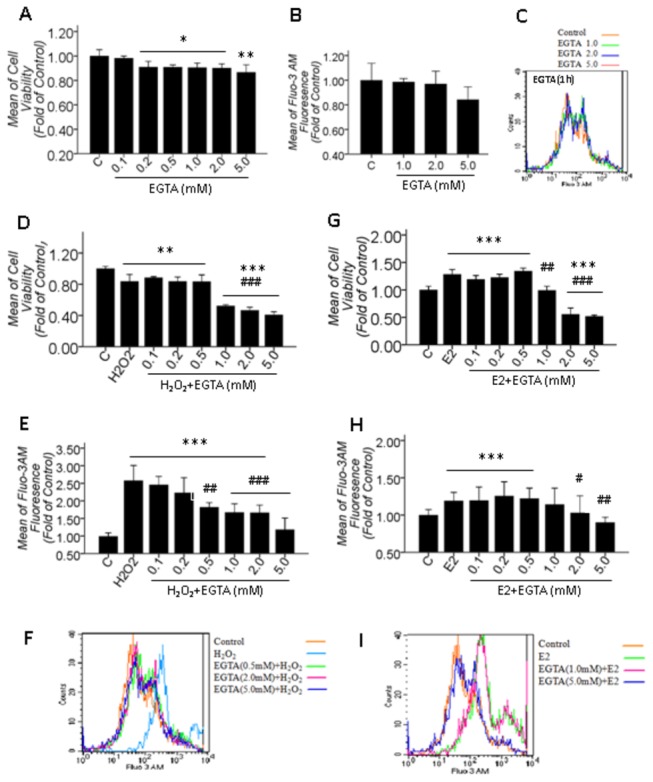
A, B: The effects of different concentrations of EGTA treatment for 24 hrs on cell viability and EGTA treatment for 1 hr on [Ca2+]i; C: The overlay figure for B; D-F and G-I: The effect of different concentrations of EGTA pretreatment for 1 hr before H2O2 or βE2 application on the alteration of cell viability and [Ca2+]i induced by H2O2 (D-F) or βE2 (G-I); F and I: The representative overlay figure for E and H. Values shown are the Mean ±SD. *represents P<0.05, **represents P<0.01 and ***represents P<0.001 compared with the control group; # represents P<0.05, ## represents P<0.01 and ### represents P<0.001 compared with the H2O2 or βE2 application groups by one-way ANOVA statistical analysis. (A, D, E: n indicates 4 independent replicates with 5 samples per condition per experiment; B, G, H: n indicates 4 independent replicates with 6 samples per condition per experiment.).
3.4: L-VGCC mediated the [Ca2+]i increase induced by 10 μM βE2 treatment for 0.5 hrs but did not mediate the [Ca2+]i increase induced by 100 μM H2O2 for 2 hrs
It has been suggested that estrogen potentiates L-VGCC in other cells [20–22]; however, it remained unknown whether L-VGCC gated the extracellular Ca2+ influx caused by 10 μM βE2 treatment for 0.5 hrs or 100 μM H2O2 treatment for 2 hrs in our model. To this end, we conducted several experiments using the L-VGCC blocker nifedipine. First, we measured the effect of nifedipine on the cell viability and found that treatment for 24 hrs with 10 μM and 20 μM nifedipine showed no effect on the cell viability, but 30 μM nifedipine significantly decreased the cell viability (Figure 4A). Second, we measured the [Ca2+]i at different time points after 20 μM nifedipine treatment and found that the [Ca2+]i increased at 0.5-1 h after 20 μM nifedipine application but later recovered (Figure 4B). When specifically blocking L-VGCC, the reactively impermanent increase in [Ca2+]i occurred at 0.5-1 h after 20 μM nifedipine application because of the Ca2+ homeostasis. Afterwards, the [Ca2+]i recovered to the resting level, and nifedipine began to develop its stable and innate effect. Third, we detected the blocking effect of nifedipine on increased [Ca2+]i under two conditions and found that 20 μM nifedipine pretreatment for 2 hrs significantly attenuated the increased [Ca2+]i induced by 10 μM βE2 treatment for 0.5 hrs (Figure 4C) but did not attenuate the increased [Ca2+]i induced by 100 μM H2O2 treatment for 2 hrs (Figure 4D). L-VGCC gated the transient [Ca2+]i increase induced by βE2 but did not gate the H2O2-induced [Ca2+]i increase. Fourth, we analyzed the impact of nifedipine on βE2-mediated retinal protection and discovered that 20 μM nifedipine pretreatment for 2 hrs significantly attenuated βE2 protection against H2O2 injury (P=0.029, Figure 4E) and also significantly attenuated the increased [Ca2+]i induced by βE2 and H2O2 co-treatment (P=0.018, Figure 4F). Therefore, βE2 protection on primary cultured SD rat retinal cells was associated with transient Ca2+ influx gated by L-VGCC.
Figure 4. The effect of the L-VGCC blocker nifedipine (N) on the alteration of [Ca2+]i during H2O2 injury and βE2 retinal protection.
