

Abstract
The striatal-enriched phosphatase (STEP) is a component of the NMDA-receptor-mediated excitotoxic signaling pathway, which plays a key role in ischemic brain injury. Using neuronal cultures and a rat model of ischemic stroke, we show that STEP plays an initial role in neuroprotection, during the insult, by disrupting the p38 MAPK pathway. Degradation of active STEP during reperfusion precedes ischemic brain damage and is associated with secondary activation of p38 MAPK. Application of a cell-permeable STEP-derived peptide that is resistant to degradation and binds to p38 MAPK protects cultured neurons from hypoxia-reoxygenation injury and reduces ischemic brain damage when injected up to 6 h after the insult. Conversely, genetic deletion of STEP in mice leads to sustained p38 MAPK activation and exacerbates brain injury and neurological deficits after ischemia. Administration of the STEP-derived peptide at the onset of reperfusion not only prevents the sustained p38 MAPK activation but also reduces ischemic brain damage in STEP KO mice. The findings indicate a neuroprotective role of STEP and suggest a potential role of the STEP-derived peptide in stroke therapy.
Introduction
The brain-enriched tyrosine phosphatase, STEP (also known as PTPN5), has been implicated in glutamate-mediated excitotoxic cell death and may serve as a downstream regulator of NMDA-dependent neuronal injury. STEP is expressed in neurons of the striatum, neocortex, hippocampus, and related structures (Lombroso et al., 1993; Boulanger et al., 1995; Pelkey et al., 2002). Alternative splicing leads to the formation of STEP isoforms, and both cytosolic (STEP46) and membrane-associated (STEP61) variants exist (Bult et al., 1996, 1997). Both isoforms contain a substrate-binding domain termed the kinase interacting motif (KIM) domain (Pulido et al., 1998).
Activation and inactivation of STEP are regulated by the phosphorylation state of a critical serine residue in the middle of the KIM domain (serine 221 in STEP61/serine 49 in STEP46), which regulates substrate binding. STEP fails to bind to its substrates after phosphorylation of this residue by dopamine/D1 receptor-mediated activation of the cAMP/protein kinase A (PKA) pathway (Paul et al., 2000). Dephosphorylation of this residue occurs after stimulation of NR1/NR2B subunit-containing NMDARs (NR2B-NMDARs) and subsequent activation of a Ca2+-dependent phosphatase, calcineurin (Paul et al., 2003; Paul and Connor, 2010). Dephosphorylation of this residue renders STEP active in terms of its ability to bind to its substrates and inhibit their activity (Paul et al., 2003; Poddar et al., 2010). This dephosphorylated form of STEP is referred to as the “active” form hereafter. Dephosphorylated STEP contributes to neuroprotection because it binds and inhibits the stress-activated kinase p38 (Poddar et al., 2010), a member of the mitogen-activated protein kinase (MAPK) family that is known to be involved in excitotoxic cell death and ischemic brain damage (Irving and Bamford, 2002; Cao et al., 2005). Indeed, recent studies in primary cultured neurons indicate that dephosphorylated STEP61 may play an initial role in neuroprotection against glutamate toxicity by preventing sustained activation of p38 MAPK (Poddar et al., 2010).
Levels of endogenous STEP can also influence the susceptibility of neurons to excitotoxic insults. The degree of poly-ubiquitination and proteasomal degradation of STEP appears to be regulated by phosphorylation at two additional sites within the kinase specificity sequence (KIS) domain. We recently showed that endogenous phosphorylation of these two sites leads to stability of STEP and that dephosphorylation of these sites leads to STEP degradation by the ubiquitin proteasome pathway. The active form of STEP is more susceptible to degradation than the inactive form (Mukherjee et al., 2011). Further evidence supporting the role of endogenous STEP in neuroprotection has come from studies in cultured neurons, demonstrating that prolonged excitotoxic insult leads to proteolysis of active STEP61, resulting in secondary activation of p38 MAPK and subsequent cell death (Poddar et al., 2010).
Based on these findings, the present study examined the temporal profile of STEP activation and subsequent degradation after cerebral ischemia and reperfusion (I/R) and evaluated the relationship with p38 MAPK activation. The study also determined the efficacy of a cell-permeable STEP-derived peptide, which is resistant to degradation and binds constitutively to its substrates, in attenuating ischemic brain injury. In addition, we determined whether genetic deletion of STEP exacerbates ischemic brain damage. The findings suggest that STEP may be a viable therapeutic target, downstream of NMDARs for intervention in diseases that involve excitotoxicity.
Materials and Methods
Reagents.
Male Sprague Dawley (SD) and Wistar rats and pregnant female SD rats were from Harlan Laboratories. STEP knock-out mice (KO or STEP−/−) were developed on a C57BL6 background (Venkitaramani et al., 2009) and were bred at the University of New Mexico Animal Care Facility. Antibodies used were as follows: polyclonal anti-p38, rabbit monoclonal anti-phospho-p38 (TPEYP) from Cell Signaling Technology, monoclonal anti-myc from Santa Cruz Biotechnology, and monoclonal anti-STEP (recognizes all STEP isoforms) from Novus Biologicals. All secondary antibodies were from Cell Signaling Technology. All tissue culture reagents were obtained from Invitrogen. All other reagents were from Sigma-Aldrich. Approval for animal experiments was given by the University of New Mexico, Health Sciences Center, Institutional Animal Care and Use Committee.
Construction and purification of TAT-STEP-myc peptide.
STEP ΔPTP cDNA encoding STEP61 173–279 amino acids was subcloned into a pTrc-His-myc-TOPO expression vector (Invitrogen). To render the peptide cell-permeable, an 11 amino acid TAT peptide (trans-activator of transcription of human immunodeficiency virus) nucleotide sequence was inserted at the N-terminal of the STEP ΔPTP cDNA (Poddar et al., 2010). A point mutation was introduced at serine 221 within the KIM domain (S221A) by site-directed mutagenesis (Pfu Turbo, Stratagene) to render the peptide constitutively active in terms of its ability to bind to its substrate. Point mutations were also introduced at threonine 231 (T231E) and serine 244 (S244E) in the KIS domain to mimic the phosphorylatable form that helps to maintain the stability of STEP (Mukherjee et al., 2011). The modified STEP ΔPTP peptide was expressed in Escherichia coli, and the TAT-STEP-myc peptide was purified using BD-Talon resin (BD Biosciences).
Cell culture.
Primary neuronal cultures were obtained from 16- to 17-d-old rat embryos as described previously (Paul et al., 2003). Briefly, the striatum and the adjoining cortex were dissected, the tissue dissociated mechanically and resuspended in DMEM/F-12 (1:1) containing 5% FCS. Cells were plated on 60 mm poly-d-lysine-coated tissue culture dishes or 2-well culture slides and grown for 12–14 d at 37°C in a humidified atmosphere (95:5% air/CO2 mixture). To inhibit proliferation of non-neuronal cells, 10 μm of cytosine d-arabinofuranoside was added to the cultures 72 h after plating.
Oxygen glucose deprivation (OGD).
For OGD challenge, neurons were placed in an anaerobic chamber (Coy Laboratories) and incubated in balanced salt solution (116 mm NaCl, 5.4 mm KCl, 1 mm NaH2PO4, 1.8 mm CaCl2, 26.2 mm NaHCO3, 5 mm HEPES, 0.01 mm glycine, pH 7.4) lacking glucose and aerated with an anaerobic gas mixture (95:5%, N2:CO2 mixture) to remove residual oxygen. Some cultures were treated with TAT-STEP-myc peptide (4 μm) during the OGD insult. At specified time periods (0, 10, and 30 min) during OGD, cells were removed from the chamber and lysed for immunoblot analysis. In some experiments at the end of the insult (2 h), the OGD medium was replaced with the original medium and then incubated for an additional 4 or 24 h in a humidified atmosphere (95:5% of air/CO2 mixture). Cells were then processed for either immunoblot analysis (4 h) or cell death assays (24 h).
Induction of transient focal cerebral ischemia.
Middle cerebral artery occlusion (MCAO) was performed in both rats and mice. Rats were used for biochemical studies and to test effects of intravenous delivery of TAT-STEP-myc peptide. As rat knock-outs are not available, mice were used to test the consequences of genetic deletion of STEP on ischemic infarct size and neurological deficits.
For rat studies, animals (270–290 g) were anesthetized by spontaneous inhalation of isoflurane (2%) in medical-grade oxygen. Rectal temperature was maintained at 37 ± 1°C with an electrical heating pad both during surgery and recovery. Focal cerebral ischemia was induced by MCAO using the previously described intraluminal method (Longa et al., 1989; Candelario-Jalil et al., 2004). Briefly, the right common carotid artery (CCA) and the external carotid artery were exposed by a ventral midline neck incision and clipped. The pterygopalatine branch of the internal carotid artery was also clipped to prevent incorrect insertion of the occluding filament. Arteriotomy was performed in the CCA ∼2 mm proximal to the bifurcation and a silicon-rubber-coated monofilament (4-0, Doccol) was inserted. The filament was advanced through the internal carotid artery to a length of 18–19 mm from the bifurcation resulting in occlusion of the origin of the anterior cerebral, middle cerebral, and posterior communicating arteries. Depending on the experiment, the occluding filament was kept in place for 15–90 min, and animals were immediately killed, or the filament was gently retracted to allow reperfusion for various durations (3–24 h). For reperfusion, the incision was closed and animals were allowed to recover from anesthesia. TAT-STEP-myc peptide, TAT-myc, or vehicle (PBS) was delivered by a single intravenous injection through the femoral vein either 20 min before MCAO, at the onset of reperfusion or 6 h after the onset of the insult.