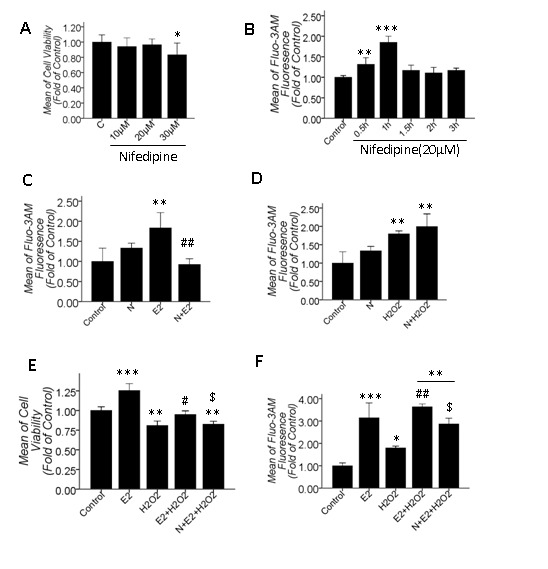
A: Cell viability under 10-30 μM nifedipine treatments for 24 hrs; B: [Ca2+]i at different time points after 20 μM nifedipine application; C, D: The effects of 20 μM nifedipine pretreatment for 2 hrs on the increase in [Ca2+]i due to 10 μM βE2 treatment for 0.5 hrs or 100 μM H2O2 treatment for 2 hrs; E, F: The attenuated effect of 20 μM nifedipine pretreatment for 2 hrs on the increased cell viability and [Ca2+]i due to βE2 and H2O2 co-treatment. N is 20 μM nifedipine in B, C, D, E, and F. Values shown are the Mean ±SD. *represents P<0.05, **represents P<0.01 and ***represents P<0.001 compared with the control group; # represents P<0.05, ## represents P<0.01 compared with the H2O2 group (E, F) and ### represents P<0.001 compared with the βE2 group (C); $ represents P<0.05 compared with the βE2 and H2O2 co-application group by one-way ANOVA statistical analysis. (A: n indicates 5 independent replicates with 5 samples per condition per experiment; B, C, D, E, F: n indicates 3 independent replicates with 4 samples per condition per experiment.).
3.5: βE2 pretreatment protected primary cultured SD rat retinal cells from H2O2-induced apoptosis by activating the PI3K pathway and then transiently up-regulating the [Ca2+]i
βE2 plays a protective role in the retina via the PI3K/Akt pathway [28]. Our results showed that βE2 protected primary cultured SD rat retinal cells from H2O2 injury, which was associated with a transient [Ca2+]i increase (Figure 2). Therefore, we hypothesized that βE2 plays a protective role in our study model by activating the PI3K pathway and then transiently increasing [Ca2+]i. To test this hypothesis, we performed the following experiments using the PI3K inhibitor LY294002. First, we confirmed that 10 μM βE2 treatment for 0.5 hrs up-regulated the p-Akt level via Western blotting (Figure 5A, B). Second, we measured the effects of LY294002 on the cell viability and the [Ca2+]i of the retinal cells and found that 1-50 μM LY294002 treatment for 24 hrs dose-dependently decreased the cell viability (Figure 5C), but treatment for 0.5 hrs had no effect on the resting [Ca2+]i (Figure 5D). Third, we detected the inhibitory effects of LY294002 on the alteration of [Ca2+]i and cell viability due to 10 μM βE2 treatment for 0.5 hrs or 100 μM H2O2 treatment for 2 hrs. Results showed that pretreatment for 0.5 hrs with 10 μM or 20 μM LY294002 significantly attenuated the increased cell viability and [Ca2+]i due to βE2 (Figure 5E, F). However, 10 μM LY294002 did not reverse the cell viability decrease induced by H2O2 but instead promoted the decrease in cell viability (Figure 5G). In addition, both 10 μM and 20 μM LY294002 had no effect on the [Ca2+]i increase induced by H2O2 (Figure 5H). PI3K was involved in the βE2-induced increase of [Ca2+]i and cell viability but was not involved in the H2O2-induced [Ca2+]i increase and cell viability decrease. Fourth, we verified that PI3K-mediated βE2 protection against H2O2 injury was associated with transiently up-regulating [Ca2+]i. As shown in Figures 5I and J, 20-50 μM LY294002 dose-dependently attenuated the βE2-mediated protective effect against H2O2 injury and dose-dependently restored the increased [Ca2+]i induced by co-treatment with 10 μM βE2 for 0.5 hrs and 100 μM H2O2 for 2 hrs.
Figure 5. The effect of the PI3K inhibitor LY294002 (LY) on the cell viability and the [Ca2+]i of primary cultured SD rat retinal cells in H2O2 injury and βE2 protection.
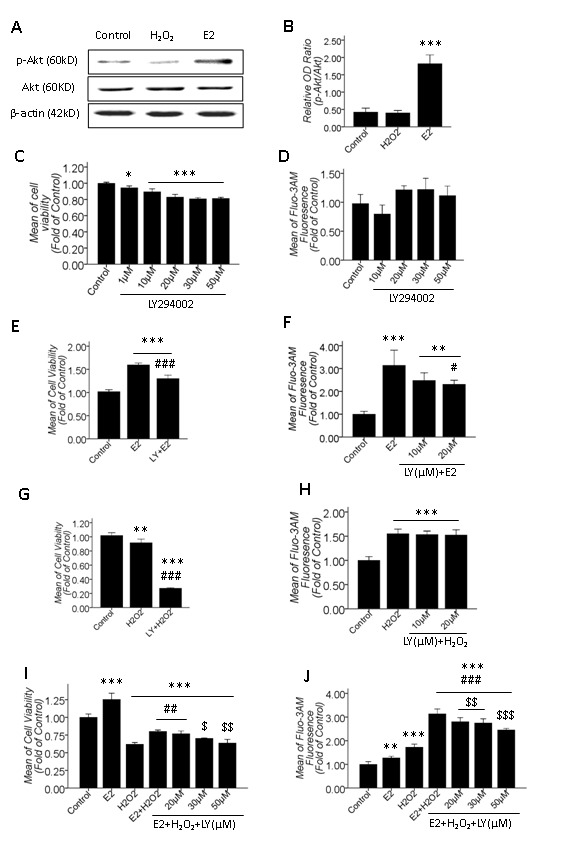
A: Western blot results of the activation of the PI3K/Akt pathway after βE2 treatment for 0.5 hrs; B: Quantitative data of A; C, E, G, and I: Cell viability quantitative data; D, F, H, and J: [Ca2+]i quantitative data; C and D: The effects of LY treatments for 24 hrs and 0.5 hrs on the cell viability and the resting [Ca2+]i; E and F: The inhibitory effect of LY pretreatment for 0.5 hrs on the increased cell viability and [Ca2+]i induced by 10 μM βE2 treatment for 0.5 hrs (10 μM LY in E, 10 μM and 20 μM LY in F); G and H: The effect of LY pretreatment for 0.5 hrs on the decreased cell viability and increased [Ca2+]i induced by 100 μM H2O2 treatment for 2 hrs (10 μM LY in G, 10 μM and 20 μM LY in H); I and J: The dose-dependent attenuating impact of 20-50 μM LY pretreatment for 0.5 hrs on the βE2 retinal protective role against H2O2 injury, which is associated with the dose-dependent attenuation of the increased [Ca2+]i (Protocol of drug application: LY for 0.5 hrs, E2 for 0.5 hrs and H2O2 for 2 hrs). Values shown are the Mean ±SD. *represents P<0.05, **represents P<0.01 and ***represents P<0.001 compared with the control group; # represents P<0.05, ## represents P<0.01 and ### represents P<0.001 compared with the βE2 (E, F) or H2O2 (G, I, J) application groups; $ represents P<0.05, $$ represents P<0.01 and $$$ represents P<0.001 compared with the βE2 and H2O2 co-application group by one-way ANOVA statistical analysis. (B: n indicates 3 independent replicates; C, E, G, I: n indicates 3 independent replicates with 4 samples per condition per experiment; D, F, H, J: n indicates 3 independent replicates with 5 samples per condition per experiment.).
Based on the results of cell viability and apoptosis assay in Figure 1D and H, 100 μM H2O2 treatment for 2 hrs led promoted retinal cell injury but not apoptosis. Therefore, we tested the role of βE2 in anti-apoptosis induced by 100 μM H2O2 for 24 hrs and the inhibitory effect of LY294002. In this experiment, we assayed the cell viability by the MTT assay and apoptosis by Annexin V/Propidium Iodide staining, and meanwhile, [Ca2+]i measurements and Western blotting were performed. The results showed that 10 μM βE2 pretreatment for 0.5 hrs effectively protected the retinal cells from injury and apoptosis induced by 100 μM H2O2-mediated stressing for 24 hrs. Moreover, application of 10 μM LY294002 for 0.5 hrs before βE2 treatment significantly inhibited the βE2-mediated retinal protection against the H2O2-induced cell viability decrease and apoptosis (Figure 6A-C). Nevertheless, the [Ca2+]i showed no alteration in all treated groups compared to the control group (Figure 6D), which further implicated that the βE2-induced increase in the [Ca2+]i is an instantaneous event and that the [Ca2+]i overload induced by H2O2 occurred during the early stage of apoptosis but did not occur at the later stages of apoptosis. Western blot results also showed that 10 μM βE2 pretreatment for 0.5 hrs markedly activated the PI3K/Akt pathway, which was significantly inhibited by 10 μM LY294002 (Figure 6E, F).
Figure 6. 10 μM βE2 pretreatment for 0.5 hrs protected primary cultured SD rat retinal cells from apoptosis induced by 100 μM H2O2 treatment for 24 hrs.