For studies using mice, MCAO was performed using male mice (25–27 g) by the intraluminal method as described above. A 6-0 monofilament (Doccol) was advanced from the CCA through the internal carotid artery to a length of 10–11 mm from the bifurcation to occlude the middle cerebral artery (Wetzel et al., 2008). Depending on the experiment, the occluding filament was kept in place for 10–30 min, and animals were immediately killed, or the filament was gently retracted after 30 min to allow reperfusion for various durations (3 or 24 h). In some experiments, a single intravenous injection of TAT-STEP peptide was administered at the onset of reperfusion. Laser Doppler flowmetry was used to monitor regional cerebral blood flow immediately before, during, and after MCAO in mice.
In a subset of rat and mice, the left femoral artery was cannulated for monitoring mean arterial blood pressure using a multichannel arterial and intracranial pressure monitoring system (TSD147B, Biopac Systems) and to obtain arterial blood samples for blood gas analysis (pO2, pCO2, and pH were measured using a portable clinical analyzer iSTAT, cartridge CG4+, Abbott Laboratories).
Cerebral vasculature.
To determine whether deletion of STEP gene can cause a phenotypic change in the cerebral vasculature, wild-type (WT) and STEP knock-out (KO) male mice were anesthetized and then transcardially perfused with India ink (5%) in an equal volume of gelatin (20%) in water. The brains were then removed, postfixed in 4% paraformaldehyde for 24 h, and the cerebral vasculature was observed under a dissecting microscope.
Behavioral studies.
Male mice were subjected to a battery of behavioral tests (locomotor, rotarod, balance beam, Y-maze, nose poke, social interaction, and swim task) to evaluate social behavior, sensory, motor, and cognitive functions.
An observer blinded to the study groups evaluated all mice. Exploratory locomotor activity was tested using an automated system (San Diego Instruments) following our previously published method (Paz et al., 2007). For the rotarod test (Allan and Harris, 1989), mice were placed on an accelerating cylinder that rotated from 0 to 50 rpm over a period of 2 min. The time and speed at which the mice fell off the cylinder were measured automatically. For the balance beam test (Crabbe et al., 2003), mice were placed on a metal beam 1.3 cm diameter and 77 cm long, suspended 2 feet above the ground, and were required to traverse the beam. They were scored on a scale of 0–6 as follows: 0, cross the beam without any slips or hesitations; 1, cross the beam with 1 or 2 slips and/or hesitation; 2, cross the beam partially with multiple slips and falls; 3, balances with steady posture (>60 s) with multiple slips and 1 fall; 4, attempts to balance on the beam and falls off (>40 s) with multiple slips and falls but is walking on feet; 5, attempts to balance on the beam but falls off (>20 s), multiple falls and fails to walk on the feet; and 6, falls off: no attempt to balance or hang on the beam (<20 s). Spatial working memory was evaluated by recording spontaneous alteration behavior in the Y-maze task (Hsiao et al., 1996). For the nose poke test, mice were placed for 10 min in a dimly lighted open field apparatus (opto-Varimex, Columbus Instruments) covered with a Plexiglas floor containing 28 evenly distributed 1.5-cm-diameter holes. A single Cheerio was placed beneath each hole. The mice were scored for latency to explore the first hole and the number of total holes. Social interaction testing was done as described previously (Allan et al., 2008). Briefly, mice were tested in place preference boxes consisting of two distinctly different chambers connected by an anteroom with a removable door. One chamber housed a novel mouse and the other chamber housed a brightly colored one-inch square cube in small wire cages. The testing mouse was placed in the anteroom with access to both the chambers for 10 min, and the time spent in both the chambers was recorded and expressed as a preference score time spent in proximity of the novel mouse/time spent in proximity of the object. The swim task was performed as described previously (Allan et al., 2008). Mice were placed in a 30-cm-diameter, 46-cm-tall cylinder of water (depth 26 cm, 22–25°C) and forced to swim for 3 min. Swimming, climbing, and thrashing behaviors were considered as escape-directed behaviors, whereas floating, kicking, and twitching behaviors represented immobility indicative of depression-like behaviors. All scores were expressed as the mean of three trials. Severity of neurologic deficit (Longa et al., 1989) was assessed on a 5-point scale as follows: 0, no observable deficits; 1, failure to extend left forepaw; 2, circling to the left; 3, falling to left; 4, no spontaneous walking with a depressed level of consciousness; 5, death.
Infarct volume measurement.
The volume of ischemic brain damage was calculated as described previously (Swanson et al., 1990). For rats, 24 hours after MCAO, animals were anesthetized, brains were removed, sliced into six 2-mm-thick sections, incubated for 30 min at 37°C in 2% 2,3,5-triphenyltetrazolium chloride monohydrate (TTC) in saline, and scanned on a Umax Powerlook scanner. Twenty-four hours after MCAO, mice were perfused intracardially with ice-cold 4% paraformaldehyde in 0.01 m PBS, brains were removed, cryoprotected, coronal forebrain sections were collected at 90 μm intervals, stained with Fluoro-Jade C, and digitized images were created using Zeiss Axiovert 200M fluorescent microscope (Applied Scientific Instruments). In each slice, the total area in the contralateral side and the noninfarcted area in the lesioned side were measured by an investigator blinded to treatment or genotype using Adobe Photoshop. The areas on each side were summed over the number of sections evaluated, and the respective volumes were calculated by multiplying each sum by 2 (thickness of each section). The percentage of infarction volume was calculated as follows: [(volume of contralateral side − noninfarcted volume of the lesioned side)/volume of contralateral side] × 100% (Swanson et al., 1990).
Immunoblotting.
For immunoblotting studies, rats were decapitated after sham surgery and at 15, 30, 60, and 90 min after MCAO or at 3, 6, and 12 h after reperfusion. Mice were decapitated after sham surgery and at 10 and 30 min after MCAO or at 3 h after reperfusion. Striatum from the ipsilateral hemisphere was rapidly dissected on ice and stored at −80°C until processed. Samples were briefly sonicated in Laemmli sample buffer, boiled at 100°C for 10 min, and processed for SDS-PAGE and immunoblotting (Paul et al., 2003, 2007). Quantification of STEP61, STEP33, and p38 protein levels as well as p38 MAPK phosphorylation level was done by computer-assisted densitometry and ImageJ analysis. Band intensities of phosphorylated p38 were normalized to total p38 levels from the same blot. Band intensities of STEP33 were normalized to total tubulin levels in the same blot. For quantification of phosphorylated and nonphosphorylated STEP61, the band intensities of both the upper phosphorylated and lower nonphosphorylated STEP bands were measured and presented as a percentage of the total (Paul et al., 2003; Paul and Connor, 2010; Poddar et al., 2010).
Immunohistochemistry, Fluoro-Jade C, and Hoechst DNA staining.
At appropriate time points after MCAO (0, 3, 12, and 24 h after reperfusion), rats or mice were rapidly anesthetized with isoflurane and perfused intracardially with ice-cold 4% PFA in 0.01 m PBS. Brains were removed and postfixed in the same fixative solution for 4 h and cryoprotected in 15% and 30% sucrose in PBS and then frozen in Optimal Cutting Temperature compound. Immunohistochemistry with anti-STEP antibody and Fluro-Jade C staining were both performed on 10–16 μm sections. For immunohistochemistry, sections were blocked with 10% normal goat serum, 3% BSA in PBS-T (PBS with 0.2% Triton X-100) for 1 h at room temperature and incubated overnight at 4°C with anti-STEP antibody (1:50). After extensive washing in PBS-T, sections were incubated with AlexaFluor-488 goat anti-mouse antibody (1:250; Invitrogen) for 1 h at room temperature. Sections were washed in PBS-T and mounted using Vectashield (Invitrogen). Corresponding sections were processed for Fluoro-Jade C staining. Briefly, sections were air-dried, dehydrated, incubated with 0.06% potassium permanganate (15 min), rinsed in water, and stained with 0.001% Fluro-Jade C (Histo-Chem) in 0.1% acetic acid for 30 min with gentle agitation on ice. Sections were rinsed in water, air-dried, and mounted using Permount mounting media. Sections were analyzed using fluorescent microscopy (Zeiss Axiovert 200M, Applied Scientific Instruments).
For assessment of nuclear damage after OGD, neuronal cultures were stained with Hoechst 33342 dye for 15 min, washed extensively with PBS, and analyzed using fluorescent microscopy. To quantitatively assess the percentage of pyknotic nuclei, a total of 1000 cells were counted for each set of experiments (Poddar et al., 2010).