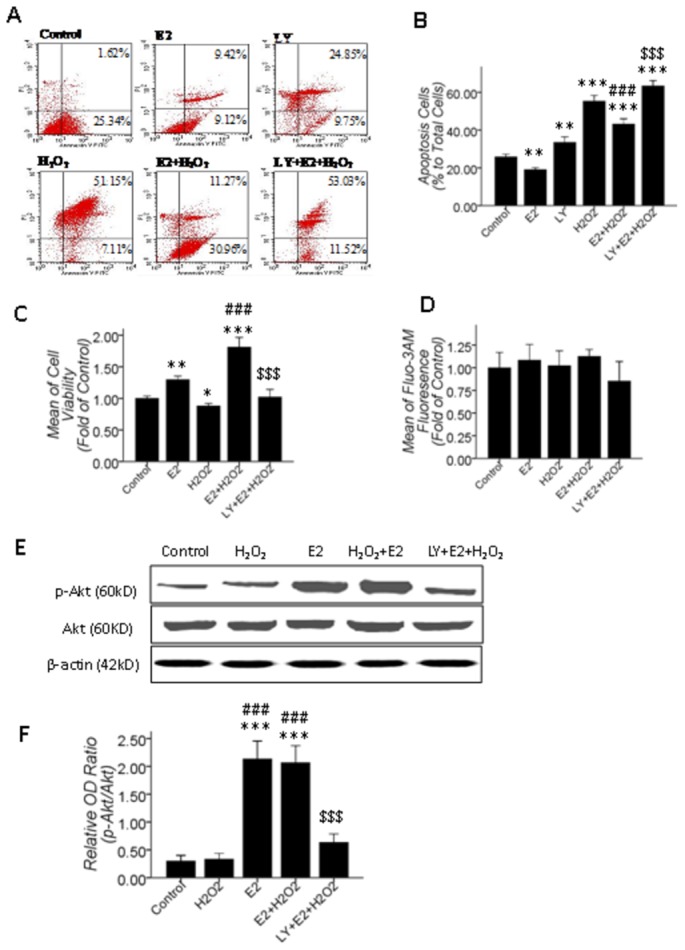
The PI3K/Akt pathway mediated this process, but the alteration in [Ca2+]i was undetectable. A: The Annexin V/Propidium Iodide staining apoptosis assay; B: Quantitative data of A; C and D: Cell viability and [Ca2+]i quantitative data; 10 μM βE2 pretreatment for 0.5 hrs significantly restored the decrease in cell viability and apoptosis, which was significantly inhibited by 10 μM LY (B, C), but the [Ca2+]i was not significantly altered in all treated groups (D); E: Western blot results, 10 μM βE2 pretreatment for 0.5 hrs promoted p-Akt level, which was inhibited by 10 μM LY pretreatment for 0.5 hrs before βE2 and H2O2 co-treatment. F: Quantitative data of E. Values shown are the Mean ±SD. *represents P<0.05, **represents P<0.01 and ***represents P<0.001 compared with the control group by the T-test or one-way ANOVA statistical analysis; ### represents P<0.001 compared with the H2O2 application group by one-way ANOVA statistical analysis; $$$ represents P<0.001 compared with the βE2 and H2O2 co-application group by one-way ANOVA statistical analysis. (B, C, D: n indicates 3 independent replicates with 4 samples per condition per experiment; F: n indicates 3 independent replicates.).
Briefly, the PI3K pathway mediated the βE2-induced [Ca2+]i increase but did not mediate the H2O2-induced [Ca2+]i increase. Pretreatment with 10 μM βE2 for 0.5 hrs protected primary cultured SD rat retinal cells from injury and apoptosis induced by H2O2 by activating the PI3K pathway, and then transiently up-regulated the [Ca2+]i, which was detectable at 2 h but not at 24 h after H2O2-induced stress.
Discussion and Conclusion
[Ca2+]i plays an important role in regulating most cellular processes and it is regulated by complex mechanisms. While brief elevations in [Ca2+]i are required to control membrane excitability and to modulate essential processes, chronic elevations in [Ca2+]i trigger toxic signaling cascades that lead to cell death [6,33–35]. Nevertheless, the selection of Ca2+ indicator and method of [Ca2+]i measurement are very important as well as. They will affect the result of [Ca2+]i measurement. Fluo-3 AM ester is a membrane-permeating form of fluo-3. It can passively diffuse across cell membranes and can be loaded into most of cells. Fluo-3 AM itself does not respond to Ca2+. However, once inside the cells, it is hydrolyzed to fluo-3 and can bind to Ca2+. Fluo-3 is one of the most suitable fluorescent Ca2+ indicators for flow cytometry. It is a good probe because of its high sensitivity, but a few limited cells can be loaded directly with Ca2+ indicators [36]. Consequently, it is feasible and reasonable that we detected the [Ca2+]i by FACS using Fluo-3 AM. The fluorescence of Fluo-3 AM precisely represents the actual [Ca2+]i.
Recent evidence indicates that [Ca2+]i is abnormal in many degenerative disorders in CNS. A number of studies suggest that alterations in [Ca2+]i may result in cell apoptosis [37], which supports the relevance of [Ca2+]i in the mechanisms leading to apoptosis. Several studies show that exposure to H2O2 induces the apoptosis of cultured neurons, which is mediated by increasing the [Ca2+]i. Several channels have been proposed to be involved in the H2O2-mediated [Ca2+]i increase, including the N-methyl-D-aspartate (NMDA) receptor, the a-amino-3-hydroxy-5-methyl-4-isoxa-zole propionic acid (AMPA) receptor and VGCC [38–40]. The Transient Receptor Potential (TRP) protein superfamily is a group of voltage-independent Ca2+-permeable cation channels expressed in mammalian cells and consists of six subfamilies: TRPC, TRPV, TRPM, TRPA, TRPP, and TRPML [41,42]. Recent evidence suggests that Ca2+ influx through TRP channels is an important mechanism through which oxidative stress mediates cell death and TRPC, and TRPM subfamily members are also activated by oxidative stress [42]. In our present study, we found that Ca2+ plays a substantial role in H2O2-induced apoptosis, and the [Ca2+]i increase occurs at the early stage of apoptosis but not during the later stages of this process. Moreover, the increased [Ca2+]i induced by H2O2 is partially caused by extracellular stores.
As for the mechanisms involved in βE2 retinal protection in our model, we speculated that βE2 resisted H2O2 stress by weakening the increased [Ca2+]i due to H2O2. Inconsistent with our hypothesis, we found that 10 μM βE2 played a protective role by immediately sharpening but not restoring the increased [Ca2+]i induced by H2O2. Furthermore, up to 2-5 mM doses of EGTA significantly attenuated the sharpening effect of βE2, indicating that this effect may be caused by a large Ca2+ transient influx. Many studies have proposed that L-VGCC plays an important role in the protective process in CNS, including retina [20–22,43]. In addition, several studies have indicated that the release of Ca2+ from the ER through the inositol 1, 4, 5-trisphosphate receptors (IP3Rs) is essential for cell survival and neuroprotection [44–46]. The members of the TRPM and TRPC subfamilies also play important roles in cell survival [47–50]. βE2 has been shown to be involved in the regulation of Ca2+ influx via the TRPV5 channels [51], and preconditioned cells with a relatively low level of Ca2+ before an excitotoxic insult experienced neuroprotection in retinal ganglion cells [52]. Therefore, we hypothesized that βE2 increased the [Ca2+]i through one or more relevant Ca2+ channels and signaling pathways. Excitedly, we discovered that the retinal protective role of βE2 through potentiating Ca2+ influx is controlled by L-VGCC and mediated by PI3K pathway.