Data analysis.
Data in the text and figures are expressed as mean ± SEM. Statistical differences between multiple groups were assessed using one-way ANOVA followed by Bonferroni's post hoc comparisons test. Two-group comparisons were analyzed by the Student's t test. Analysis of nonparametric data (beam balance, Y-maze, and NSS test) in both rat and mice were performed using Mann–Whitney U test. Differences were considered statistically significant when p < 0.05.
Results
Alteration of p38 MAP kinase and STEP activity after transient focal ischemia
To determine the effect of transient ischemia on p38 MAPK and STEP phosphorylation, adult male SD rats were subjected to MCAO for varying time periods (15, 30, 60, or 90 min). Striatal punches obtained from the ipsilateral side were processed for immunoblot analysis. A rapid increase in p38 MAPK phosphorylation was observed within 15 min of the ischemic onset. p38 MAPK phosphorylation progressively decreased to basal levels over the next 30–90 min, whereas total p38 MAPK level remained unaltered (Fig. 1a). Analyzing the same samples with an anti-STEP antibody showed that STEP61 was phosphorylated in sham tissues (Fig. 1b, lane 1), detected by an upward shift in the mobility of the STEP band (Paul et al., 2003). Using a phospho-specific antibody that recognizes STEP only when it is phosphorylated at the PKA site in the KIM domain (Paul et al., 2003; Snyder et al., 2005; Valjent et al., 2005), we further confirmed that STEP61 is basally phosphorylated at this site (Fig. 1c, top, lane 1). Ischemia led to a time-dependent dephosphorylation of STEP, as determined both by downward shift in mobility of the STEP band (Fig. 1b, lanes 2–5) and lack of detection of any protein band with the phospho-specific antibody (Fig. 1c, top, lane 2).
Figure 1.

Activation of p38 MAPK and STEP during MCAO and reperfusion. SD (a–e) and Wistar (g–j) rats were subjected to MCAO for (a, b, g, h) 15, 30, 60, or 90 min or (d, e, i, j) 90 min followed by reperfusion for 3, 6, or 12 h. Tissue punches from the ipsilateral striatum were analyzed by immunoblotting with (a, d, g, i) anti-phospho-p38 MAPK (top) and reprobed with anti-p38 MAPK (bottom); (b, e, h, j) anti-STEP antibody (top, STEP61; middle, STEP33) and then reprobed with anti-tubulin antibody (bottom). c, STEP was immunoprecipitated from striatal lysates (sham and 90 min MCAO) using anti-STEP antibody and processed for immunoblot analysis with p-ser 221 antibody (top). The blot was then reprobed with anti-STEP antibody (bottom). f, SD rats were subjected to 90 min MCAO followed by reperfusion for 6 h and then processed for immunohistochemistry with anti-phospho-p38 MAPK and NeuN antibodies and DAPI staining. Bar diagrams represent mean ± SEM obtained from 4 animals per group. a, *p < 0.01, from 15 min MCAO. **p < 0.001, from 15 min MCAO. b, *p < 0.001, from phosphorylated STEP in sham. #p < 0.001, from dephosphorylated form of STEP in sham. d, *p < 0.001, from I/R-6 h. e, *p < 0.05, from I/R-6 h.
Figure 1d shows the temporal profile of p38 MAPK phosphorylation during reperfusion, after 90 min of MCAO. An increase in phosphorylation of p38 MAPK was observed at 6 h, which returned to basal levels by 12 h after reperfusion. A time increase in degradation of active STEP61 was seen in the same preparations from 6 h of reperfusion, resulting in the appearance of a cleaved product of ∼33 kDa (STEP33) (Gurd et al., 1999; Braithwaite et al., 2008) (Fig. 1e). Immunohistochemical staining of coronal sections through the striatum with anti-phospho-p38 and NeuN antibodies confirmed that the activation of p38 MAPK at 6 h after reperfusion was neuron-specific (Fig. 1f). The responses shown here in SD rats were also observed in Wistar rats, suggesting that these effects are not specific to one rat strain (Fig. 1g–j).
This pattern of rapid but transient activation of p38 MAPK followed by its inactivation in conjunction with the activation of STEP, as well as the secondary activation of p38 MAPK associated with subsequent degradation of active STEP are similar to what we have observed earlier in cultured neurons exposed to an excitotoxic dose of glutamate (Poddar et al., 2010). Because p38 MAPK is a substrate of STEP, it raised the possibility that, after an ischemic insult, STEP plays an initial neuroprotective role by downregulating the activity of p38 MAPK.
To determine whether downregulation of active STEP precedes ischemic brain damage, Fluoro-Jade C staining and STEP immunohistochemistry were compared in rat brain sections after ischemia (90 min MCAO) and reperfusion (3, 6, and 12 h). Figure 2 shows that STEP was unilaterally and progressively downregulated in the striatum. Loss of STEP staining was evident even at the earliest time point (3 h reperfusion) and was substantial at 6 h of reperfusion (Fig. 2a, right). In contrast, no damage was visible with Fluoro-Jade C staining at early time points (3 and 6 h of reperfusion), when STEP decreases were already well established and positive Fluoro-Jade C cells were only seen beginning at 12 h (Fig. 2b, right).
Figure 2.

Downregulation of STEP precedes ischemic brain damage. SD rats were subjected to right MCAO for 90 min followed by reperfusion for specified time periods (3, 6, and 12 h). a, Immunohistochemistry of coronal sections through the striatum with anti-STEP antibody. b, Fluro-Jade C staining (a marker for cellular degeneration) of adjacent sections.
A cell-permeable TAT-STEP peptide is neuroprotective against OGD
To examine the possible consequences of STEP downregulation on neuronal vulnerability to injury, we exposed corticostriatal neuronal cultures to OGD to mimic ischemic injury. OGD was performed either briefly without recovery (10 or 30 min challenge) to study acute effects, or for a longer duration (2 h) followed by recovery to assess delayed STEP degradation and MAPK activation.
Brief OGD exposure resulted in a rapid increase in phosphorylation of p38 MAPK within 10 min that decreased substantially by 30 min of the insult (Fig. 3a). Total p38 MAPK level remained unaltered by this treatment. The dephosphorylation of p38 MAPK was associated with dephosphorylation and subsequent activation of STEP observed at 30 min (Fig. 3b, lane 3), and is consistent with our findings in the in vivo stroke model (Fig. 1a,b). Following a more extended OGD and recovery (2 h OGD, 4 h recovery), a secondary activation of p38 MAPK was observed (Fig. 3c, lane 3), which coincided with proteolytic cleavage and downregulation of STEP (Fig. 3d, lane 3).
Figure 3.
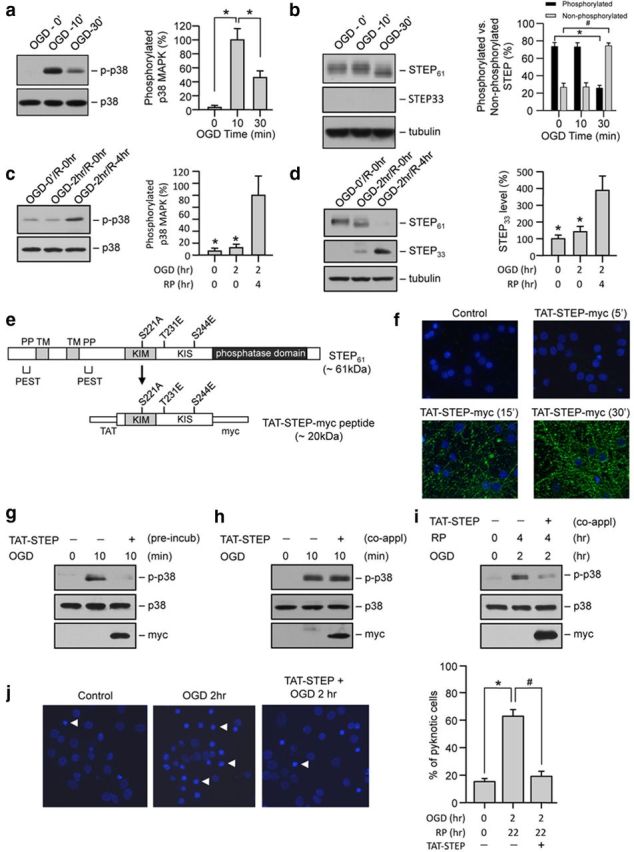
TAT-STEP-myc peptide blocks phosphorylation of p38 MAPK and attenuates OGD-induced neuronal cell death. Neuron cultures were exposed to (a, b) OGD for 10 and 30 min or (c, d) OGD for 2 h (OGD/R-0 h) followed by reoxygenation for 4 h (OGD/R-4 h). Immunoblot analysis of cell lysates with (a, c) anti-phospho-p38 MAPK (top) and reprobed with anti-p38 MAPK (bottom), (b, d) anti-STEP antibody (top- STEP61; middle, STEP33), and then reprobed with anti-tubulin antibody (bottom). e, Schematic diagram of the TAT-STEP-myc peptide (TAT-STEP) indicating the positions of the three phosphorylation sites in the KIM and the KIS domains that were mutated either to alanine (S221A) or to glutamic acid (T231E and S244E). f, Neuronal cultures treated with TAT-STEP-myc peptide for the specified time periods were processed for immunocytochemical staining with anti-myc antibody and DAPI. g, Immunoblot analysis of neuron cultures preincubated (incub) with TAT-STEP-myc before exposure to OGD for 10 min. h, Immunoblot analysis of neuron cultures exposed to OGD for 10 min where TAT-STEP-myc peptide was applied at the onset of OGD (co-appl). i, Immunoblot analysis of neuron cultures exposed to OGD for 2 h followed by reoxygenation for 4 h, where TAT-STEP-myc peptide was applied at the onset of OGD (co-appl). g–i, Blots were analyzed using anti-phospho-p38 MAPK antibody (top) and then reprobed with anti-p38 (middle) or anti-myc (bottom) antibodies. a–d, Bar diagrams represent mean ± SEM (n = 4). a, *p < 0.001 from 10 min OGD. b, *p < 0.01 from phosphorylated STEP at 0 min OGD. #p < 0.01 from dephosphorylated form of STEP at 0 min OGD. c, *p < 0.0001 from OGD/R-4 h. d, *p < 0.05 from OGD/R-4 h. j, Representative photomicrographs of neurons exposed to OGD for 2 h in the presence of TAT-STEP-myc peptide (4 μm) showing pyknotic DNA stained with Hoechst 33342, 24 h later. Arrows indicate pyknotic nuclei. Percentage of neurons with pyknotic nuclei is represented as mean ± SEM from 12 cultures in four separate experiments (control 15.4 ± 1.1%, OGD 2 h 62.9 ± 2.5%, and TAT-STEP-myc + OGD 2 h 19.1 ± 1%). *p < 0.001 from untreated control. #p < 0.001 from OGD alone.
To directly test the hypothesis that interaction of STEP with p38 MAPK regulates neuronal injury, we generated a cell-permeable TAT-STEP-myc peptide that constitutively binds to p38 MAPK and is resistant to ubiquitin-mediated proteasomal degradation (Fig. 3e). Immunocytochemical studies with an anti-myc antibody demonstrated that the TAT-STEP-myc peptide was detectable in neurons within 15 min of application and peaked at 30 min (Fig. 3f). We next examined whether preincubation with the TAT-STEP-myc peptide or its coapplication during the OGD insult can block the phosphorylation of p38 MAPK. Preincubation of neurons with the peptide (30 min) before OGD insult blocked the phosphorylation of p38 MAPK at 10 min (Fig. 3g). Application of the peptide at the onset of OGD failed to block the initial phosphorylation of p38 MAPK by 10 min (Fig. 3h) but blocked the delayed phosphorylation of p38 MAPK at 4 h after the insult (Fig. 3i). Coapplication of the peptide during the OGD insult also significantly reduced neuronal cell death assessed 24 h after a 2 h OGD challenge (Fig. 3j). Thus, TAT-STEP-myc peptide, when applied during the OGD insult, failed to block the initial p38 MAPK activation but significantly reduced the delayed p38 MAPK activation and neuronal injury. This is consistent with our hypothesis that neuronal injury after an ischemic insult is primarily caused by secondary activation of p38 MAPK after the downregulation of STEP.
TAT-STEP peptide attenuates MCAO-induced ischemic brain damage
We next investigated whether the TAT-STEP-myc peptide could attenuate ischemic brain injury in vivo. In initial experiments, we examined the feasibility of delivering the TAT-STEP-myc peptide in a noninvasive manner into the brain of intact animals. Rats were injected intravenously with either the peptide (3 nmol/g of body weight) or the vehicle (PBS). Coronal brain sections from animals injected with TAT-STEP-myc peptide showed strong fluorescence with anti-myc antibody in both the cortex and striatum within 1 h of injection of the peptide, whereas those treated with saline showed no staining, confirming the peptide uptake (Fig. 4a). To determine whether the TAT-STEP-myc peptide could bind to endogenous p38 MAPK in vivo, cortical and striatal lysates from rats injected with either the peptide or vehicle were processed for immunoprecipitation with anti-myc antibody. Immunoblotting with anti-p38 antibody showed that the peptide forms a stable complex with p38 MAPK (Fig. 4b).
Figure 4.
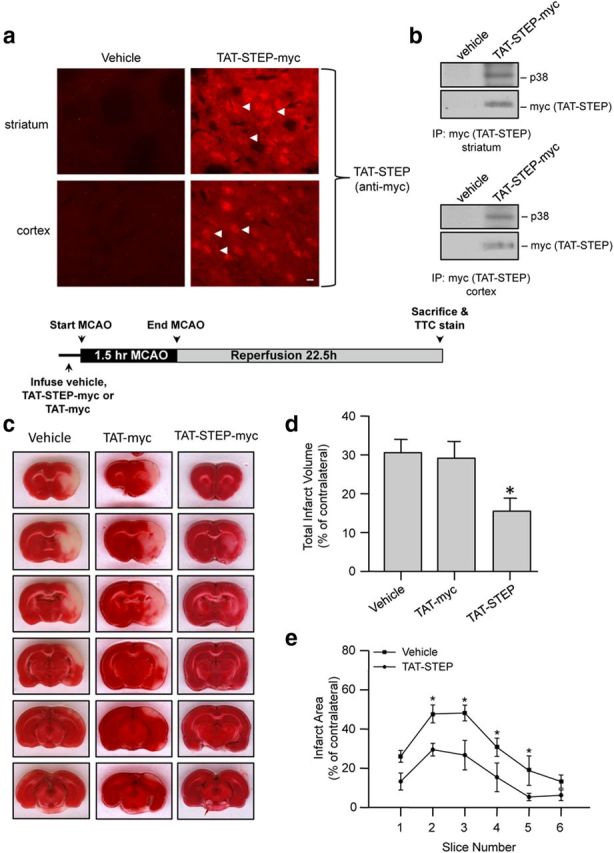
Administration of TAT-STEP peptide at the onset of insult significantly reduces ischemic brain damage. a, Immunohistochemical analysis with anti-myc antibody for detection of the STEP peptide in the brain after intravenous injection of vehicle or TAT-STEP-myc peptide (3 nmol/g). Representative photomicrographs, demonstrating myc-positive cells (indicated by arrows) in coronal sections through the striatum (top) and the cortex (bottom). Scale bar, 20 μm. b, Intravenous administration of TAT-STEP peptide followed by immunoprecipitation of the peptide from striatal and cortical lysates with anti-myc antibody. The immune complex was processed for immunoblot analysis with anti-p38 MAPK antibody (top) and then reprobed with anti-myc antibody (bottom). c–e, Rats were preinjected with TAT-STEP-myc (n = 8), TAT-myc (n = 4), or vehicle (n = 10), before 90 min of MCAO. c, Representative photomicrographs of TTC-stained brain slices (2 mm) showing brain infarct 24 h after the onset of ischemia. d, Quantitative analysis of the total infarct volume (vehicle 30.6 ± 3.5%, TAT-myc 29.1 ± 4.3%, vs TAT-STEP-myc 15.4 ± 3.3%). e, Total infarct area within each slice is represented as mean ± SEM. *p < 0.05 from vehicle-treated control.
We then examined whether pretreatment with the peptide would reduce ischemic brain damage. A single dose of the vehicle, TAT-myc (3 nmol/g) or TAT-STEP-myc peptide (3 nmol/g), was injected intravenously 20 min before MCAO. Rats were then subjected to MCAO for 90 min followed by reperfusion for 22.5 h. Figure 4c–e shows that pretreatment with the TAT-STEP-myc peptide significantly reduced ischemic brain damage compared with vehicle-treated controls. In contrast, treatment with TAT-myc alone had no significant effect on ischemic brain damage. Physiological parameters monitored before, during, and after MCAO in both TAT-STEP-myc-treated and vehicle-treated controls did not show significant difference between the treatment groups (Table 1).
Table 1.
Comparison of physiological parameters in vehicle versus TAT-STEP-myc peptide-treated rats (n = 4)a
| Before MCAO |
During MCAO |
After MCAO |
||||
|---|---|---|---|---|---|---|
| Control | TAT-STEP | Control | TAT-STEP | Control | TAT-STEP | |
| MABP (mmHg) | 102 ± 1.4 | 107 ± 1.6 | 124 ± 3.0 | 106 ± 1.4 | 110 ± 1.5 | 119 ± 1.2 |
| PO2 (mmHg) | 106 ± 4.5 | 122 ± 2.7 | 96 ± 4.5 | 105 ± 9 | 95 ± 10 | 119 ± 7.5 |
| PCO2 (mmHg) | 36.8 ± 1.5 | 38.1 ± 1.4 | 39.1 ± 8.1 | 38.1 ± 4.6 | 37 ± 4.9 | 36.9 ± 0.2 |
| pH | 7.4 ± 0.1 | 7.3 ± 0.3 | 7.4 ± 0.1 | 7.2 ± 0.1 | 7.4 ± 0.2 | 7.4 ± 0.5 |
aNo significant difference was observed between any two groups (p > 0.05; ANOVA, Bonferroni's multiple-comparison test). MABP, Mean arterial blood pressure.