Perplexedly, the results in our present study showed that both H2O2 injury and βE2 protection are mediated by increasing the [Ca2+]i sourced from extracellular Ca2+ influx. These findings can be explained by the following ideas. First, Ca2+ exerts a biphasic effect on cellular growth, and a modest increase in [Ca2+]i promotes cell proliferation, whereas relatively high [Ca2+]i leads to increased mitochondrial Ca2+ and accounts for the release of pro-apoptotic factors resulting in cell death [8,9]. Second, a short increase in [Ca2+]i is tolerated and may be needed to modulate biological functions, but the sustained increase in [Ca2+]i leads to various degrees of cell damage until cell death. Third, under the two treatment conditions, the increased [Ca2+]i may be due to different channels, and Ca2+ influx through different routes may perform different biological functions [53]. For example, equally high Ca2+ loads are toxic when entering via the NMDA channels but not when entering via the VGCC [54]. Our present results showed that 2-12 hrs of a sustained [Ca2+]i increase induced by H2O2 is harmful, but a transient [Ca2+]i increase induced by βE2 for only 0.5 hrs is protective. Furthermore, the favorable [Ca2+]i increase due to βE2 was gated by L-VGCC and was mediated by the PI3K pathway, but the harmful [Ca2+]i increase caused by H2O2 was not gated by L-VGCC or mediated by the PI3K pathway.
The majority of the results in this study are easily interpreted; nevertheless, several results are difficult to understand. For example, EGTA attenuated the increase of [Ca2+]i induced by the 100 μM H2O2-induced injury (Figure 3E and F) but did not attenuate and inversely aggravated the decrease in cell viability (Figure 3D), which is most likely because extracellular Ca2+ is necessary for cell growth and chelating the extracellular Ca2+ leads to a decrease in cell viability. In our present study, we chelated the extracellular Ca2+, but we did not chelate the increased intracellular Ca2+, and we did not specifically block the channels controlling the extracellular Ca2+ influx due to the H2O2 injury. Further specific chelating and blocking experiments are being performed. Surprisingly, 20 μM nifedipine treatment for 0.5-1 hr increased the [Ca2+]i significantly (Figure 4B); however, it was the reactively impermanent [Ca2+]i increase. In this phenomenon, [Ca2+]i may have reactively increased through other channels when L-VGCC was specifically blocked due to Ca2+ homeostasis at resting condition. After 0.5-1 hr of an impermanent [Ca2+]i increase, the nifedipine developed its innate effect. However, this finding is novel and needs to be further investigated.
In summary, 100 μM H2O2-induced stress led to primary cultured SD rat retinal cell injury and apoptosis; however, 10 μM βE2 played a protective role on retinal cells. Both completely different roles were mediated by increasing the [Ca2+]i, which occurred at the early stage of 100 μM H2O2-induced apoptosis and 10 μM βE2 treatment for 0.5 hrs. Furthermore, the increase in [Ca2+]i under completely opposite conditions were partially due to extracellular Ca2+ stores. Meaningfully, the transient [Ca2+]i increase induced by βE2 was gated by L-VGCC, and the PI3K pathway was found to be involved but was not found to be involved in the H2O2-induced [Ca2+]i increase. This finding may be due to different sources of Ca2+ through different channels activating pro-apoptotic or pro-survival pathways, thus performing the injury or the protective roles. Our present findings are very important for understanding the mechanism of retina degeneration and the search for preventative treatment targets. The detailed mechanisms and downstream signaling pathways of Ca2+ are mostly unknown; therefore, it is important to direct future efforts towards the mechanisms and pathways of βE2-mediated anti-apoptosis through regulating [Ca2+]i and the downstream signals of Ca2+.
The data from our present study were based on a primary mixed cell culture of retinal cell population. The in vitro model is widely used for studying the pathogenesis of diseases. The primary retinal cell culture began in the late 1950’s. Today, it is routinely used for studies and remains the most widely-used form of retinal cell culture [55]. Moreover, mixed primary culture of retinal cell population includes various retinal cells and may better represent the in vivo condition than a cell line. Besides, H2O2 triggers apoptosis and becomes a well-established in vitro model for studying the pathology of oxidative stress in degenerative disorders of CNS such as AMD, which is relative to the producing of reactive oxygen species (ROS) [11,13]. Therefore, the model of H2O2-induced apoptosis of primary cultured retinal cells represents the pathogenesis of multiple retinal degenerative diseases. Certainly, in our future studies, we will do some research using in vivo model to obtain results that are more closely applicable to in vivo conditions.
Acknowledgments
We should express gratitude to Mr. Dianzeng Zhang, Ms. Yan Han and Ms. Shuhong Wang for their expertise and assistance in the experiments.