We next evaluated whether the peptide could attenuate ischemic brain damage when applied after an ischemic insult. Rats were subjected to MCAO for 90 min followed by a single intravenous injection of vehicle or TAT-STEP-myc peptide (3 nmol/g) at the onset of reperfusion. The extent of brain damage was measured 24 h after the onset of MCAO. Similar to the pretreatment study, a significant reduction in total brain infarct size was observed in animals treated with TAT-STEP-myc compared with vehicle-treated controls (Fig. 5a–c). We then determined whether the peptide when administered after the ischemic insult could block the phosphorylation of p38 MAPK at 6 h of reperfusion. The extent of p38 MAPK phosphorylation was analyzed by immunoblot analysis of striatal punches obtained from the ipsilateral side. Figure 5d shows a significant decrease in p38 MAPK phosphorylation in the TAT-STEP peptide-treated animals (lane 3) compared with the vehicle-treated controls (lane 2).
Figure 5.
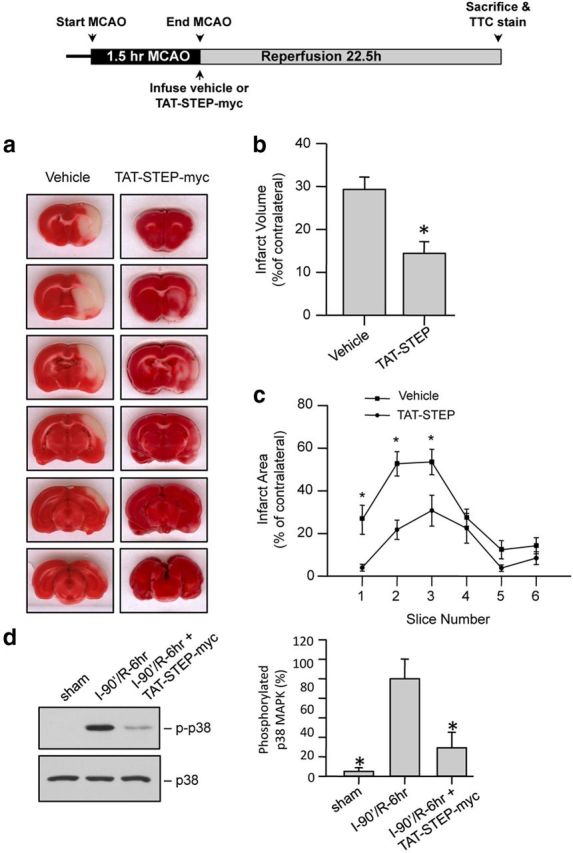
Administration of TAT-STEP-myc peptide at the onset of reperfusion significantly reduces ischemic brain damage. a–c, Rats were subjected to MCAO for 90 min followed by injection of vehicle or TAT-STEP-myc (3 nmol/g). a, Representative photomicrographs of TTC-stained brain slices (2 mm) showing brain infarct 24 h after the onset of ischemia. Quantitative analysis of the (b) total infarct volume (vehicle 29.3 ± 2.5% vs TAT-STEP-myc 14.4 ± 2.7%) and (c) area of infarction within each slice is represented as mean ± SEM (n = 7). *p < 0.05 from vehicle-treated control. d, Rats subjected to MCAO for 90 min were injected with vehicle (I-90′/R-6 h) or TAT-STEP-myc peptide (I-90′/R-6 h + TAT-STEP-myc) at the onset of reperfusion. Immunoblot analysis of striatal lysates obtained from the ipsilateral side, 6 h after the insult. Blots were probed with anti-phospho-p38 MAPK (top) and reprobed with anti-p38 MAPK (bottom). Bar diagram represents mean ± SEM obtained from 4 animals. *p < 0.01 from phosphorylated p38 MAPK in vehicle (I-90′/R-6 h) treated animals.
To determine whether delayed treatment with the peptide could attenuate ischemic brain damage, rats were subjected to MCAO for 90 min followed by a single intravenous injection of the vehicle or TAT-STEP-myc peptide (3 nmol/g) 6 h after the onset of MCAO. A significant reduction in total brain infarct size was observed in animals treated with TAT-STEP-myc compared with vehicle-treated controls (Fig. 6a–c). However, the extent of reduction in ischemic brain damage with the delayed treatment was lesser than that observed with the treatment at the onset of reperfusion (27.5% with delayed treatment at 6 h vs 51% with treatment at the onset of reperfusion).
Figure 6.
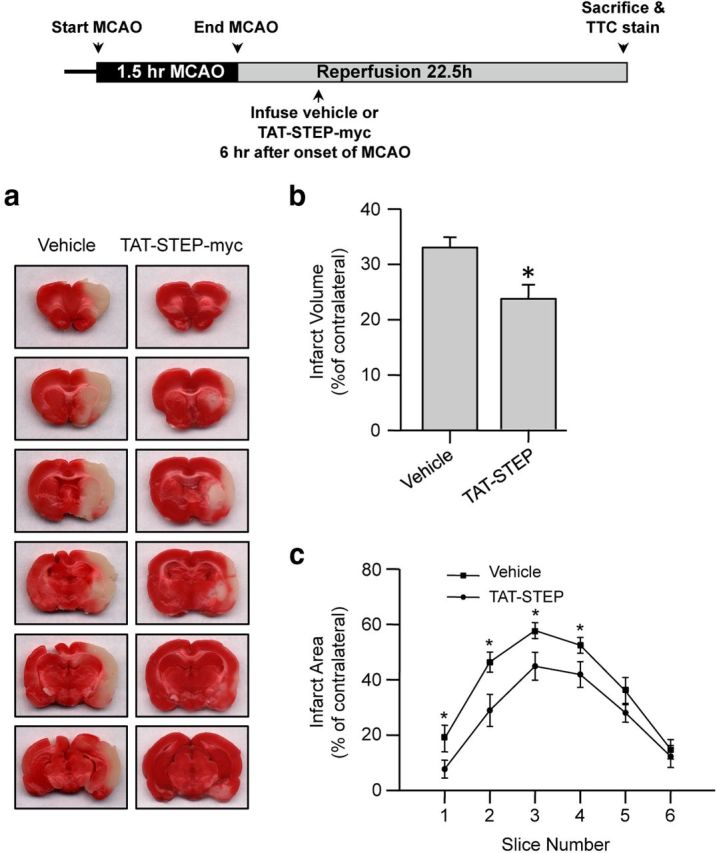
Delayed administration of TAT-STEP-myc peptide significantly reduced ischemic brain damage. Rats were subjected to 90 min MCAO, and the peptide was administered 6 h after the onset of the insult. a, Representative photomicrographs of TTC-stained brain slices (2 mm) showing brain infarct at 24 h after the onset of ischemia. Quantitative analysis of the (b) total infarct volume (vehicle 33 ± 1.9% vs TAT-STEP-myc 23.6 ± 2.6%) and (c) area of infarction within each slice is represented as mean ± SEM (n = 10). *p < 0.05 from vehicle-treated control.
Deletion of STEP exacerbates ischemic brain damage and neurological deficits
We next examined whether deletion of endogenous STEP would lead to an increase in ischemic brain damage. In initial studies, a series of behavioral tests showed that there were no inherent neurological deficits in the STEP KO mice (Fig. 7). Studies on cerebral vasculature in both WT and STEP KO mice revealed no major differences between the anatomy of the circle of Willis, anterior cerebral, middle cerebral, and posterior arteries in the two genotypes (Fig. 8a). WT and STEP KO mice were then subjected to mild focal ischemia induced by 30 min of MCAO followed by reperfusion for 24 h. Cerebral blood flow during MCAO was monitored by laser Doppler flowmetry, and a comparable reduction in regional blood flow was observed in both genotypes (Fig. 8b). Physiological parameters also remained comparable between the two genotypes (Table 2). At 24 h after reperfusion, WT mice showed minimal decline in neurological and motor function, as assessed by the modified neurological severity score, beam balance, and rotarod test. However, STEP KO mice showed a severe decline in neurological and motor function (Fig. 8c–e). Consistent with earlier findings, Fluoro-Jade C-labeled cells were limited to the striatum in WT mice (Zhan et al., 2009). In contrast, Fluoro-Jade C-positive cells were observed in the striatum, cortex, and hippocampus of STEP KO mice (Fig. 8f,g). Together, these observations suggest that deletion of STEP can exaggerate ischemic brain damage resulting in severe neurological deficits.
Figure 7.

Neurobehavioral evaluation of WT and STEP KO mice. a, Spontaneous locomotion in the WT and STEP KO mice was assessed for 180 min. The number of photobeam breaks are represented as mean ± SEM (n = 11–18). p > 0.259. b, Forced swim task test (3 min) to assess fatigue-ability and motivation to escape. The initial length of active escape swimming (p > 0.915) and the total duration of escape (p > 0.275) directed behaviors across the duration of the test are represented as mean ± SEM (assessed in 11 WT and 9 KO mice). c, Balance and coordinated alternation of fore and hindpaw were evaluated using the rotarod. The mean ± SEM of three rotarod performances are presented for n = 6–8 for each strain. p > 0.713. d, Gait and balance were evaluated using a balance beam task. Performance scores were quantified as described in Materials and Methods. A score of 1 indicated strong coordination and gait, whereas a score of 6 indicated very poor performance. Scores for three traverses across the beam are represented as mean ± SEM (n = 9–12). p > 0.394 (Mann–Whitney). e, Spontaneous alteration in a Y-maze was used to evaluate working motor memory. Ability to spontaneously alternate in three consecutive sessions within the Y-maze is represented as mean ± SEM (n = 11 or 12). p > 0.974 (Mann–Whitney). f, Exploratory and general anxiety was measured using a nose poke exploration task. Latency to first explore a hole (p > 0.280) and the number of holes explored (p > 0.608) in the 5 min test are represented as mean ± SEM (n = 7–11). g, Social avoidance was assessed using a social interaction test. Time spent in the 10 min test in the side containing the novel mouse relative to time spent in the side containing a novel object is represented as mean ± SEM (n = 6–9). p > 0.891.
Figure 8.
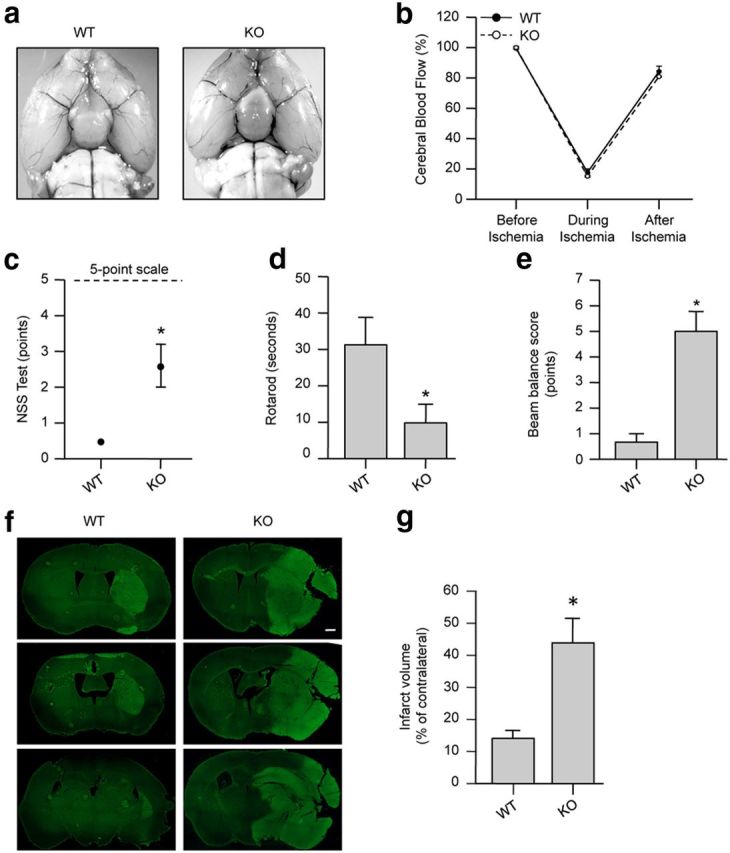
Genetic deletion of STEP exacerbates ischemic brain damage in mice. a, Representative photomicrographs of WT and STEP KO mice brain subjected to MCAO for 30 min and then perfused transcardially with India ink for visualization of cerebral vasculature. b, Quantitative analysis of changes in cerebral blood flow in WT and STEP KO mice before, during, and after 30 min of MCAO (n = 4 per genotype). c–g, WT and KO mice were subjected to 30 min of MCAO followed by 24 h of reperfusion. c–e, Postischemic neurological dysfunctions tested at 24 h after MCAO are represented as mean ± SEM (n = 7). c, Neurological severity score (NSS) tested on a 5 point scale was observed to be 0.5 ± 0.1 in WT mice compared with 2.6 ± 0.6 in STEP KO mice. *p < 0.05 (Mann–Whitney). d, Rotarod testing was done to measure the latency time for a mouse to fall from a rotating cylinder and was observed to be 31.25 ± 7.5 s for WT mice compared with 9.8 ± 5.14 s for STEP KO mice. *p < 0.05. e, The ability to stay and walk on a beam was tested using the beam balance test with a score range of 1–6 and was observed to be 0.66 ± 0.3 for WT mice compared with 5 ± 0.77 for STEP KO mice. *p < 0.05 (Mann–Whitney). f, Representative photomicrographs of coronal sections stained with Fluoro-Jade C, a marker for cellular degeneration. g, Total infarct volume was 15.03 ± 2.64% for WT mice compared with 43.9 ± 7.07% in STEP KO mice. *p < 0.003. n = 7.
Table 2.
Comparison of physiological parameters in WT versus STEP KO mice (n = 4)a
| Before MCAO |
During MCAO |
After MCAO |
||||
|---|---|---|---|---|---|---|
| WT | KO | WT | KO | WT | KO | |
| MABP (mmHg) | 96 ± 5.7 | 94 ± 6.7 | 107 ± 6.9 | 94 ± 15 | 106 ± 13 | 84 ± 3.4 |
| PO2 (mmHg) | 116 ± 7 | 107 ± 12 | 121 ± 10 | 110 ± 14 | 110 ± 6 | 114 ± 10 |
| PCO2 (mmHg) | 44.3 ± 2.6 | 36.4 ± 3.4 | 35.6 ± 4.9 | 33.3 ± 2.2 | 36.2 ± 1.3 | 34.7 ± 3.5 |
| pH | 7.3 ± 0.08 | 7.4 ± 0.03 | 7.3 ± 0.01 | 7.2 ± 0.05 | 7.2 ± 0.1 | 7.3 ± 0.3 |
aNo significant difference was observed between any two groups (p > 0.05; ANOVA, Bonferroni's multiple-comparison test). MABP, Mean arterial blood pressure.
TAT-STEP peptide attenuates MCAO-induced ischemic brain damage in STEP KO mice
To evaluate the role of p38 MAPK signaling pathway in the exacerbation of ischemic brain injury in STEP KO mice, WT and STEP KO mice were subjected to MCAO for 10 or 30 min. Figure 9a, b shows that, during the insult, p38 MAPK phosphorylation increased in both WT and KO mice within 10 min of ischemia. However, p38 MAPK phosphorylation decreased to near basal levels by 30 min in the WT mice, although it remained sustained in STEP KO mice. Figure 9c shows that phosphorylation of p38 MAPK remained elevated in STEP KO mice even at 3 h after reperfusion, following 30 min MCAO. In WT mice, ischemia also led to dephosphorylation of STEP by 30 min (Fig. 9a), and it remained dephosphorylated during reperfusion (Fig. 9c), as evident by downward shift in mobility of the STEP band. As expected, no STEP protein was detectable in the STEP KO mice (Fig. 9b, c, bottom). We then determined whether administration of the STEP-derived peptide at the onset of reperfusion could block the phosphorylation of p38 MAPK in STEP KO mice at 3 h of reperfusion. Figure 9d shows a significant reduction in p38 MAPK phosphorylation in the TAT-STEP peptide-treated STEP KO mice (lane 2) compared with vehicle-treated littermates (lane 1). We next evaluated whether the TAT-STEP peptide could attenuate ischemic brain damage in STEP KO mice when administered at the onset of reperfusion. The extent of brain damage was measured at 24 h after the onset of MCAO. Representative photomicrograph (Fig. 9e), and the corresponding bar diagram (Fig. 9f) shows a significant reduction in ischemic brain damage in TAT-STEP peptide-treated STEP KO mice compared with vehicle-treated STEP KO controls.
Figure 9.