Funding Statement
This work was supported by the National Natural Science Foundation of China (No: 30672286 and No: 81271013) and the National Research Foundation for the Doctoral Program of Higher Education of China (No: 20120201110051). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Selvaraj S, Sun YY, Singh BB (2010) TRPC Channels and their Implications for Neurological Diseases. CNs Neurol Disorddrugs Targets 9: 94-104. doi: 10.2174/187152710790966650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bezprozvanny I (2009) Calcium signaling and neurodegenerative diseases. Trends Mol Med 15: 89-100. doi: 10.1016/j.molmed.2009.01.001. PubMed: 19230774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marambaud P, Dreses-Werringloer U, Vingtdeux V (2009) Calcium signaling in neurodegeneration. Mol Neurodegener 4: 20-34. doi: 10.1186/1750-1326-4-20. PubMed: 19419557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Putney JW (1986) A Model for Receptor-Regulated Calcium Entry. Cell Calcium 7: 1-12. doi: 10.1016/0143-4160(86)90026-6. PubMed: 2420465. [DOI] [PubMed] [Google Scholar]
- 5. Berridge MJ, Bootman MD, Lipp P (1998) Calcium - a life and death signal. Nature 395: 645-648. doi: 10.1038/27094. PubMed: 9790183. [DOI] [PubMed] [Google Scholar]
- 6. Arundine M, Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34: 325-337. doi: 10.1016/S0143-4160(03)00141-6. PubMed: 12909079. [DOI] [PubMed] [Google Scholar]
- 7. Yoshida I, Monji A, Tashiro K, Nakamura K, Inoue R et al. (2006) Depletion of intracellular Ca2+ store itself may be a major factor in thapsigargin-induced ER stress and apoptosis in PC12 cells. Neurochem Int 48: 696-702. doi: 10.1016/j.neuint.2005.12.012. PubMed: 16481070. [DOI] [PubMed] [Google Scholar]
- 8. Paschen W (2000) Role of calcium in neuronal cell injury: Which subcellular compartment is involved? Brain. Res Bull 53: 409-413. doi: 10.1016/S0361-9230(00)00369-5. [DOI] [PubMed] [Google Scholar]
- 9. Beech DJ (2005) TRPC1: store-operated channel and more. Pflugers Arch_Eur J Physiol 451: 53-60. doi: 10.1007/s00424-005-1441-3. PubMed: 15965706. [DOI] [PubMed] [Google Scholar]
- 10. Moosmann B, Behl C (2002) Antioxidants as treatment for neurodegenerative disorders. Expert Opin Invest Drugs 11: 1407-1435. doi: 10.1517/13543784.11.10.1407. PubMed: 12387703. [DOI] [PubMed] [Google Scholar]
- 11. Song KS, Nguyen TTH, Cho SO, Ban JY, Kim JY et al. (2008) Neuroprotective Effect of Sanguisorbae Radix against Oxidative Stress-Induced Brain Damage: in Vitro and in Vivo. Biol Pharm Bull 31: 2028-2035. doi: 10.1248/bpb.31.2028. PubMed: 18981568. [DOI] [PubMed] [Google Scholar]
- 12. Guarneri P, Cascio C, Russo D, D'Agostino S, Drago G et al. (2003) Neurosteroids in the retina: neurodegenerative and neuroprotective agents in retinal degeneration. Ann N Y Acad Sci 1007: 117-128. doi: 10.1196/annals.1286.012. PubMed: 14993046. [DOI] [PubMed] [Google Scholar]
- 13. Remé CE, Grimm C, Hafezi F, Marti A, Wenzel A (1998) Apoptotic cell death in retinal degenerations. Prog Retin Eye Res 17: 443-464. doi: 10.1016/S1350-9462(98)00009-3. PubMed: 9777646. [DOI] [PubMed] [Google Scholar]
- 14. Arundine M, Tymianski M (2004) Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci 61: 657-668. doi: 10.1007/s00018-003-3319-x. PubMed: 15052409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berliocchi L, Bano D, Nicotera P (2005) Ca2+ signals and death programmes in neurons. Philos Trans R Soc Lond B-Biol Sci 360: 2255-2258. doi: 10.1098/rstb.2005.1765. PubMed: 16321795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu SZ, Hyrc KL, Moulder KL, Lin Y, Warmke T et al. (2009) Cellular calcium deficiency plays a role in neuronal death caused by proteasome inhibitors. J Neurochem 109: 1225-1236. doi: 10.1111/j.1471-4159.2009.06037.x. PubMed: 19476541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Darios F, Muriel MP, Khondiker ME, Brice A, Ruberg M (2005) Neurotoxic calcium transfer from endoplasmic reticulum to mitochondria is regulated by cyclin-dependent kinase 5-dependent phosphorylation of tau. J Neurosci 25: 4159-4168. doi: 10.1523/JNEUROSCI.0060-05.2005. PubMed: 15843619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Behl C (2002) Oestrogen as a neuroprotective hormone. Nat Rev Neurosci 3: 433-442. PubMed: 12042878. [DOI] [PubMed] [Google Scholar]
- 19. Prince RL (1994) Counterpoint - Estrogen Effects on Calcitropic Hormones and Calcium Homeostasis. Endocr Rev 15: 301-309. doi: 10.1210/edrv-15-3-301. PubMed: 8076583. [DOI] [PubMed] [Google Scholar]