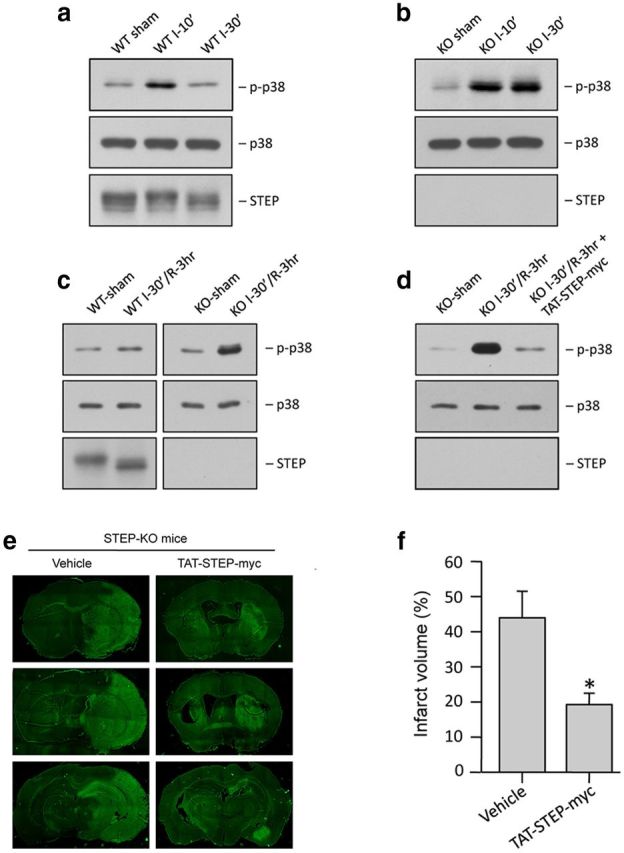
Administration of TAT-STEP peptide at the onset of reperfusion reduces p38 MAPK phosphorylation and ischemic brain damage in STEP KO mice. a, Immunoblot analysis of striatal lysates from the ipsilateral side of WT mice, after 10 or 30 min MCAO (n = 3/group). b, Immunoblot analysis of striatal lysates from the ipsilateral side of STEP KO mice, after 10 or 30 min MCAO (n = 3/group). c, Immunoblot analysis of striatal lysates from the ipsilateral side of WT mice and STEP KO mice, after 30 min MCAO and 3 h reperfusion (n = 3/group). d, Immunoblot analysis of striatal lysates from the ipsilateral side of STEP KO mice subjected to 30 min MCAO followed by administration of TAT-STEP-myc peptide (3 nmol/g) and reperfusion for 3 h (n = 3/group). a–d, Blots were probed with anti-phospho-p38 MAPK (top) and reprobed with anti-p38 MAPK (bottom) or anti-STEP antibody (bottom). e, STEP KO mice were subjected to 30 min MCAO, and the peptide was administered at the onset of reperfusion. Representative photomicrographs of coronal brain sections, 24 h after the onset of ischemia, stained with Fluoro-Jade C. f, Total infarct volume was 43.9 ± 7.07% for STEP KO mice compared with 19.3 ± 2.87% in STEP KO mice. *p < 0.05. n = 5.
Discussion
The present study provides the first evidence that a tyrosine phosphatase can be targeted to limit stroke injury. A role of endogenous STEP in neuroprotection is evident from activation of STEP during an ischemic insult that appears to inhibit the p38 MAPK signaling pathway. However, degradation of active STEP during reperfusion limits the efficacy of STEP and results in upregulation of the p38 MAPK pathway. Based on these observations, we developed a novel strategy that involves a peptide mimetic that constitutively binds to p38 MAPK and is resistant to proteasomal degradation. Transduction of neurons with this peptide is sufficient to inhibit the p38 MAPK phosphorylation during recovery and prevent neuronal injury in an in vitro model of ischemic injury. Intravenous injection of the peptide is also able to block the secondary increase in p38 MAPK phosphorylation during reperfusion and attenuate ischemic brain damage in vivo. Complementary studies in mice with a targeted deletion of STEP gene show that phosphorylation of p38 MAPK remains sustained, when exposed to a mild ischemic insult. The STEP KO mice also exhibit a dramatic increase in brain injury that encompasses the striatum, cortex, and hippocampus. Restoration of the STEP signaling pathway with the STEP-derived peptide inhibits sustained p38 MAPK phosphorylation and reduces ischemic brain damage in the STEP KO mice. Together, the study demonstrates the neuroprotective role of endogenous STEP and further establishes the blood–brain barrier-permeable TAT-STEP-peptide as a promising new tool for stroke therapy.
Previous studies showed that endogenous STEP undergoes proteasomal degradation after exposure to an excitotoxic dose of glutamate (Poddar et al., 2010). It is evident from the current study that STEP is degraded in vitro during OGD in neurons as well as in vivo after an ischemic insult and reperfusion. These findings provided the basis for testing whether a brain-permeable STEP peptide could be effective in limiting stroke injury. It is important to note that the STEP peptide was effective in reducing brain damage even when administered 6 h after the onset of stroke. The efficacy of the peptide in the poststroke treatment paradigm suggests that it may have clinical implications and justifies further evaluation of the peptide in providing long-term protection when administered in the poststroke time window.
p38 MAPK is a critical mediator of neuronal injury in both acute and chronic neurological disorders (Beal, 1996; Mielke and Herdegen, 2000). Several studies have shown that p38 MAPK plays a role in glutamate-mediated excitotoxic cell death (Cao et al., 2005; Poddar et al., 2010), and selective inhibition of p38 MAPK signaling pathway provides protection against excitotoxicity (Poddar et al., 2010). A marked increase in extracellular glutamate levels has been observed in experimental models of ischemic stroke and is responsible for NMDAR-induced brain damage (Choi and Rothman, 1990; Dirnagl et al., 1999). Activation of p38 MAPK has also been observed after ischemic stroke and plays a crucial role in ischemic brain injury (Barone et al., 2001a, b; Irving and Bamford, 2002; Cao et al., 2005; Nito et al., 2008). Using affinity chromatography and cell culture studies, we have shown earlier that p38 MAPK is a substrate of STEP. Using recombinant p38 MAPK and STEP proteins, we further demonstrated that the critical serine residue within the KIM domain of STEP regulates STEP-p38 MAPK interaction. Phosphorylation of this serine residue inhibits the interaction of STEP with p38 MAPK, whereas dephosphorylation of this residue allows STEP to bind to and dephosphorylate p38 MAPK (Poddar et al., 2010). These findings formed the basis of our hypothesis that a STEP-derived peptide, where the serine residue in the KIM domain has been mutated, may bind irreversibly to p38 MAPK in the absence of the phosphatase domain and thereby act as a dominant-negative regulator of p38 MAPK phosphorylation. Consistent with this hypothesis, we further showed that the STEP-derived peptide when transduced into neurons was bound constitutively to p38 MAPK, prevented glutamate-mediated p38 MAPK phosphorylation and nuclear translocation, and greatly reduced neuronal injury (Poddar et al., 2010).
The present study confirms the neuroprotective efficacy of this peptide through the disruption of the p38 MAPK signaling pathway in a cell culture model of hypoxia-reoxygenation injury. The findings also demonstrate the ability of the peptide to cross the blood–brain barrier, remain resistant to proteasomal degradation, and bind to p38 MAPK in brain tissue. This unique function of the peptide allowed us to use it as a novel tool for validating the neuroprotective role of STEP through regulation of p38 MAPK, in ischemic brain injury. Restoring STEP signaling with intravenous administration of the peptide prevents the secondary increase in phosphorylation of p38 MAPK that enhances neuronal survival and reduces ischemic brain damage.
The sustained phosphorylation of p38 MAPK and the dramatic increase in ischemic brain damage in the STEP KO mice suggest that knocking out STEP cannot be fully compensated by upregulation of other tyrosine phosphatases in the brain. Earlier studies have shown that extracellular regulated kinase 1 and 2 (ERK1/2) activity is significantly elevated in the STEP KO brain and provides additional support for the lack of any compensatory mechanisms in these mice (Venkitaramani et al., 2009). It has also been reported that deletion of the STEP gene does not generate a lethal phenotype, and these mice appear to have normal neuroanatomy with no gross changes in brain morphology compared with their wild-type littermates (Venkitaramani et al., 2009). Consistent with these findings, we have not observed any apparent neurological differences between the WT and STEP KO mice. However, the susceptibility of these mice to increased brain damage and severe neurological deficits after a mild ischemia confirms the neuroprotective role of endogenous STEP and also establishes it as a key regulator of the progression of the injury process. The ability of the STEP-derived peptide to prevent the sustained increase in p38 MAPK phosphorylation as well as reducing ischemic brain damage further substantiates the neuroprotective role of STEP through its regulation of the p38 MAPK signaling pathway.
Although the focus of the present study is on the p38 MAPK pathway, previous studies have identified additional substrates of STEP that include ERK 1/2 and the nonreceptor tyrosine kinases Pyk2 and Fyn (Nguyen et al., 2002; Paul et al., 2003, 2007; Xu et al., 2012). Although the consequences of ERK MAPK activation after cerebral ischemia are still unclear (Alessandrini et al., 1999; Park et al., 2004; Jover-Mengual et al., 2007), several studies have implicated a role of Pyk2 and Fyn in ischemic brain damage (Tian et al., 2000; Paul et al., 2001; Hou et al., 2007). However, the binding of STEP with Pyk2 and Fyn involves multiple domains in STEP (Nguyen et al., 2002; Xu et al., 2012), suggesting that they may not be relevant targets of the STEP-derived peptide that lacks most of these domains. To further our understanding of additional pathways that may be regulated by the STEP peptide, future studies will focus on identification of other pathways that are upregulated in the STEP-deficient mice after an ischemic insult and evaluate the precise contribution of the STEP peptide in regulating them.
In conclusion, it appears that an ischemic insult not only triggers a multitude of cytotoxic pathways in the brain, but also triggers some endogenous protective responses capable of limiting injury. Here we identify STEP as one such protein that is activated after an excitotoxic/ischemic insult and may act to provide initial neuroprotection against neuronal vulnerability to injury. However, prolonged insults may lead to loss of endogenous STEP and its neuroprotective efforts, resulting in secondary activation of the cytotoxic pathways. Accordingly, delayed administration of the TAT-STEP peptide, even 6 h after the ischemic insult, attenuates the progression of brain damage and might provide a promising new tool for treatment of stroke.