- 20. Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH et al. (2008) Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci U S A 105: 15148-15153. doi: 10.1073/pnas.0802379105. PubMed: 18815371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu TW, Wang JM, Chen S, Brinton RD (2005) 17 beta-Estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-amp response element binding protein signal pathway and Bcl-2 expression in rat hippocampal neurons: A potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience 135: 59-72. doi: 10.1016/j.neuroscience.2004.12.027. PubMed: 16084662. [DOI] [PubMed] [Google Scholar]
- 22. Farkas I, Sárvári M, Aller M, Okada N, Okada H et al. (2012) Estrogen receptor alpha and beta differentially mediate C5aR agonist evoked Ca2+-influx in neurons through L-type voltage-gated Ca2+ channels. Neurochem Int 60: 631-639. doi: 10.1016/j.neuint.2012.02.024. PubMed: 22406418. [DOI] [PubMed] [Google Scholar]
- 23. Sribnick EA, Del Re AM, Ray SK, Woodward JJ, Banik NL (2009) Estrogen attenuates glutamate-induced cell death by inhibiting Ca2+ influx through L-type voltage-gated Ca2+ channels. Brain Res 1276: 159-170. doi: 10.1016/j.brainres.2009.04.022. PubMed: 19389388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lapanantasin S, Chongthammakun S, Floyd CL, Berman RF (2006) Effects of 17 beta-estradiol on intracellular calcium changes and neuronal survival after mechanical strain injury in neuronal-glial cultures. Synapse 60: 406-410. doi: 10.1002/syn.20308. PubMed: 16856173. [DOI] [PubMed] [Google Scholar]
- 25. Kobayashi K, Kobayashi H, Ueda M, Honda Y (1998) Estrogen receptor expression in bovine and rat retinas. Invest Ophthalmol Vis Sci 39: 2105-2110. PubMed: 9761289. [PubMed] [Google Scholar]
- 26. Munaut C, Lambert V, Noël A, Frankenne F, Deprez M et al. (2001) Presence of oestrogen receptor type beta in human retina. Br J Ophthalmol 85: 877-882. doi: 10.1136/bjo.85.7.877. PubMed: 11423466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu X, Tang Y, Li F, Frank MB, Huang H et al. (2005) Protection against hydrogen peroxide-induced cell death in cultured human retinal pigment epithelial cells by 17beta-estradiol: a differential gene expression profile. Mech Ageing Dev 126: 1135-1145. doi: 10.1016/j.mad.2005.05.005. PubMed: 16029884. [DOI] [PubMed] [Google Scholar]
- 28. Yu XR, Rajala RVS, McGinnis JF, Li F, Anderson RE et al. (2004) Involvement of insulin/phosphoinositide 3-kinase/Akt signal pathway in 17 beta-estradiol-mediated neuroprotection. J Biol Chem 279: 13086-13094. PubMed: 14711819. [DOI] [PubMed] [Google Scholar]
- 29. Mo MS, Li HB, Wang BY, Wang SL, Zhu ZL et al. (2012) PI3K/Akt and NF-kappaB activation following intravitreal administration of 17beta-estradiol: Neuroprotection of the rat retina from light-induced apoptosis. Neuroscience 228: 1-12. PubMed: 23069760. [DOI] [PubMed] [Google Scholar]
- 30. Arroba AI, Wallace D, Mackey A, de la Rosa EJ, Cotter TG (2009) IGF-I maintains calpastatin expression and attenuates apoptosis in several models of photoreceptor cell death. Eur J Neurosci 30: 975-986. doi: 10.1111/j.1460-9568.2009.06902.x. PubMed: 19723289. [DOI] [PubMed] [Google Scholar]
- 31. Sokolova T, Gutterer JM, Hirrlinger J, Hamprecht B, Dringen R (2001) Catalase in astroglia-rich primary cultures from rat brain: immunocytochemical localization and inactivation during the disposal of hydrogen peroxide. Neurosci Lett 297: 129-132. doi: 10.1016/S0304-3940(00)01689-X. PubMed: 11121887. [DOI] [PubMed] [Google Scholar]
- 32. Halliwell B, Clement MV, Long LH (2000) Hydrogen peroxide in the human body. FEBS Lett 486: 10-13. doi: 10.1016/S0014-5793(00)02197-9. PubMed: 11108833. [DOI] [PubMed] [Google Scholar]
- 33. Brzyska M, Elbaum D (2003) Dysregulation of calcium in Alzheimer's disease. Acta Neurobiol Exp 63: 171-183. PubMed: 14518509. [DOI] [PubMed] [Google Scholar]
- 34. Chen KC, Blalock EM, Thibault O, Kaminker P, Landfield PW (2000) Expression of alpha(1D) subunit mRNA is correlated with L-type Ca2+ channel activity in single neurons of hippocampal "zipper" slices. Proc Natl Acad Sci U S A 97: 4357-4362. doi: 10.1073/pnas.070056097. PubMed: 10759553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DeLorenzo RJ, Sun DA, Deshpande LS (2005) Cellular mechanisms underlying acquired epilepsy: The calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol Therapeutics 105: 229-266. doi: 10.1016/j.pharmthera.2004.10.004. PubMed: 15737406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takahashi A, Camacho P, Lechleiter JD, Herman B (1999) Measurement of intracellular calcium. Physiol Rev 79: 1089-1125. PubMed: 10508230. [DOI] [PubMed] [Google Scholar]