Footnotes
This work was supported by the National Institutes of Health Grant NS059962 to S.P., Grant 3P20RR015636-09 S1 to K.J. Liu, Grant MH052711 to P.J.L., and Grant NS065343 to R.P. We thank Dr. William Shuttleworth (University of New Mexico) for helpful discussions and critical reading of the manuscript, and Samantha Goggin, Matthew Labrecque, Eduardo Estrada, Dennis Bragin, and Edwin Nemoto for technical help with some experiments.
The authors declare no competing financial interests.
References
- Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci U S A. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan AM, Harris RA. A new alcohol antagonist: phaclofen. Life Sci. 1989;45:1771–1779. doi: 10.1016/0024-3205(89)90516-X. [DOI] [PubMed] [Google Scholar]
- Allan AM, Liang X, Luo Y, Pak C, Li X, Szulwach KE, Chen D, Jin P, Zhao X. The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum Mol Genet. 2008;17:2047–2057. doi: 10.1093/hmg/ddn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, Legos JJ, Erhardt JA, Ohlstein EH, Hunter AJ, Harrison DC, Philpott K, Smith BR, Adams JL, Parsons AA. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev. 2001a;21:129–145. doi: 10.1002/1098-1128(200103)21:2<129::AID-MED1003>3.0.CO%3B2-H. [DOI] [PubMed] [Google Scholar]
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, White RF, McVey MJ, Legos JJ, Erhardt JA, Nelson AH, Ohlstein EH, Hunter AJ, Ward K, Smith BR, Adams JL, Parsons AA. SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther. 2001b;296:312–321. [PubMed] [Google Scholar]
- Beal MF. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/S0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neurosci. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Xu J, Leung J, Urfer R, Nikolich K, Oksenberg D, Lombroso PJ, Shamloo M. Expression and function of striatal enriched protein tyrosine phosphatase is profoundly altered in cerebral ischemia. Eur J Neurosci. 2008;27:2444–2452. doi: 10.1111/j.1460-9568.2008.06209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Jr, Sharma E, Lukacsi E, Solimena M, Naegele JR, Lombroso PJ. STEP61: a member of a family of brain-enriched PTPs is localized to the endoplasmic reticulum. J Neurosci. 1996;16:7821–7831. doi: 10.1523/JNEUROSCI.16-24-07821.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult A, Zhao F, Dirkx R, Jr, Raghunathan A, Solimena M, Lombroso PJ. STEP: a family of brain-enriched PTPs. Alternative splicing produces transmembrane, cytosolic and truncated isoforms. Eur J Cell Biol. 1997;72:337–344. [PubMed] [Google Scholar]
- Candelario-Jalil E, González-Falcón A, García-Cabrera M, Léon OS, Fiebich BL. Wide therapeutic time window for nimesulide neuroprotection in a model of transient focal cerebral ischemia in the rat. Brain Res. 2004;1007:98–108. doi: 10.1016/j.brainres.2004.01.078. [DOI] [PubMed] [Google Scholar]
- Cao J, Viholainen JI, Dart C, Warwick HK, Leyland ML, Courtney MJ. The PSD95-nNOS interface: a target for inhibition of excitotoxic p38 stress-activated protein kinase activation and cell death. J Cell Biol. 2005;168:117–126. doi: 10.1083/jcb.200407024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Metten P, Yu CH, Schlumbohm JP, Cameron AJ, Wahlsten D. Genotypic differences in ethanol sensitivity in two tests of motor incoordination. J Appl Physiol. 2003;95:1338–1351. doi: 10.1152/japplphysiol.00132.2003. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/S0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Gurd JW, Bissoon N, Nguyen TH, Lombroso PJ, Rider CC, Beesley PW, Vannucci SJ. Hypoxia-ischemia in perinatal rat brain induces the formation of a low molecular weight isoform of striatal enriched tyrosine phosphatase (STEP) J Neurochem. 1999;73:1990–1994. doi: 10.1046/j.1471-4159.1999.01990.x. [DOI] [PubMed] [Google Scholar]
- Hou XY, Liu Y, Zhang GY. PP2, a potent inhibitor of Src family kinases, protects against hippocampal CA1 pyramidal cell death after transient global brain ischemia. Neurosci Lett. 2007;420:235–239. doi: 10.1016/j.neulet.2007.03.048. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Jover-Mengual T, Zukin RS, Etgen AM. MAPK signaling is critical to estradiol protection of CA1 neurons in global ischemia. Endocrinology. 2007;148:1131–1143. doi: 10.1210/en.2006-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombroso PJ, Naegele JR, Sharma E, Lerner M. A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. J Neurosci. 1993;13:3064–3074. doi: 10.1523/JNEUROSCI.13-07-03064.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.STR.20.1.84. [DOI] [PubMed] [Google Scholar]
- Mielke K, Herdegen T. JNK and p38 stress kinases: degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol. 2000;61:45–60. doi: 10.1016/S0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Poddar R, Deb I, Paul S. Dephosphorylation of specific sites in the kinase-specificity sequence domain leads to ubiquitin-mediated degradation of the tyrosine phosphatase STEP. Biochem J. 2011;440:115–125. doi: 10.1042/BJ20110240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Liu J, Lombroso PJ. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J Biol Chem. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH. Role of the p38 mitogen-activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood–brain barrier disruption after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2008;28:1686–1696. doi: 10.1038/jcbfm.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EM, Joh TH, Volpe BT, Chu CK, Song G, Cho S. A neuroprotective role of extracellular signal-regulated kinase in N-acetyl-O-methyldopamine-treated hippocampal neurons after exposure to in vitro and in vivo ischemia. Neuroscience. 2004;123:147–154. doi: 10.1016/j.neuroscience.2003.08.023. [DOI] [PubMed] [Google Scholar]
- Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, Chopp M, Cheresh DA. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 2001;7:222–227. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]
- Paul S, Connor JA. NR2B-NMDA receptor-mediated increases in intracellular Ca2+ concentration regulate the tyrosine phosphatase, STEP, and ERK MAP kinase signaling. J Neurochem. 2010;114:1107–1118. doi: 10.1111/j.1471-4159.2010.06835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Snyder GL, Yokakura H, Picciotto MR, Nairn AC, Lombroso PJ. The dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. J Neurosci. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- Paul S, Olausson P, Venkitaramani DV, Ruchkina I, Moran TD, Tronson N, Mills E, Hakim S, Salter MW, Taylor JR, Lombroso PJ. The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biol Psychiatry. 2007;61:1049–1061. doi: 10.1016/j.biopsych.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz R, Barsness B, Martenson T, Tanner D, Allan AM. Behavioral teratogenicity induced by nonforced maternal nicotine consumption. Neuropsychopharmacology. 2007;32:693–699. doi: 10.1038/sj.npp.1301066. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, Salter MW, Lombroso PJ. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/S0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Poddar R, Deb I, Mukherjee S, Paul S. NR2B-NMDA receptor mediated modulation of the tyrosine phosphatase STEP regulates glutamate induced neuronal cell death. J Neurochem. 2010;115:1350–1362. doi: 10.1111/j.1471-4159.2010.07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido R, Zúñiga A, Ullrich A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998;17:7337–7350. doi: 10.1093/emboj/17.24.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Tian D, Litvak V, Lev S. Cerebral ischemia and seizures induce tyrosine phosphorylation of PYK2 in neurons and microglial cells. J Neurosci. 2000;20:6478–6487. doi: 10.1523/JNEUROSCI.20-17-06478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Herve D, Girault JA. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaramani DV, Paul S, Zhang Y, Kurup P, Ding L, Tressler L, Allen M, Sacca R, Picciotto MR, Lombroso PJ. Knockout of striatal enriched protein tyrosine phosphatase in mice results in increased ERK1/2 phosphorylation. Synapse. 2009;63:69–81. doi: 10.1002/syn.20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel M, Li L, Harms KM, Roitbak T, Ventura PB, Rosenberg GA, Khokha R, Cunningham LA. Tissue inhibitor of metalloproteinases-3 facilitates Fas-mediated neuronal cell death following mild ischemia. Cell Death Differ. 2008;15:143–151. doi: 10.1038/sj.cdd.4402246. [DOI] [PubMed] [Google Scholar]
- Xu J, Kurup P, Bartos JA, Patriarchi T, Hell JW, Lombroso PJ. Striatal-enriched protein-tyrosine phosphatase (STEP) regulates Pyk2 kinase activity. J Biol Chem. 2012;287:20942–20956. doi: 10.1074/jbc.M112.368654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan S, Zhao H, White AJ, Minami M, Pignataro G, Yang T, Zhu X, Lan J, Xiong Z, Steiner DF, Simon RP, Zhou A. Defective neuropeptide processing and ischemic brain injury: a study on proprotein convertase 2 and its substrate neuropeptide in ischemic brains. J Cereb Blood Flow Metab. 2009;29:698–706. doi: 10.1038/jcbfm.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]