- 37. Noguchi A, Takada M, Nakayama K, Ishikawa T (2008) cGMP-independent anti-apoptotic effect of nitric oxide on thapsigargin-induced apoptosis in the pancreatic beta-cell line INS-1. Life Sci 83: 865-870. doi: 10.1016/j.lfs.2008.10.002. PubMed: 18957297. [DOI] [PubMed] [Google Scholar]
- 38. Lee HJ, Ban JY, Seong YH (2005) Blockade of 5-HT3 receptor with MDL7222 and Y25130 reduces hydrogen peroxide-induced neurotoxicity in cultured rat cortical cells. Life Sci 78: 294-300. doi: 10.1016/j.lfs.2005.04.043. PubMed: 16112139. [DOI] [PubMed] [Google Scholar]
- 39. Duncan RS, Goad DL, Grillo MA, Kaja S, Payne AJ et al. (2010) Control of Intracellular Calcium Signaling as a Neuroprotective Strategy. Molecules 15: 1168-1195. doi: 10.3390/molecules15031168. PubMed: 20335972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Herson PS, Lee K, Pinnock RD, Hughes J, Ashford MLJ (1999) Hydrogen peroxide induces intracellular calcium overload by activation of a non-selective cation channel in an insulin-secreting cell line. J Biol Chem 274: 833-841. doi: 10.1074/jbc.274.2.833. PubMed: 9873022. [DOI] [PubMed] [Google Scholar]
- 41. Clapham DE (2003) TRP channels as cellular sensors. Nature 426: 517-524. doi: 10.1038/nature02196. PubMed: 14654832. [DOI] [PubMed] [Google Scholar]
- 42. Clapham DE (2007) Calcium signaling. Cell 131: 1047-1058. doi: 10.1016/j.cell.2007.11.028. PubMed: 18083096. [DOI] [PubMed] [Google Scholar]
- 43. Molnar T, Barabas P, Birnbaumer L, Punzo C, Kefalov V et al. (2012) Store-operated channels regulate intracellular calcium in mammalian rods. J Physiol 590: 3465-3481. doi: 10.1113/jphysiol.2012.234641. PubMed: 22674725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cárdenas C, Miller RA, Smith I, Bui T, Molgó J et al. (2010) Essential Regulation of Cell Bioenergetics by Constitutive InsP(3) Receptor Ca2+ Transfer to Mitochondria. Cell 142: 270-283. doi: 10.1016/j.cell.2010.06.007. PubMed: 20655468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vicencio JM, Ibarra C, Estrada M, Chiong M, Soto D et al. (2006) Testosterone induces an intracellular calcium increase by a nongenomic mechanism in cultured rat cardiac myocytes. Endocrinology 147: 1386-1395. PubMed: 16339199. [DOI] [PubMed] [Google Scholar]
- 46. Hwang JY, Duncan RS, Madry C, Singh M, Koulen P (2009) Progesterone potentiates calcium release through IP3 receptors by an Akt-mediated mechanism in hippocampal neurons. Cell Calcium 45: 233-242. doi: 10.1016/j.ceca.2008.10.006. PubMed: 19081133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang W, Chu X, Tong Q, Cheung JY, Conrad K et al. (2003) A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem 278: 16222-16229. doi: 10.1074/jbc.M300298200. PubMed: 12594222. [DOI] [PubMed] [Google Scholar]
- 48. Fonfria E, Marshall ICB, Boyfield I, Skaper SD, Hughes JP et al. (2005) Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J Neurochem 95: 715-723. doi: 10.1111/j.1471-4159.2005.03396.x. PubMed: 16104849. [DOI] [PubMed] [Google Scholar]
- 49. Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M et al. (2003) A key role for TRPM7 channels in anoxic neuronal death. Cell 115: 863-877. doi: 10.1016/S0092-8674(03)01017-1. PubMed: 14697204. [DOI] [PubMed] [Google Scholar]
- 50. Yao HH, Peng FW, Dhillon N, Callen S, Bokhari S et al. (2009) Involvement of TRPC Channels in CCL2-Mediated Neuroprotection against Tat Toxicity. J Neurosci 29: 1657-1669. doi: 10.1523/JNEUROSCI.2781-08.2009. PubMed: 19211873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Irnaten M, Blanchard-Gutton N, Praetorius J, Harvey BJ (2009) Rapid effects of 17 beta-estradiol on TRPV5 epithelial Ca2+ channels in rat renal cells. Steroids 74: 642-649. doi: 10.1016/j.steroids.2009.02.002. PubMed: 19463684. [DOI] [PubMed] [Google Scholar]
- 52. Brandt SK, Weatherly ME, Ware L, Linn DM, Linn CL (2011) Calcium Preconditioning Triggers Neuroprotection in Retinal Ganglion Cells. Neuroscience 172: 387-397. doi: 10.1016/j.neuroscience.2010.10.071. PubMed: 21044663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiang GX, Pan LB, Xing WL, Zhang L, Huang LH et al. (2007) Identification of activity-dependent gene expression profiles reveals specific subsets of genes induced by different routes of Ca2+ entry in cultured rat cortical neurons. J Cell Physiol 212: 126-136. doi: 10.1002/jcp.21008. PubMed: 17443680. [DOI] [PubMed] [Google Scholar]
- 54. Chen Q, Surmeier DJ, Reiner A (1999) NMDA and non-NMDA receptor-mediated excitotoxicity are potentiated in cultured striatal neurons by prior chronic depolarization. Exp Neurol 159: 283-296. doi: 10.1006/exnr.1999.7135. PubMed: 10486197. [DOI] [PubMed] [Google Scholar]
- 55. Seigel GM (1999) The golden age of retinal cell culture. Mol Vis 5: 4 PubMed: 10209197. [PubMed] [Google Scholar]